AVLAYAH- tividenofusp alfa-eknm injection, powder, lyophilized, for solution
Drug Labeling and Warnings
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AVLAYAH™ safely and effectively. See full prescribing information for AVLAYAH.
AVLAYAH (tividenofusp alfa-eknm) for injection, for intravenous use
Initial U.S. Approval: 2026WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
See full prescribing information for complete boxed warning.
- Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy. (5.1)
- Initiate AVLAYAH in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. (5.1)
- If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue AVLAYAH and immediately initiate appropriate medical treatment, including use of epinephrine. (5.1)
INDICATIONS AND USAGE
AVLAYAH is a hydrolytic lysosomal glycosaminoglycan (GAG)-specific enzyme indicated for the treatment of neurologic manifestations of Hunter syndrome (Mucopolysaccharidosis type II, MPS II) when initiated in presymptomatic or symptomatic pediatric patients weighing at least 5 kg prior to advanced neurologic impairment. (1)
This indication is approved under accelerated approval based on reduction of cerebrospinal fluid heparan sulfate observed in patients treated with AVLAYAH. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). (1)
Limitations of Use
AVLAYAH is not recommended for use in combination with other enzyme replacement therapies. (1)
DOSAGE AND ADMINISTRATION
- Administration of AVLAYAH should be supervised by a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. (2.1)
- Obtain a baseline hemoglobin value in all patients. (2.1)
- Recommended AVLAYAH maintenance dosage for pediatric patients who weigh at least 5 kg is 15 mg/kg administered once weekly as an intravenous infusion over approximately 4 hours. (2.2, 2.6)
- Initiate AVLAYAH treatment with a dose escalation regimen. (2.2)
- See the full prescribing information for dosage and administration modifications and monitoring. (2.3)
- See the full prescribing information for preparation and administration instructions. (2.4, 2.6)
DOSAGE FORMS AND STRENGTHS
For injection: 150 mg of tividenofusp alfa-eknm as a lyophilized powder in a single-dose vial for reconstitution and further dilution. (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Infusion-Associated Reactions (IARs): If a severe IAR occurs, discontinue AVLAYAH and initiate appropriate medical treatment. (5.2)
- Anemia: Obtain baseline hemoglobin levels in all patients and monitor 3 months after initiation, and as clinically indicated. Administer appropriate supportive measures for anemia based on clinical judgment. (5.3)
- Membranous Nephropathy: Monitor serum creatinine and urinary protein to creatinine ratio. If membranous nephropathy is suspected, conduct diagnostic evaluation and initiate appropriate treatment. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥20%) were IAR, upper respiratory tract infection, ear infection, pyrexia, anemia, cough, vomiting, diarrhea, rash, COVID-19, rhinorrhea, nasal congestion, fall, headache, skin abrasion, and urticaria. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Denali Therapeutics toll-free at 1-833-ONE-DNLI (1-833-663-3654) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendations Prior to AVLAYAH Treatment Initiation
2.2 Recommended Dosage
2.3 Dosage and Administration Modifications and Monitoring
2.4 Preparation Instructions
2.5 Storage Instructions for the Reconstituted and Diluted Solutions
2.6 Administration Instructions
2.7 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
5.2 Infusion-Associated Reactions
5.3 Anemia
5.4 Membranous Nephropathy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HYPERSENSITIVITY REACTIONS INCLUDING ANAPHYLAXIS
Patients treated with enzyme replacement therapies, including AVLAYAH, have experienced life-threatening hypersensitivity reactions, including anaphylaxis. Anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Initiate AVLAYAH in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment. If a severe hypersensitivity reaction (e.g., anaphylaxis) occurs, discontinue AVLAYAH and immediately initiate appropriate medical treatment, including use of epinephrine. Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
AVLAYAH is indicated for the treatment of neurologic manifestations of Hunter syndrome (Mucopolysaccharidosis type II, MPS II) when initiated in presymptomatic or symptomatic pediatric patients weighing at least 5 kg prior to advanced neurologic impairment.
This indication is approved under accelerated approval based on the reduction of cerebrospinal fluid heparan sulfate [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Limitations of Use
AVLAYAH is not recommended for use in combination with other enzyme replacement therapies for the treatment of Hunter syndrome.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendations Prior to AVLAYAH Treatment Initiation
Administer AVLAYAH under the supervision of a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis [see Warnings and Precautions (5.1)].
Initiate AVLAYAH in a healthcare setting with appropriate medical monitoring and support measures, including access to cardiopulmonary resuscitation equipment [see Warnings and Precautions (5.1)].
Consider pretreatment with antihistamines, antipyretics, and/or corticosteroids [see Warnings and Precautions (5.1, 5.2)].
Obtain a baseline hemoglobin value in all patients [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage
The recommended starting dosage of AVLAYAH for pediatric patients weighing at least 5 kg is 3 mg/kg administered once weekly via intravenous infusion.
To reduce the risk of infusion-associated reactions (IARs), follow the dose escalation regimen in Table 1 [see Warnings and Precautions (5.2)]. Administer each dosage level for at least 4 weeks before escalating to the next dosage level.
The recommended maintenance dosage of AVLAYAH for pediatric patients who weigh at least 5 kg is 15 mg/kg administered once weekly via intravenous infusion.
Table 1: Recommended AVLAYAH Dosage for Pediatric Patients Weighing ≥5 kg a Dosing Week Dosage Level a Do not escalate the dosage level if the current dosage level is not tolerated [see Dosage and Administration (2.3)]. Week 1 to Week 4 3 mg/kg once weekly Week 5 to Week 8 7.5 mg/kg once weekly Week 9 and beyond 15 mg/kg once weekly
(maintenance dosage)2.3 Dosage and Administration Modifications and Monitoring
In the event of a severe hypersensitivity reaction (e.g., anaphylaxis) or a severe IAR, discontinue AVLAYAH and immediately initiate appropriate medical treatment. Consider the risks and benefits of re-administering AVLAYAH following a severe reaction. If the decision is made to re-administer AVLAYAH, re-evaluate pre-treatment medications, slow the infusion rate, and/or reduce the AVLAYAH dose. Monitor patients closely upon re-administration of AVLAYAH [see Warnings and Precautions (5.1, 5.2)].
In the event of a mild to moderate hypersensitivity reaction or a mild to moderate IAR, temporarily hold the infusion and/or reduce the infusion rate by at least 50% from the current rate, then titrate up to the recommended infusion rate as tolerated (see Table 3) [see Warnings and Precautions (5.1, 5.2)].
If the dose has been decreased due to an adverse reaction, evaluate when it is appropriate to increase the dose and follow the recommended dose escalation regimen to achieve the maintenance dosage of 15 mg/kg once weekly [see Dosage and Administration (2.2)].
2.4 Preparation Instructions
Prepare AVLAYAH using polypropylene syringes and infusion bags composed of polyvinylchloride (PVC) or polyolefins (PO) such as polyethylene (PE) and polypropylene (PP); infusion sets composed of PVC or PE; and filter membranes composed of polyethersulfone (PES).
Use aseptic technique during preparation. Reconstitute and dilute AVLAYAH in the following manner:
Reconstitution Instructions
- 1) Determine the number of AVLAYAH vials to be reconstituted based on the patient's weight in kg and the recommended dosage [see Dosage and Administration (2.2)]. Round the number of vials up to the next whole number.
- 2) Remove the required number of AVLAYAH vials from the refrigerator and set aside for 15 to 30 minutes to allow vials to reach room temperature 20°C to 25°C (68°F to 77°F). Do not use an external heat source.
- 3) Reconstitute each vial with 5.2 mL of Sterile Water for Injection by slowly injecting the diluent onto the inside wall of each vial to avoid foaming. Do not inject forcefully or directly onto the lyophilized powder.
- 4) Gently swirl each vial to completely dissolve the lyophilized powder. Do not invert or shake the vial. Each reconstituted vial will yield a concentration of 30 mg/mL of tividenofusp alfa-eknm.
- 5) Visually inspect the reconstituted solution in the vial(s) for particulate matter and discoloration. The solution should be clear to slightly opalescent and colorless to slightly brown/yellow, and free of visible particles. Discard the reconstituted AVLAYAH solution if it is discolored, cloudy, or contains visible particulates.
Dilution Instructions
Dilute the reconstituted AVLAYAH solution with 0.9% Sodium Chloride Injection to a final concentration between 0.6 mg/mL and 15 mg/mL [see Dosage and Administration (2.6)] in an infusion bag as follows:
- 1) Determine the appropriate volume of the infusion bag based on patient weight (see Table 2) and determine the volume of reconstituted AVLAYAH solution required for the calculated dose.
- 2)
Prepare the infusion bag:
- a. Remove any airspace within the infusion bag.
- b. Withdraw a volume of 0.9% Sodium Chloride Injection from the infusion bag equivalent to the volume of AVLAYAH to be added.
- 3) Slowly withdraw the required volume of reconstituted solution from the AVLAYAH vial(s). Discard unused portion after each use; do not administer more than one dose from the vial.
- 4) Slowly inject AVLAYAH into the infusion bag of 0.9% Sodium Chloride Injection. Avoid introducing air into the infusion bag.
- 5) Gently invert the infusion bag to mix the solution. Do not shake.
Table 2: Recommended Total Infusion Volumes for AVLAYAH Based on Patient Weight and Dose Patient Weight Range AVLAYAH Dose 3 mg/kg 7.5 mg/kg 15 mg/kg Recommended Total Infusion Volumes a a Ensure the final concentration of the diluted AVLAYAH solution is between 0.6 mg/mL and 15 mg/mL. 5 kg to less than 10 kg 25 mL 25 mL or 50 mL 25 mL or 50 mL 10 kg to less than 20 kg 25 mL or 50 mL 25 mL, 50 mL, or 100 mL 25 mL, 50 mL, or 100 mL 20 kg to less than 25 kg 25 mL, 50 mL, or 100 mL 25 mL, 50 mL, or 100 mL 25 mL, 50 mL, or 100 mL 25 kg to less than 50 kg 25 mL, 50 mL, or 100 mL 25 mL, 50 mL, or 100 mL 50 mL or 100 mL 50 kg to less than 60 kg 50 mL or 100 mL 50 mL or 100 mL 100 mL 60 kg to less than 100 kg 50 mL, 100 mL, or 250 mL 50 mL, 100 mL, or 250 mL 100 mL or 250 mL 100 kg or greater 100 mL or 250 mL 100 mL or 250 mL 250 mL 2.5 Storage Instructions for the Reconstituted and Diluted Solutions
Reconstituted Solution
Do not shake. Do not freeze.
If the reconstituted AVLAYAH vials are not diluted immediately, store at controlled room temperature between 20°C to 25°C (68°F to 77°F) for up to 4 hours.
Diluted Solution
If the diluted AVLAYAH solution is not used immediately, store refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
After removal of the diluted solution from the refrigerator:
- Completely infuse within 10 hours.
- Do not store back into the refrigerator.
Discard the diluted solution if refrigerated more than 24 hours or if the diluted solution cannot be completely infused within 10 hours after removal from the refrigerator.
Do not shake. Do not freeze.
2.6 Administration Instructions
- 1) Administer AVLAYAH without delay as an intravenous infusion using only infusion sets composed of PVC or PE and filter membranes composed of PES.
- 2) If the diluted solution was refrigerated, allow solution to equilibrate to room temperature prior to infusion.
- 3) Use a dedicated infusion line equipped with a sterile, non-pyrogenic, low protein-binding, 0.2 micron, in-line filter to administer AVLAYAH.
- 4) Infuse AVLAYAH over approximately 4 hours per the recommended infusion rates in Table 3. Increase the initial infusion rate to the subsequent infusion rate every hour based on patient tolerance. Total infusion time should not exceed 8 hours.
- 5) In the absence of hypersensitivity reactions and IARs, AVLAYAH infusion rate can be gradually increased to complete the infusion in a minimum infusion duration of 3 hours based on patient tolerance.
Table 3: AVLAYAH Infusion Rate Based on Total Infusion Volume Total Infusion Volume Infusion Duration First hour Second hour Third hour to completion Infusion Rate (mL/hour) 25 mL 2.5 mL/hour 5 mL/hour 10 mL/hour 50 mL 5 mL/hour 10 mL/hour 20 mL/hour 100 mL 10 mL/hour 20 mL/hour 40 mL/hour 250 mL 25 mL/hour 50 mL/hour 100 mL/hour - 6) At the end of the infusion, flush the infusion line with 0.9% Sodium Chloride Injection using the final infusion rate that was used to administer AVLAYAH.
- 7) Do not infuse AVLAYAH in the same intravenous infusion line with other products.
Home Infusion
If a patient reaches and tolerates the maintenance AVLAYAH dosage, the patient may receive home infusion under the supervision of a healthcare provider [see Dosage and Administration (2.1, 2.7)]. The decision to have patients move to home infusion should be made after evaluation and recommendation by a healthcare provider.
In case of a missed dose or an IAR, contact a healthcare provider.
2.7 Missed Dose
If an AVLAYAH dose is missed, skip the missed dose. Do not double a dose to compensate for a missed dose. Restart AVLAYAH treatment as soon as possible, maintaining the one-week interval between infusions thereafter. Resume dosing at the last administered dosage following the recommended infusion rate [see Dosage and Administration (2.6)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions Including Anaphylaxis
Life-threatening hypersensitivity reactions, including anaphylaxis, have been reported in patients treated with enzyme replacement therapies (ERTs), including AVLAYAH [see Adverse Reactions (6)]. Symptoms of anaphylaxis that have occurred with AVLAYAH have included tachycardia, hypotension, wheezing, vomiting, hives, and lip and tongue swelling. Anaphylaxis has occurred during the early course of ERT and after extended duration of therapy.
Administer AVLAYAH under the supervision of a healthcare provider knowledgeable in the management of hypersensitivity reactions including anaphylaxis. Initiate AVLAYAH in a healthcare setting with appropriate medical monitoring and support measures including access to cardiopulmonary resuscitation equipment.
Prior to AVLAYAH administration, consider pre-treatment with antihistamines, antipyretics, and/or corticosteroids.
- If a severe hypersensitivity reaction (including anaphylaxis) occurs, discontinue AVLAYAH and immediately initiate appropriate medical treatment, including use of epinephrine. Consider the risks and benefits of re-administering AVLAYAH following a severe hypersensitivity reaction (including anaphylaxis). If the decision is made to re-administer AVLAYAH, re-evaluate pre-treatment medications (e.g., antihistamines, antipyretics, and/or corticosteroids), slow the infusion rate, and/or reduce the AVLAYAH dose. Monitor patients closely upon re-administration of AVLAYAH.
- Inform patients of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis, and to seek immediate medical care should symptoms occur.
- If a mild or moderate hypersensitivity reaction occurs, temporarily hold the infusion and/or reduce the infusion rate by at least 50% from the current rate, then titrate up to the recommended infusion rate as tolerated (see Table 3). Re-evaluate the pre-treatment medication regimen [see Dosage and Administration (2.3)].
If the dose has been decreased due to an adverse reaction, evaluate when it is appropriate to increase the dose and follow the recommended dose escalation regimen to achieve the maintenance dosage of 15 mg/kg once weekly [see Dosage and Administration (2.2)].
5.2 Infusion-Associated Reactions
Infusion-associated reactions (IARs) have been reported in patients treated with AVLAYAH [see Adverse Reactions (6.1)]. IARs are defined as adverse reactions occurring during or within 24 hours of the infusion. Symptoms of IARs observed with AVLAYAH can include (but are not limited to) chills, angioedema, hypotension, tachycardia, urticaria, vomiting, wheezing, pyrexia, flushing, erythema, rash, cough, diarrhea, abdominal pain, retching, headache, irritability, and papules. IARs have been reported more frequently in ERT-naïve patients compared to ERT-experienced patients. Cases of infusion-associated reactions occurring 2 hours or more after completion of the infusion have occurred with AVLAYAH.
Prior to AVLAYAH administration, consider pre-treatment with antihistamines, antipyretics, and/or corticosteroids to reduce the risk of IARs. IARs may still occur in patients after receiving pre-treatment. Onset of IARs was most common during the first 8 weeks of treatment with a median time to onset of approximately 2 weeks for the first IAR; IARs declined in frequency with continued use of AVLAYAH. IARs may still occur despite extended duration of AVLAYAH treatment. Appropriate medical monitoring and support measures, including cardiopulmonary resuscitation equipment, should be readily available during AVLAYAH administration.
- If a severe IAR occurs, discontinue AVLAYAH and immediately initiate appropriate medical treatment. Consider the risks and benefits of re-administering AVLAYAH following a severe IAR. If the decision is made to re-administer AVLAYAH, re-evaluate pre-treatment medications, slow the infusion rate, and/or reduce the AVLAYAH dose. Monitor patients closely upon re-administration of AVLAYAH.
- If a mild or moderate IAR occurs, temporarily hold the infusion, and/or reduce the infusion rate by at least 50% from the current rate, then titrate up to the recommended infusion rate as tolerated.
If the dose has been decreased due to an adverse reaction, evaluate when it is appropriate to increase the dose and follow the recommended dose escalation regimen to achieve the maintenance dosage of 15 mg/kg once weekly [see Dosage and Administration (2.2)].
Patients with Hunter syndrome may have compromised cardiac and respiratory function which may predispose them to a higher risk of severe complications from IARs. Closely monitor patients with compromised cardiac and respiratory function following AVLAYAH administration.
5.3 Anemia
Anemia has been reported in patients treated with AVLAYAH [see Adverse Reactions (6.1)].
The incidence of anemia after initiation of AVLAYAH was higher in patients with pre-existing anemia compared to those without pre-existing anemia. Reductions in hemoglobin levels were generally observed by Week 13, though the occurrence was observed up to one year in some patients. Overall, the incidence and severity of anemia decreased over time, with the majority of patients recovering by Week 24. Anemia did not result in treatment discontinuation; management may include supplementation with iron.
Obtain hemoglobin levels prior to initiating AVLAYAH, at 3 months after initiation, and periodically thereafter as clinically indicated. Administer appropriate supportive measures for anemia based on clinical judgment.
5.4 Membranous Nephropathy
A case of steroid-refractory membranous nephropathy with immune complex deposits in the kidney was reported in an AVLAYAH-treated patient [see Adverse Reactions (6.1)]. Monitor serum creatinine and urinary protein to creatinine ratio. If membranous nephropathy is suspected, conduct diagnostic evaluation and initiate appropriate treatment. Consider risks and benefits of continuing AVLAYAH in patients who develop membranous nephropathy.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity Reactions Including Anaphylaxis [see Warnings and Precautions (5.1)]
- Infusion-Associated Reactions [see Warnings and Precautions (5.2)]
- Anemia [see Warnings and Precautions (5.3)]
- Membranous Nephropathy [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of AVLAYAH was evaluated in male pediatric patients with Hunter syndrome in Trial 1 [see Clinical Studies (14)]. A total of 47 male patients (age range: 3 months to 13 years) received intravenous AVLAYAH at 3 mg/kg to 30 mg/kg (0.2 to 2 times the approved recommended maintenance dose) weekly, and the majority of patients received 15 mg/kg intravenously weekly after Week 24. The median (minimum, maximum) duration of exposure was 117 (19, 219) weeks.
In Trial 1, the most common adverse reactions (≥20%) reported in AVLAYAH-treated patients were infusion-associated reaction (IAR), upper respiratory tract infection, ear infection, pyrexia, anemia, cough, vomiting, diarrhea, rash, COVID-19, rhinorrhea, nasal congestion, fall, headache, skin abrasion, and urticaria.
Dose interruptions of AVLAYAH due to an adverse reaction occurred in 91% of patients. The most frequently reported adverse reaction leading to dose interruption was IAR (31 [66%] patients). Other frequently reported adverse reactions leading to dose interruption were COVID-19 (18 [38%] patients), pyrexia (16 [34%]), upper respiratory tract infection (16 [34%]), nasal congestion (6 [13%]), and vomiting (6 [13%]). Dose interruption included skipped infusions due to an adverse reaction as well as temporary infusion pauses with subsequent completion during the same visit.
Dose reductions of AVLAYAH due to adverse reactions occurred in 57% of patients; the majority of these reactions were IARs.
In Trial 1, one (2%) AVLAYAH-treated patient experienced anaphylaxis, which occurred in the first month of treatment.
Table 4 summarizes adverse reactions that occurred in >15% of AVLAYAH-treated pediatric patients with Hunter syndrome.
Table 4: Adverse Reactions That Occurred in >15% in AVLAYAH-treated Pediatric Patients With Hunter Syndrome (Trial 1) Adverse Reaction Any Severity
N (%)
(N = 47)a Infusion-associated reaction includes infusion-related reaction. b Ear infection includes ear infection, otitis media, otitis media acute, otitis externa. c Anemia includes anemia, iron deficiency anemia, and decreased hemoglobin. Infusion-associated reaction a 41 (87%) Upper respiratory tract infection 28 (60%) Ear infection b 26 (55%) Pyrexia 26 (55%) Anemia c 24 (51%) Cough 22 (47%) Vomiting 20 (43%) Diarrhea 19 (40%) Rash 19 (40%) COVID-19 18 (38%) Rhinorrhea 18 (38%) Nasal congestion 17 (36%) Fall 11 (23%) Headache 11 (23%) Skin abrasion 11 (23%) Urticaria 10 (21%) Constipation 8 (17%) Contusion 8 (17%) Gastroenteritis 8 (17%) Infusion site extravasation 8 (17%) Insomnia 8 (17%) Neutropenia 8 (17%) Description of Selected Adverse Reactions
Infusion-Associated Reaction
Three (6%) AVLAYAH-treated patients experienced severe IARs. One patient permanently discontinued treatment due to an IAR.
Anemia
Two (4%) AVLAYAH-treated patients experienced severe anemia (defined as hemoglobin <8 g/dL) prior to Week 24. One (2%) AVLAYAH-treated patient, aged 0.5 years, experienced moderate anemia (hemoglobin 9.2 g/dL), which was considered serious due to the patient's age.
Membranous Nephropathy
A case of biopsy-confirmed, steroid-refractory membranous nephropathy with immune complex deposits in the kidney was reported in an AVLAYAH-treated patient.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of AVLAYAH during pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Animal studies to evaluate the potential for embryofetal developmental toxicity and pre- and postnatal developmental toxicity of tividenofusp alfa-eknm have not been conducted.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, and other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of tividenofusp alfa-eknm in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for AVLAYAH and any potential adverse effects on the breastfed infant from AVLAYAH or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of AVLAYAH have been established under accelerated approval for the treatment of neurologic manifestations of Hunter syndrome (Mucopolysaccharidosis type II, MPS II) when initiated in presymptomatic or symptomatic pediatric patients weighing at least 5 kg prior to advanced neurologic impairment and the information on this use is discussed throughout the labeling [see Indications and Usage (1)]. The safety and effectiveness of AVLAYAH have not been established in pediatric patients weighing less than 5 kg.
-
11 DESCRIPTION
Tividenofusp alfa-eknm is a fusion protein consisting of the hydrolytic lysosomal glycosaminoglycan (GAG)-specific enzyme iduronate-2-sulfatase (IDS) fused to the N-terminus of an immunoglobulin G1 (IgG1) fragment, crystallizable (Fc). It is produced by recombinant DNA technology in Chinese Hamster Ovary (CHO) cells. The approximate molecular weight of tividenofusp alfa-eknm is 110 kDa.
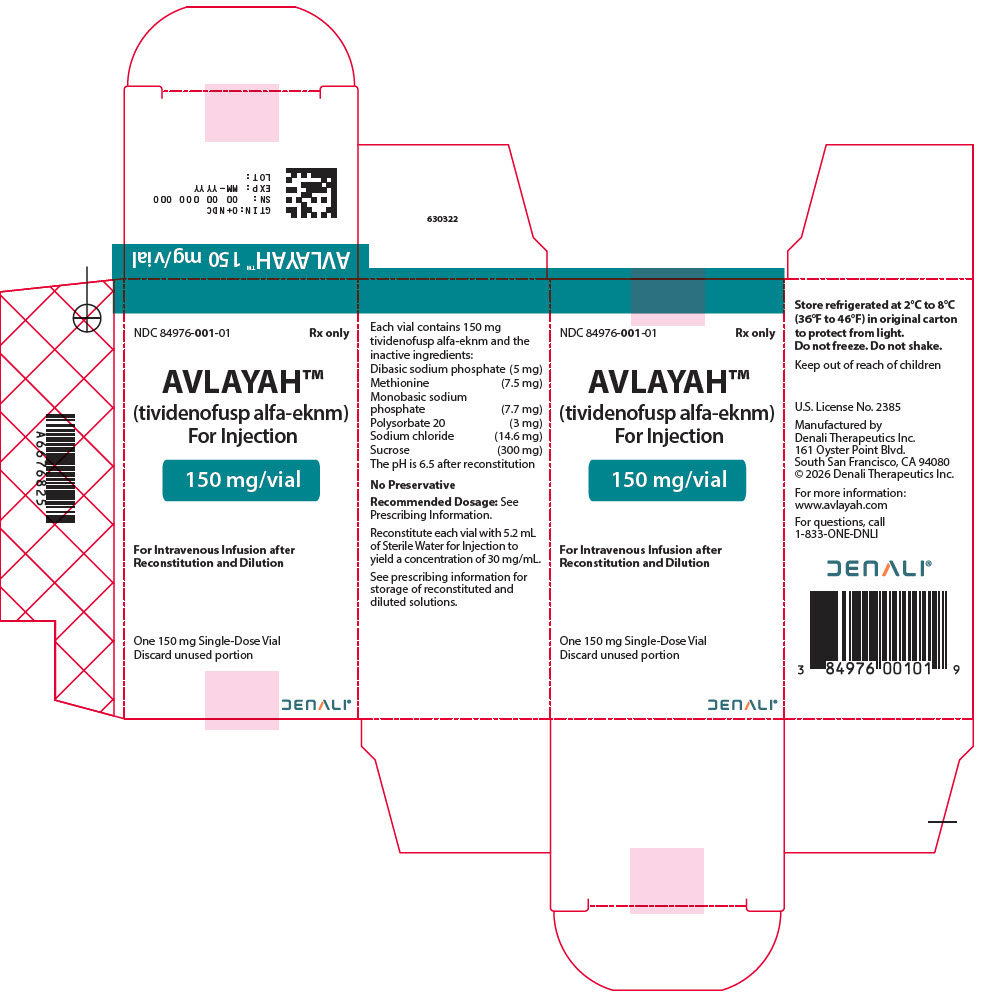
AVLAYAH (tividenofusp alfa-eknm) for injection is a sterile, preservative-free, white to off-white lyophilized powder with a cake-like appearance for intravenous infusion after reconstitution and dilution. Each single-dose vial contains 150 mg tividenofusp alfa-eknm, and the inactive ingredients dibasic sodium phosphate (5 mg), methionine (7.5 mg), monobasic sodium phosphate (7.7 mg), polysorbate 20 (3 mg), sodium chloride (14.6 mg), and sucrose (300 mg). The pH is 6.5 after reconstitution.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Hunter syndrome is an inherited X-linked recessive lysosomal storage disease caused by a deficiency of iduronate-2-sulfatase (IDS), a lysosomal enzyme, that degrades heparan sulfate (HS) and dermatan sulfate (DS), the two primary glycosominoglycans (GAGs) in the lysosome. Insufficiency or absence of IDS leads to accumulation of GAGs, including HS and DS, and subsequent lysosome dysfunction in multiple organs and tissues, including the central nervous system (CNS).
Tividenofusp alfa-eknm provides an exogenous source of IDS. The fragment, crystallizable (Fc) component of tividenofusp alfa-eknm binds to the apical domain of the transferrin receptor (TfR) and delivers IDS to peripheral tissues and to the CNS through receptor-mediated transcytosis across the blood-brain barrier. Tividenofusp alfa-eknm is internalized via binding to the mannose-6-phosphate receptor on the cell surface and transported into lysosomes where it is thought to exert enzymatic activity and reduce accumulated GAGs. In addition, since TfR is ubiquitously expressed, it is expected that the interaction of tividenofusp alfa-eknm and TfR will contribute to its uptake into cells in the brain and peripheral tissues.
12.2 Pharmacodynamics
In clinical studies with AVLAYAH, the relative concentrations of HS or DS in human cerebrospinal fluid (CSF) and urine were estimated based on an assessment of selected disaccharides following enzymatic digestion of HS or DS. It is not possible to directly quantify intact HS or DS concentrations in human CSF or urine using currently available bioanalytical methods. Differences in bioanalytical methods preclude meaningful comparison of the pharmacodynamic results based on HS or DS concentrations in clinical studies with AVLAYAH with the results in other clinical studies.
In Trial 1, reductions in CSF HS from baseline were observed in AVLAYAH-treated pediatric patients with Hunter syndrome [see Clinical Studies (14)]. Reductions in urine HS (86%), urine DS (91%), and total urine GAGs (57%) from baseline were observed at Week 24 in AVLAYAH-treated pediatric patients with Hunter syndrome. At baseline, 2 of 47 (4%) patients had total urine GAG levels below the upper limit of normal (ULN). At Week 24, 26 of 38 (68%) patients had total urine GAG levels below the ULN. The relationship between changes in CSF HS, urine HS, urine DS, and total urine GAG levels to clinical response in patients with Hunter syndrome has not been established.
In Trial 1, higher serum tividenofusp alfa-eknm concentrations appeared to be associated with greater reductions of CSF HS and urine HS concentrations from baseline, with maximum effect achieved at 15 mg/kg of AVLAYAH once weekly (the recommended maintenance dosage).
The time to maximum effect on pharmacodynamic response and the exposure-response relationship for the safety and effectiveness of tividenofusp alfa-eknm have not been fully characterized.
12.3 Pharmacokinetics
The pharmacokinetics of tividenofusp alfa-eknm were evaluated in pediatric patients with Hunter syndrome aged 3 months to 13 years (age at baseline).
The maximum serum concentration (Cmax) increased proportionally with dose, while the area under the serum concentration-time curve (AUCtau) increased in a greater-than-dose-proportional manner across the dose range of 3 mg/kg to 30 mg/kg (0.2 to 2 times the approved recommended maintenance dosage). Table 5 shows the Cmax and AUCtau of tividenofusp alfa-eknm following the recommended starting dosage and recommended maintenance dosage [see Dosage and Administration (2.2)].
Table 5: Cmax and AUCtau of Tividenofusp Alfa-eknm in Pediatric Patients With Hunter Syndrome Pharmacokinetic Parameter Week 1 (3 mg/kg weekly) Week 24 (15 mg/kg weekly) Geometric Mean (Range) Geometric Mean (Range) Abbreviations: AUCtau = area under the serum concentration-time curve from 0 to 168 hours after the start of infusion; Cmax = maximum serum concentration. Cmax (mcg/mL) 33.1 (19.9 – 50.3) 204 (19.5 – 615) AUCtau (h∙mcg/mL) 277 (82.4 – 446) 3,000 (839 – 12,100) Distribution
The geometric mean (range) volume of distribution of tividenofusp alfa-eknm was 2.7 (1.4 to 9.6) L.
Elimination
Tividenofusp alfa-eknm is cleared via linear and nonlinear mechanisms and the total clearance was increased in the presence of anti-tividenofusp alfa-eknm antibodies [see Clinical Pharmacology (12.6)]. The geometric mean (range) total clearance of tividenofusp alfa-eknm was 0.14 (0.05 to 0.45) L/h following 15 mg/kg weekly dosing of AVLAYAH at Week 24.
Patients are predicted to have a 97% reduction from Cmax in tividenofusp alfa-eknm concentrations at a median time of 40 hours (5th to 95th percentile: 18.6 to 74.1 hours) after the end of the first 3 mg/kg AVLAYAH infusion at Week 1 and 41 hours (5th to 95th percentile: 23.3 to 100 hours) after the end of the first 15 mg/kg AVLAYAH infusion at Week 9.
Metabolism
Tividenofusp alfa-eknm is expected to be metabolized into small peptides via catabolic pathways.
Specific Populations
Following the approved recommended weight-based dosage, no clinically significant differences in tividenofusp alfa-eknm serum Cmax or AUCtau were observed based on age (3 months to 16 years) or body weight (7 kg to 80 kg).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADAs) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADAs in the studies described below with the incidence of ADAs in other studies, including those of AVLAYAH or of other tividenofusp alfa products.
In Trial 1 [see Clinical Studies (14)], anti-tividenofusp alfa-eknm antibodies (referred to as ADAs) were detected in 100% (47/47) of AVLAYAH-treated patients with Hunter syndrome following 19 to 219 weeks of treatment. In Trial 1, neutralizing antibodies (NAbs) that inhibit enzyme activity of tividenofusp alfa-eknm were detected in 87% (41/47) of AVLAYAH-treated patients with Hunter syndrome. NAbs that inhibit cellular uptake of tividenofusp alfa-eknm were not characterized in this trial.
Among the 10 enzyme replacement therapy (ERT)-experienced and 13 ERT-naïve patients with Hunter syndrome who were ADA negative at baseline, all of the patients became ADA positive following AVLAYAH treatment.
ADA responses in both ERT-experienced and ERT-naïve patients with Hunter syndrome were more frequently directed against the IDS component of tividenofusp alfa-eknm than the Fc component. ADA titers peaked at approximately Week 13, remained elevated through Week 24, and declined thereafter in the majority of patients.
Antibodies against other IDS ERTs are expected to be cross-reactive to tividenofusp alfa-eknm.
Prevalence for ADAs and NAbs that inhibited enzyme activity in AVLAYAH-treated pediatric patients with Hunter syndrome are summarized in Table 6.
Table 6: ADA and NAb Prevalence at Baseline and Post-AVLAYAH Treatment in Pediatric Patients With Hunter Syndrome ERT-Experienced a
(N = 32)ERT-Naïve b
(N = 15)Abbreviations: ADA = anti-drug antibody (anti-tividenofusp alfa-eknm antibody); ERT = enzyme replacement therapy; NAb = neutralizing antibody.
Data are presented as n/N (%). Prevalence reflects ADA detected at baseline or post-baseline.a ERT-experienced was defined as patients who had ≥4 months of ERT treatment at any time prior to AVLAYAH treatment initiation. b ERT-naïve was defined as patients who had <4 months of continuous ERT treatment at any time prior to AVLAYAH treatment initiation. ADA Baseline 22/32 (69%) 2/15 (13%) Following AVLAYAH Treatment 32/32 (100%) 15/15 (100%) NAb Baseline 10/32 (31%) 0/15 (0%) Following AVLAYAH Treatment 26/32 (81%) 15/15 (100%) Anti-Drug Antibody Effects on Pharmacokinetics, Pharmacodynamics, Safety, and Efficacy
Development of ADAs was associated with reduced serum tividenofusp alfa-eknm concentrations. AVLAYAH-treated patients who developed higher ADA titers had greater reductions in serum tividenofusp alfa-eknm concentrations. The observed ADA-associated pharmacokinetic changes did not result in significant effects on the reduction in CSF HS or urine HS at the recommended dosage [see Dosage and Administration (2.2)].
The effect of ADAs on the safety of AVLAYAH in patients with Hunter syndrome has not been fully characterized.
The effect of ADAs on the effectiveness of AVLAYAH in the treatment of Hunter syndrome is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Animal studies to evaluate the carcinogenic potential of tividenofusp alfa-eknm have not been conducted.
Mutagenesis
Studies to evaluate the mutagenic potential of tividenofusp alfa-eknm have not been conducted.
Impairment of Fertility
In a fertility and embryonic development study in transgenic mice, twice weekly intravenous tividenofusp alfa-eknm doses were administered to females for two weeks prior to mating and through day 4 of gestation, and to males two weeks prior to mating. No adverse effects on fertility parameters were observed in either female or male transgenic mice at exposures approximately 12-fold greater than those observed in patients at the maintenance dosage level of 15 mg/kg (based on AUC).
-
14 CLINICAL STUDIES
Trial 1 (NCT04251026) was a Phase 1/2 multi-center, international, multi-cohort, single-arm, open-label trial of AVLAYAH in 47 pediatric patients with Hunter syndrome including 44 patients with neuronopathic Hunter syndrome and 3 patients with non-neuronopathic Hunter syndrome. Of the 47 patients enrolled, 46 patients completed part 1 of the study (up to Week 24) and 1 patient discontinued the study due to an adverse reaction prior to Week 24. In part 1 of Trial 1, patients received a starting dosage of AVLAYAH ranging from 3 mg/kg to 15 mg/kg weekly and a maximum dosage ranging from 3 mg/kg to 30 mg/kg weekly (0.2 to 2 times the approved recommended maintenance dosage) [see Dosage and Administration (2.2)]. Patients who received 30 mg/kg weekly dosage (2 times the approved recommended maintenance dosage) did not show additional reductions from baseline in cerebrospinal fluid (CSF) or urine heparan sulfate (HS) concentration compared to patients who received 15 mg/kg weekly [see Clinical Pharmacology (12.2)]. The most common AVLAYAH dosage (57% of the patients) in Trial 1 was 15 mg/kg administered once weekly.
All 47 patients in Trial 1 were male, with a baseline median age of 5 years (range: 3 months to 13 years of age). The patient population consisted of 27 White (57%), 4 Black or African American (9%), 4 Asian (9%), 3 who were more than one race (6%), 1 of another race (2%), and 8 of an unknown race (17%). Ethnicity consisted of 7 patients who were Hispanic or Latino (15%), 38 who were Not Hispanic or Latino (81%), and 2 of an unknown ethnicity (4%). Of the 47 patients, 15 patients were enzyme replacement therapy (ERT)-naïve, and 32 patients were ERT-experienced, 2 of whom had a history of prior treatment for Hunter syndrome with hematopoietic stem cell transplantation (HSCT), and 2 of whom had a history of prior treatment for Hunter syndrome with gene therapy. The median duration of previous ERT treatment was 26 months (range: 1 to 134 months).
In Trial 1, treatment with AVLAYAH resulted in a significant reduction of CSF HS. For the 44 patients who had measurements at Week 24, the CSF HS mean percent reduction from baseline was 91% (95% CI: 89%, 92%); the minimum and maximum percent change in CSF HS from baseline were 72% and 98%, respectively. At baseline, 0% (0 of 47) of patients had CSF HS levels below the upper limit of normal (ULN). At Week 24, 93% (41 of 44) of AVLAYAH-treated patients had CSF HS levels below the ULN.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
AVLAYAH (tividenofusp alfa-eknm) for injection is supplied as a sterile, preservative-free, white to off-white lyophilized powder with a cake-like appearance in a single-dose vial. Each vial contains 150 mg of tividenofusp alfa-eknm. AVLAYAH is available as one carton containing a 150 mg single-dose vial (NDC: 84976-001-01).
-
17 PATIENT COUNSELING INFORMATION
Hypersensitivity Reactions Including Anaphylaxis, and Infusion-Associated Reactions
Advise the patient and/or caregiver that life-threatening hypersensitivity reactions, including anaphylaxis, and infusion-associated reactions (IARs) may occur with AVLAYAH treatment.
Advise the patient and/or caregiver that anaphylaxis has occurred during the early course of enzyme replacement therapy and after extended duration of therapy.
Inform the patient and/or caregiver of the symptoms of life-threatening hypersensitivity reactions, including anaphylaxis, and IARs and to seek immediate medical care should symptoms occur [see Warnings and Precautions (5.1, 5.2)].
Anemia
Inform the patient and caregiver that anemia may occur during AVLAYAH treatment. Advise the patient and/or caregiver of the symptoms of anemia (e.g., fatigue, pallor). Instruct the patient and/or caregiver to contact their healthcare provider if those symptoms occur [see Warnings and Precautions (5.3)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 150 mg Vial Label
Rx only
NDC: 84976-001-01AVLAYAH™
(tividenofusp alfa-eknm)
For Injection150 mg/vial
For Intravenous Infusion after
Reconstitution and Dilution
Single-Dose Vial
Discard unused portion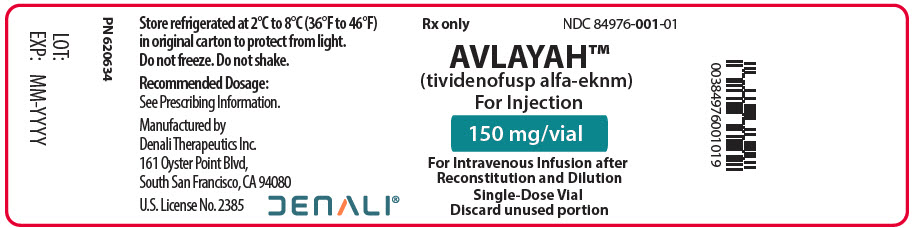
-
PRINCIPAL DISPLAY PANEL - 150 mg Vial Carton
NDC: 84976-001-01
Rx onlyAVLAYAH™
(tividenofusp alfa-eknm)
For Injection150 mg/vial
For Intravenous Infusion after
Reconstitution and DilutionOne 150 mg Single-Dose Vial
Discard unused portionDEnALI®
-
INGREDIENTS AND APPEARANCE
AVLAYAH
tividenofusp alfa-eknm injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 84976-001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIVIDENOFUSP ALFA (UNII: QLD7UJN8CF) (TIVIDENOFUSP ALFA - UNII:QLD7UJN8CF) TIVIDENOFUSP ALFA 150 mg in 5 mL Inactive Ingredients Ingredient Name Strength METHIONINE (UNII: AE28F7PNPL) 7.5 mg in 5 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 3 mg in 5 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 14.6 mg in 5 mL SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) 9.5 mg in 5 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 8.9 mg in 5 mL SUCROSE (UNII: C151H8M554) 300 mg in 5 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 84976-001-01 1 in 1 CARTON 04/06/2026 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761485 04/06/2026 Labeler - Denali Therapeutics Inc. (079861913)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.