These highlights do not include all the information needed to use ORTIKOS™ safely and effectively. See full prescribing information for ORTIKOS. ORTIKOS™ (budesonide) extended-release capsules, for oral use Initial U.S. Approval: 1997
Ortikos by
Drug Labeling and Warnings
Ortikos by is a Prescription medication manufactured, distributed, or labeled by FERRING PHARMACEUTICALS INC., Sun Pharmaceutical Industries, Inc., Sun Pharmaceutical Industries Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
ORTIKOS - budesonide capsule
FERRING PHARMACEUTICALS INC.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ORTIKOS™ safely and effectively. See full prescribing information for ORTIKOS.
ORTIKOS™ (budesonide) extended-release capsules, for oral use Initial U.S. Approval: 1997 INDICATIONS AND USAGEORTIKOS is a corticosteroid indicated for: DOSAGE AND ADMINISTRATIONAdministration Instructions (2.1):
Recommended Dosage: Mild to moderate active Crohn’s disease (2.2):
Maintenance of clinical remission of mild to moderate Crohn’s disease (2.3):
DOSAGE FORMS AND STRENGTHSExtended-Release Capsules: 6 mg and 9 mg (3) CONTRAINDICATIONSHypersensitivity to budesonide or any of the ingredients in ORTIKOS. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (≥ 5%) in adults are: headache, respiratory infection, nausea, back pain, dyspepsia, dizziness, abdominal pain, flatulence, vomiting, fatigue, and pain. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-818-4555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSPregnancy: Based on animal data, may cause fetal harm. (8.1) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 1/2020 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
- Take ORTIKOS once daily in the morning.
- Swallow ORTIKOS whole. Do not chew or crush.
- Avoid consumption of grapefruit juice for the duration of therapy with ORTIKOS [see Drug Interactions (7.1)].
2.2 Treatment of Mild to Moderate Active Crohn's Disease
The recommended dosage of ORTIKOS is:
Adults: 9 mg orally once daily for up to 8 weeks. Repeated 8 week courses of ORTIKOS can be given for recurring episodes of active disease.
Pediatric patients 8 to 17 years who weigh more than 25 kg: 9 mg orally once daily for up to 8 weeks, followed by 6 mg once daily for 2 weeks.
2.3 Maintenance of Clinical Remission of Mild to Moderate Crohn's Disease
The recommended dosage in adults, following an 8 week course(s) of treatment for active disease and once the patient’s symptoms are controlled (CDAI less than 150), is ORTIKOS 6 mg orally once daily for maintenance of clinical remission up to 3 months. If symptom control is still maintained at 3 months an attempt to taper to complete cessation is recommended. Continued treatment with ORTIKOS 6 mg for more than 3 months has not been shown to provide substantial clinical benefit.
Patients with mild to moderate active Crohn’s disease involving the ileum and/or ascending colon have been switched from oral prednisolone to ORTIKOS with no reported episodes of adrenal insufficiency. Since prednisolone should not be stopped abruptly, tapering should begin concomitantly with initiating ORTIKOS treatment.
3 DOSAGE FORMS AND STRENGTHS
Extended-Release Capsules:
- 6 mg: hard gelatin capsules with light grey colored cap and pink colored body imprinted with “061” on cap and body in black ink containing white to off-white pellets.
- 9 mg: hard gelatin capsules with pink colored cap and pink colored body imprinted with “062” on cap and body in black ink containing white to off-white pellets.
4 CONTRAINDICATIONS
ORTIKOS is contraindicated in patients with hypersensitivity to budesonide or any of the ingredients of the capsules. Serious hypersensitivity reactions, including anaphylaxis have occurred [see Adverse Reactions (6.2)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypercorticism and Adrenal Axis Suppression
When corticosteroids are used chronically, systemic effects such as hypercorticism and adrenal axis suppression may occur. Corticosteroids can reduce the response of the hypothalamus-pituitary-adrenal (HPA) axis to stress. In situations where patients are subject to surgery or other stress situations, supplementation with a systemic corticosteroid is recommended. Since ORTIKOS contains a corticosteroid, general warnings concerning corticosteroids should be followed [see Warnings and Precautions (5.2), (5.3), (5.4)].
Pediatric patients with Crohn’s disease have a slightly higher systemic exposure of budesonide and increased cortisol suppression than adults with Crohn’s disease [see Use in Specific Populations (8.4), Clinical Pharmacology (12.2)].
Patients with moderate to severe hepatic impairment (Child-Pugh Class B and C respectively) could be at an increased risk of hypercorticism and adrenal axis suppression due to an increased systemic exposure of oral budesonide. Avoid use of ORTIKOS in patients with moderate and severe hepatic impairment. [see Use in Specific Populations (8.6)].
5.2 Symptoms of Steroid Withdrawal in Patients Transferred from Other Systemic Corticosteroids
Monitor patients who are transferred from corticosteroid treatment with high systemic effects to corticosteroids with lower systemic availability, such as budesonide, since symptoms attributed to withdrawal of steroid therapy, including those of acute adrenal axis suppression or benign intracranial hypertension, may develop. Adrenocortical function monitoring may be required in these patients and the dose of corticosteroid treatment with high systemic effects should be reduced cautiously.
Replacement of systemic corticosteroids with budesonide may unmask allergies (e.g., rhinitis and eczema), which were previously controlled by the systemic drug.
5.3 Increased Risk of Infection
Patients who are on drugs that suppress the immune system are more susceptible to infection than healthy individuals. Chicken pox and measles, for example, can have a more serious or even fatal course in susceptible patients or patients on immunosuppressant doses of corticosteroids. In patients who have not had these diseases, particular care should be taken to avoid exposure.
How the dose, route and duration of corticosteroid administration affect the risk of developing a disseminated infection is not known. The contribution of the underlying disease and/or prior corticosteroid treatment to the risk is also not known. If exposed, therapy with varicella zoster immune globulin (VZIG) or pooled intravenous immunoglobulin (IVIG), as appropriate, may be indicated. If exposed to measles, prophylaxis with pooled intramuscular immunoglobulin (IG) may be indicated. (See prescribing information for VZIG and IG). If chicken pox develops, treatment with antiviral agents may be considered.
Corticosteroids should be used with caution, if at all, in patients with active or quiescent tuberculosis infection, untreated fungal, bacterial, systemic viral or parasitic infections, or ocular herpes simplex.
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hypercorticism and adrenal axis suppression [see Warnings and Precautions (5.1)]
- Symptoms of steroid withdrawal in those patients transferred from other systemic corticosteroids [see Warnings and Precautions (5.2)]
- Increased risk of infection [see Warnings and Precautions (5.3)]
- Other corticosteroid effects [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ORTIKOS has been established from adequate and well-controlled studies of another oral budesonide product [see Clinical Studies (14)]. Below is a display of the adverse reactions of budesonide in these adequate and well-controlled studies.
Adults
The data described below reflect exposure to budesonide in 520 patients with Crohn’s disease, including 520 exposed to 9 mg per day (total daily dose) for 8 weeks and 145 exposed to 6 mg per day for one year in placebo controlled clinical trials. Of the 520 patients, 38% were males and the age range was 17 to 74 years.
Treatment of Mild to Moderate Active Crohn’s Disease
The safety of budesonide was evaluated in 651 adult patients in five clinical trials of 8 weeks duration in patients with active mild to moderate Crohn’s disease. The most common adverse reactions, occurring in greater than or equal to 5% of the patients, are listed in Table 1.
Table 1: Common Adverse Reactions1 in 8-Week Treatment Clinical Trials
|
Adverse Reaction |
Budesonide 9 mg n=520 |
Placebo n=107 |
Prednisolone2 40 mg n=145 |
Comparator3 n=88 |
|
Number (%) |
Number (%) |
Number (%) |
Number (%) |
|
|
Headache |
107(21) |
19(18) |
31(21) |
11(13) |
|
Respiratory Infection |
55 (11) |
7(7) |
20(14) |
5(6) |
|
Nausea |
57(11) |
10(9) |
18(12) |
7(8) |
|
Back Pain |
36(7) |
10(9) |
17(12) |
5(6) |
|
Dyspepsia |
31(6) |
4(4) |
17(12) |
3(3) |
|
Dizziness |
38(7) |
5(5) |
18(12) |
5(6) |
|
Abdominal Pain |
32(6) |
18(17) |
6(4) |
10(11) |
|
Flatulence |
30(6) |
6(6) |
12(8) |
5(6) |
|
Vomiting |
29(6) |
6(6) |
6(4) |
6(7) |
|
Fatigue |
25(5) |
8(7) |
11(8) |
0(0) |
|
Pain |
24(5) |
8(7) |
17(12) |
2(2) |
1 Occurring in greater than or equal to 5% of the patients in any treated group.
2 Prednisolone tapering scheme: either 40 mg in week 1 to 2, thereafter tapering with 5 mg per week; or 40 mg in week 1 to 2, 30 mg in week 3 to 4, thereafter tapering with 5 mg per week.
3 This drug is not approved for the treatment of Crohn’s disease in the United States.
The incidence of signs and symptoms of hypercorticism reported by active questioning of patients in 4 of the 5 short-term clinical trials are displayed in Table 2.
Table 2: Summary and Incidence of Signs/Symptoms of Hypercorticism in 8-Week Treatment Clinical Trials
| Signs/Symptom
| Budesonide 9 mg
n=427 | Placebo
n=107 | Prednisolone1 40 mg
n=145 |
| Number (%)
| Number (%)
| Number (%)
|
|
|
Total | 145 (34%) | 29 (27%) | 69 (48%) |
|
Acne | 63(15) | 14(13) | 33(23)2
|
|
Bruising Easily | 63(15) | 12(11) | 13(9) |
|
Moon Face | 46(11) | 4(4) | 53(37)2
|
|
Swollen Ankles | 32(7) | 6(6) | 13(9) |
|
Hirsutism3 | 22(5) | 2(2) | 5(3) |
|
Buffalo Hump | 6(1) | 2(2) | 5(3) |
|
Skin Striae | 4(1) | 2(2) | 0(0) |
1 Prednisolone tapering scheme: either 40 mg in week 1-2, thereafter tapering with 5 mg/week; or 40 mg in week 1 to 2, 30 mg in week 3 to 4, thereafter tapering with 5 mg/week.
2 Statistically significantly different from budesonide 9 mg
3 Including hair growth increased, local and hair growth increased, general
Maintenance of Clinical Remission of Mild to Moderate Crohn’s Disease
The safety of budesonide was evaluated in 233 adult patients in four long-term clinical trials (52 weeks) of maintenance of clinical remission in patients with mild to moderate Crohn’s disease. A total of 145 patients were treated with budesonide 6 mg once daily.
The adverse reaction profile of budesonide 6 mg once daily in maintenance of Crohn’s disease was similar to that of short-term treatment with budesonide 9 mg once daily in active Crohn’s disease. In the long-term clinical trials, the following adverse reactions occurred in greater than or equal to 5% and are not listed in Table 1: diarrhea (10%); sinusitis (8%); infection viral (6%); and arthralgia (5%).
Signs/symptoms of hypercorticism reported by active questioning of patients in the long-term maintenance clinical trials are displayed in Table 3.
Table 3: Summary and Incidence of Signs/Symptoms of Hypercorticism in Long-Term Clinical Trials
| Signs/Symptom
| Budesonide
6 mg n=145 | Placebo
n=143 |
|
| Number (%)
| Number (%)
|
| Bruising Easily | 15(10) | 5(4) |
| Acne | 14(10) | 3(2) |
| Moon Face | 6(4) | 0 |
| Hirsutism | 5(3) | 1(1) |
| Swollen Ankles | 3(2) | 3(2) |
| Buffalo Hump | 1(1) | 0 |
| Skin Striae | 0 | 0 |
The incidence of signs/symptoms of hypercorticism as described above in long-term maintenance clinical trials was similar to that seen in the short-term treatment clinical trials.
Less Common Adverse Reactions in Treatment and Maintenance Clinical Trials
Less common adverse reactions (less than 5%), occurring in adult patients treated with budesonide 9 mg (total daily dose) in short-term treatment clinical studies and/or budesonide 6 mg (total daily dose) in long-term maintenance clinical trials, with an incidence are listed below by system organ class:
Cardiac disorders: palpitation, tachycardia
Eye disorders: eye abnormality, vision abnormal
General disorders and administration site conditions: asthenia, chest pain, dependent edema, face edema, flu-like disorder, malaise, fever
Gastrointestinal disorders: anus disorder, enteritis, epigastric pain, gastrointestinal fistula, glossitis, hemorrhoids, intestinal obstruction, tongue edema, tooth disorder
Infections and infestations: Ear infection -not otherwise specified, bronchitis, abscess, rhinitis, urinary tract infection, thrush
Investigations: weight increased
Metabolism and nutrition disorders: appetite increased
Musculoskeletal and connective tissue disorders: arthritis, cramps, myalgia
Nervous system disorders: hyperkinesia, paresthesia, tremor, vertigo, somnolence, amnesia
Psychiatric disorders: agitation, confusion, insomnia, nervousness, sleep disorder
Renal and urinary disorders: dysuria, micturition frequency, nocturia
Reproductive system and breast disorders: intermenstrual bleeding, menstrual disorder
Respiratory, thoracic and mediastinal disorders: dyspnea, pharynx disorder
Skin and subcutaneous tissue disorders: alopecia, dermatitis, eczema, skin disorder, sweating increased, purpura
Vascular disorders: flushing, hypertension
Bone Mineral Density
A randomized, open, parallel-group multicenter safety clinical trial specifically compared the effect of budesonide (less than 9 mg per day) and prednisolone (less than 40 mg per day) on bone mineral density over 2 years when used at doses adjusted to disease severity. Bone mineral density decreased significantly less with budesonide than with prednisolone in steroid-naïve patients, whereas no difference could be detected between treatment groups for steroid-dependent patients and previous steroid users. The incidence of symptoms associated with hypercorticism was significantly higher with prednisolone treatment.
Clinical Laboratory Test Findings
The following potentially clinically significant laboratory changes in clinical trials, irrespective of relationship to budesonide, were reported in greater than or equal to 1% of patients: hypokalemia, leukocytosis, anemia, hematuria, pyuria, erythrocyte sedimentation rate increased, alkaline phosphatase increased, atypical neutrophils, c-reactive protein increased and adrenal insufficiency.
Pediatric Patients --Treatment of Mild to Moderate Active Crohn’s Disease
Adverse reactions reported in pediatric patients 8 to 17 years of age, who weigh more than 25 kg, were similar to those reactions described above in adult patients.
6.2 Postmarketing Experience
The following adverse reactions have been reported during post-approval use of another oral formulation of budesonide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Anaphylactic reactions
Nervous System Disorders: Benign intracranial hypertension
Psychiatric Disorders: Mood swings
7 DRUG INTERACTIONS
7.1 CYP3A4 Inhibitors
Budesonide is a substrate for CYP3A4. Avoid use with CYP3A4 inhibitors. Concomitant oral administration of a strong CYP3A4 inhibitor (ketoconazole) caused an eight-fold increase of the systemic exposure to oral budesonide. Inhibitors of CYP3A4 (e.g., ketoconazole, itraconazole, ritonavir, indinavir, saquinavir, erythromycin, and cyclosporine) can increase systemic budesonide concentrations [see Clinical Pharmacology (12.3)].
Grapefruit Juice Avoid ingestion of grapefruit juice with ORTIKOS. Intake of grapefruit juice which inhibits CYP3A4 activity can increase the systemic exposure to budesonide [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited published studies report on the use of budesonide in pregnant women; however, the data are insufficient to inform a drug-associated risk for major birth defects and miscarriage. There are clinical considerations (see Clinical Considerations). In animal reproduction studies with pregnant rats and rabbits, administration of subcutaneous budesonide during organogenesis at doses approximately 0.5 times or 0.05 times, respectively, the maximum recommended human dose, resulted in increased fetal loss, decreased pup weights, and skeletal abnormalities. Maternal toxicity was observed in both rats and rabbits at these dose levels [see Data]. Based on animal data, advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage of the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Some published epidemiological studies show an association of adverse pregnancy outcomes in women with Crohn’s disease, including preterm birth and low birth weight infants, during periods of increased disease activity (including increased stool frequency and abdominal pain). Pregnant women with Crohn’s disease should be counseled regarding the importance of controlling disease.
Fetal/Neonatal adverse reactions
Hypoadrenalism may occur in infants born of mothers receiving corticosteroids during pregnancy. Infants should be carefully observed for signs of hypoadrenalism, such as poor feeding, irritability, weakness, and vomiting, and managed accordingly [see Warnings and Precautions (5.1)].
Data
Animal Data
Budesonide was teratogenic and embryolethal in rabbits and rats.
In an embryo-fetal development study in pregnant rats dosed subcutaneously with budesonide during the period of organogenesis from gestation days 6-15 there were effects on fetal development and survival at subcutaneous doses up to approximately 500 mcg/kg in rats (approximately 0.5 times the maximum recommended human dose on a body surface area basis). In an embryo-fetal development study in pregnant rabbits dosed during the period of organogenesis from gestation days 6-18, there was an increase in maternal abortion, and effects on fetal development and reduction in litter weights at subcutaneous doses up to approximately 25 mcg/kg in rabbits (approximately 0.05 times the maximum recommended human dose on a body surface area basis). Maternal toxicity, including reduction in body weight gain, was observed at subcutaneous doses of 5 mcg/kg in rabbits (approximately 0.01 times the maximum recommended human dose on a body surface area basis) and 500 mcg/kg in rats (approximately 0.5 times the maximum recommended human dose on a body surface area basis).
In a peri- and post-natal development study, rats dosed subcutaneously with budesonide during the period of Day 15 post coitum to Day 21 postpartum, budesonide had no effects on delivery but did have an effect on growth and development of offspring. In addition, offspring survival was reduced and surviving offspring had decreased mean body weights at birth and during lactation at exposures 0.02 times the MRHD (on a mg/m2 basis at maternal subcutaneous doses of 20 mcg/kg/day and higher). These findings occurred in the presence of maternal toxicity.
8.2 Lactation
Risk Summary
Lactation studies have not been conducted with oral budesonide, including ORTIKOS, and no information is available on the effects of the drug on the breastfed infant or the effects of the drug on milk production. One published study reports that budesonide is present in human milk following maternal inhalation of budesonide (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ORTIKOS and any potential adverse effects on the breastfed infant from ORTIKOS, or from the underlying maternal condition.
Data
One published study reports that budesonide is present in human milk following maternal inhalation of budesonide which resulted in infant doses approximately 0.3% to 1% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 0.4 and 0.5. Budesonide plasma concentrations were not detected and no adverse events were noted in the breastfed infants following maternal use of inhaled budesonide. The recommended daily dose of ORTIKOS is higher (up to 9 mg daily) compared with inhaled budesonide (up to 800 mcg daily) given to mothers in the above described study. The maximum budesonide plasma concentration following a 9 mg daily dose (in both single-and repeated-dose pharmacokinetic studies) of oral budesonide is approximately 5 nmol/L to 10 nmol/L which is up to 10 times higher than the 1 nmol/L to 2 nmol/L for a 800 mcg daily dose of inhaled budesonide at steady state in the above inhalation study. Assuming the coefficient of extrapolation between the inhaled and oral doses is constant across all dose levels, at therapeutic doses of ORTIKOS, budesonide exposure to the nursing child may be up to 10 times higher than that by budesonide inhalation.
8.4 Pediatric Use
The safety and effectiveness of ORTIKOS have been established in pediatric patients 8 to 17 years of age who weigh more than 25 kg for the treatment of mild to moderate active Crohn’s disease involving the ileum and/or the ascending colon. Use of ORTIKOS in this age group is supported by evidence from adequate and well controlled studies of oral budesonide in adults, with additional data from 2 clinical studies in 149 pediatric patients treated up to 8 weeks and one pharmacokinetic study in 8 pediatric patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
The observed safety profile of oral budesonide in pediatric patients is consistent with its known safety profile in adults and no new safety concerns were identified [see Adverse Reactions (6.1)].
The safety and effectiveness of ORTIKOS have not been established in pediatric patients less than 8 years of age for the treatment of mild to moderate active Crohn’s disease involving the ileum and/or the ascending colon.
The safety and effectiveness of ORTIKOS have not been established in pediatric patients for the maintenance of clinical remission of mild to moderate Crohn’s disease. An open-label study to evaluate the safety and tolerability of oral budesonide as maintenance treatment in pediatric patients aged 5 to 17 years was conducted, and did not establish the safety and efficacy of maintenance of clinical remission.
Systemic corticosteroids, including ORTIKOS, may cause a reduction of growth velocity in pediatric patients. Pediatric patients with Crohn’s disease have a 17% higher mean systemic exposure and cortisol suppression than adults with Crohn’s disease [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
8.5 Geriatric Use
Clinical studies of oral budesonide did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Of the 651 patients treated with oral budesonide in clinical studies, 17 (3%) were greater than or equal to 65 years of age and none were greater than 74 years of age. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
Patients with moderate to severe hepatic impairment (Child-Pugh Class B and C, respectively) could be at an increased risk of hypercorticism and adrenal axis suppression due to an increased systemic exposure to budesonide [see Warnings and Precautions (5.1)]. Avoid use of ORTIKOS in patients with moderate and severe hepatic impairment. No dosage adjustment is needed in patients with mild hepatic impairment (Child-Pugh Class A).
10 OVERDOSAGE
Reports of acute toxicity and/or death following overdosage of glucocorticoids are rare. Treatment consists of immediate gastric lavage or emesis followed by supportive and symptomatic therapy.
If corticosteroids are used at excessive doses for prolonged periods, systemic corticosteroid effects such as hypercorticism and adrenal axis suppression may occur. For chronic overdosage in the case of severe disease requiring continuous steroid therapy, the dosage may be reduced temporarily.
Single oral doses of 200 and 400 mg/kg were lethal in female and male mice, respectively. The signs of acute toxicity were decreased motor activity, piloerection and generalized edema.
11 DESCRIPTION
Budesonide, the active ingredient of budesonide capsules, is a synthetic corticosteroid. Budesonide is designated chemically as (RS)-11β, 16α, 17,21tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with butyraldehyde. Budesonide is provided as a mixture of two epimers (22R and 22S). The molecular formula of budesonide is C25H34O6 and its molecular weight is 430.5. Its structural formula is:
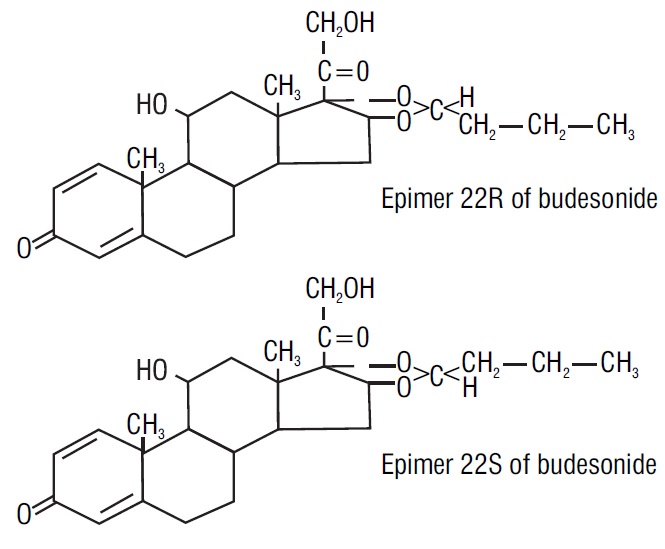
Budesonide is a white to off-white powder that is practically insoluble in water and heptane, sparingly soluble in ethanol, and freely soluble in chloroform. Its partition coefficient between octanol and water at pH 5 is 1.6 x 103 ionic strength 0.01.
Each extended-release capsule for oral administration contains 6 mg or 9 mg of budesonide, USP (micronized) with the following inactive ingredients: acetyl tributyl citrate, corn starch, ethylcellulose aqueous dispersion, methacrylic acid and ethyl acrylate copolymer dispersion, polysorbate 80, simethicone emulsion, sucrose, talc, and triethyl citrate.
Capsule shell contains gelatin, iron oxide black (for 6 mg), iron oxide red, iron oxide yellow, sodium lauryl sulphate and titanium dioxide.
The imprinting ink contains black iron oxide, potassium hydroxide and shellac.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Budesonide is an anti-inflammatory corticosteroid and has a high glucocorticoid effect and a weak mineralocorticoid effect, and the affinity of budesonide to glucocorticoid receptors, which reflects the intrinsic potency of the drug, is about 200-fold that of cortisol and 15-fold that of prednisolone.
12.2 Pharmacodynamics
Treatment with glucocorticoids, including ORTIKOS is associated with a suppression of endogenous cortisol concentrations and an impairment of the hypothalamus-pituitary-adrenal (HPA) axis function. There was a positive correlation between the percent (%) reduction of AUC0-24 of plasma cortisol and systemic exposure to budesonide both in pediatric and adult patients.
Adults
Plasma cortisol suppression was compared following five days’ administration of oral budesonide and prednisolone in a crossover study in healthy volunteers. The mean decrease in the area under the plasma cortisol concentration-time curve over 24 hour (AUC0-24) was greater (78%) with prednisolone 20 mg per day compared to 45% with budesonide 9 mg per day.
Pediatric Patients
The effect of budesonide on endogenous cortisol concentrations was compared between pediatric patients (n=8, aged 9 to 14 years) and adults (n=6) with active Crohn’s disease following administration of oral budesonide 9 mg once daily for 7 days. Compared to baseline values before treatment, the mean decrease in the AUC0-24 of cortisol was 64% (±18%) in pediatric patients and 50% (±27%) in adults after budesonide treatment [see Warnings and Precautions (5.1), Adverse Reactions (6.1) and Use in Specific Populations (8.4)].
The responses to adrenocorticotropin challenge (i.e., ACTH stimulation test) was studied in pediatric patients aged 8 to 17 years, with mild to moderate active Crohn’s disease in randomized, double-blind, active control study [see Clinical Studies (14.1)]. After 8 weeks of treatment with oral budesonide 9 mg once daily or with prednisolone, administered at tapering doses starting from 1 mg/kg, the proportion of patients with normal response to the ACTH challenge was 6% in the budesonide group compared to none in the prednisolone group; the proportion of patients with morning p-cortisol of greater than 5 mcg/dL was 50% in the budesonide group compared to 22% in the prednisolone group. The mean morning p-cortisol was 6.3 mcg/dL in the budesonide group and 2.6 mcg/dL in the prednisolone group (Table 4).
Table 4: Proportion of Pediatric Patients 8 to 17 years old with Peak Endogenous Cortisol Levels (above 18 mcg/dL) after ACTH Stimulation and Normal Response1 to ACTH Challenge Following Administration of Oral Budesonide or Prednisolone for 8 weeks
| Budesonide | Prednisolone |
|
| Peak plasma cortisol above 18 mcg/dL |
||
| At baseline | 91% (20/22) | 91% (21/23) |
| At week 8 | 25% (4/16) | 0% (0/18) |
| Normal response* to ACTH challenge |
||
| At baseline | 73% (16/22) | 78% (18/23) |
| At week 8 | 6% (1/16) | 0% (0/18) |
12.3 Pharmacokinetics
Absorption
Following administration of oral budesonide, the time to peak concentration varied in individual patients between 2.5 to 8 hours. Mean oral bioavailability of budesonide ranged from 9% to 21% both in patients and in healthy subjects, demonstrating a high first-pass elimination of the drug.
Budesonide pharmacokinetics were dose-proportional following repeated administration in the dose range of 3 mg to 15 mg. No accumulation of budesonide was observed following repeated dosing.
Following administration of oral budesonide 9 mg for five days in healthy subjects, the mean peak plasma concentration and the steady state area under the plasma concentration time curve for budesonide were 5.3 ± 1.8 nmol/L and 37.0 ±14.6 nmolhr/L, respectively.
Following administration of oral budesonide 9 mg once daily in patients with active Crohn’s disease, the mean peak plasma concentration and AUC were 4.0 ±2.1 nmol/L and 35.0 ±19.8 nmolh/L, respectively.
Concomitant administration of a high-fat meal delayed the time to peak concentration of budesonide by 1 hour and overall exposure was increased by about 25%.
Distribution
The mean volume of distribution (Vss) of budesonide varied between 2.2 L/kg and 3.9 L/kg in healthy subjects and in patients. Plasma protein binding was estimated to be 85% to 90% in the concentration range 1 nmol/L to 230 nmol/L, independent of gender. The erythrocyte/plasma partition ratio at clinically relevant concentrations was about 0.8.
Elimination
Budesonide had a plasma clearance, 0.9 L/min to 1.8 L/min in healthy adults. Mean plasma clearance after intravenous administration of budesonide in patients with Crohn’s disease was 1.0 L/min. These plasma clearance values approached the estimated liver blood flow, and, accordingly, suggest that budesonide is a high hepatic clearance drug. The plasma elimination half-life, after administration of intravenous doses ranged between 2 and 3.6 hours, and did not differ between healthy adults and patients with Crohn’s disease.
Metabolism
Following absorption, budesonide is subject to high first pass metabolism (80% to 90%). In vitro experiments in human liver microsomes demonstrated that budesonide is rapidly and extensively biotransformed, mainly by CYP3A4, to its 2 major metabolites, 6β-hydroxy budesonide and 16α-hydroxy prednisolone. The corticosteroid activity of these metabolites was negligible (less than 1/100) in relation to that of the parent compound. In vivo investigations with intravenous doses in healthy subjects were in agreement with the in vitro findings.
Excretion
Budesonide was excreted in urine and feces in the form of metabolites. After oral as well as intravenous administration of micronized [3H]-budesonide, approximately 60% of the recovered radioactivity was found in urine. The major metabolites, including 6β-hydroxy budesonide and 16α-hydroxy prednisolone, are mainly renally excreted, intact or in conjugated forms. No unchanged budesonide was detected in urine.
Specific Populations
Age: Pediatric Population (8 years and older)
The pharmacokinetics of budesonide were investigated in pediatric patients aged 9 to 14 years (n=8) after oral administration of budesonide and intravenous administration of budesonide. Following administration of 9 mg oral budesonide once daily for 7 days, the median time to peak plasma concentration of budesonide was 5 hours and the mean peak plasma concentration was 6.0 ± 3.5 nmol/L. The mean AUC was 41.3 ±12.2 nmolh/L and 17% higher than that in adult patients with Crohn’s disease in the same study. The mean absolute oral availability was 9.2% (3 to 17%; n=4) in pediatric patients.
After single dose administration of intravenous budesonide (n=4), the mean volume of distribution (Vss) was 2.2 ± 0.4 L/kg and mean clearance was 0.81 ± 0.2 L/min. The mean elimination half-life was 1.9 hours in pediatric patients. The body-weight normalized clearance in pediatric patients was 20.5 mL/min/kg in comparison to 15.9 mL/min/kg in adult patients after intravenous administration [see Warnings and Precautions (5.1), Use in Specific Population (8.4)].
Patients with Hepatic Impairment
In patients with mild (Child-Pugh Class A, n=4) or moderate (Child-Pugh Class B, n=4) hepatic impairment, budesonide 4 mg was administered orally as a single dose. The patients with moderate hepatic impairment had a 3.5-fold higher AUC compared to the healthy subjects with normal hepatic function while the patients with mild hepatic impairment had an approximately 1.4-fold higher AUC. The Cmax values demonstrated similar increases [see Warnings and Precautions (5.1)]. The increased systemic exposure in patients with mild hepatic impairment was not considered to be clinically relevant. Patients with severe liver impairment (Child-Pugh Class C) were not studied [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Budesonide is metabolized via CYP3A4. Potent inhibitors of CYP3A4 can increase the plasma concentrations of budesonide several-fold. Conversely, induction of CYP3A4 potentially could result in the lowering of budesonide plasma concentrations.
Effects of Other Drugs on Budesonide
Ketoconazole
In an open, non-randomized, cross-over study, 6 healthy subjects were given budesonide 10 mg as a single dose, either alone or concomitantly with the last ketoconazole dose of 3 days treatment with ketoconazole 100 mg twice daily. Coadministration of ketoconazole resulted in an eight-fold increase in AUC of budesonide, compared to budesonide alone [see Drug Interactions (7.1)].
Grapefruit Juice
In an open, randomized, cross-over study, 8 healthy subjects were given oral budesonide 3 mg, either alone, or concomitantly with 600 mL concentrated grapefruit juice (which inhibits CYP3A4 activity predominantly in the intestinal mucosa), on the last of 4 daily administrations. Concomitant administration of grapefruit juice resulted in a 2-fold increase of the bioavailability of budesonide compared to budesonide alone [see Drug Interactions (7.1)].
Oral Contraceptives (CYP3A4 Substrates)
In a parallel study, the pharmacokinetics of budesonide were not significantly different between healthy female subjects who received oral contraceptives containing desogestrel 0.15 mg and ethinyl estradiol 30 mcg and healthy female subjects who did not receive oral contraceptives. Budesonide 4.5 mg once daily (one-half the recommended dose) for one week did not affect the plasma concentrations of ethinyl estradiol, a CYP3A4 substrate. The effect of budesonide 9 mg once daily on the plasma concentrations of ethinyl estradiol was not studied.
Omeprazole
In a study in 11 healthy subjects, performed in a double-blind, randomized, placebo controlled manner, the effect of 5 to 6 days treatment with omeprazole 20 mg once daily on the pharmacokinetics of budesonide administered as oral budesonide 9 mg as a single dose was investigated. Omeprazole 20 mg once daily did not affect the absorption or pharmacokinetics of budesonide.
Cimetidine
In an open, non-randomized, cross-over study, the potential effect of cimetidine on the pharmacokinetics of budesonide was studied. Six healthy subjects received cimetidine 1 gram daily (200 mg with meals and 400 mg at night) for 2 separate 3-day periods. Budesonide 4 mg was administered either alone or on the last day of one of the cimetidine treatment periods. Coadministration of cimetidine resulted in a 52% and 31% increase in the budesonide peak plasma concentration and the AUC of budesonide, respectively.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with budesonide were conducted in rats and mice. In a two-year study in Sprague-Dawley rats, budesonide caused a statistically significant increase in the incidence of gliomas in male rats at an oral dose of 50 mcg/kg (approximately 0.05 times the maximum recommended human dose on a body surface area basis). In addition, there were increased incidences of primary hepatocellular tumors in male rats at 25 mcg/kg (approximately 0.023 times the maximum recommended human dose on a body surface area basis) and above. No tumorigenicity was seen in female rats at oral doses up to 50 mcg/kg (approximately 0.05 times the maximum recommended human dose on a body surface area basis). In an additional two-year study in male Sprague-Dawley rats, budesonide caused no gliomas at an oral dose of 50 mcg/kg (approximately 0.05 times the maximum recommended human dose on a body surface area basis). However, it caused a statistically significant increase in the incidence of hepatocellular tumors at an oral dose of 50 mcg/kg (approximately 0.05 times the maximum recommended human dose on a body surface area basis). The concurrent reference corticosteroids (prednisolone and triamcinolone acetonide) showed similar findings. In a 91-week study in mice, budesonide caused no treatment-related carcinogenicity at oral doses up to 200 mcg/kg (approximately 0.1 times the maximum recommended human dose on a body surface area basis).
Budesonide was not genotoxic in the Ames test, the mouse lymphoma cell forward gene mutation (TK+/-) test, the human lymphocyte chromosome aberration test, the Drosophila melanogaster sex-linked recessive lethality test, the rat hepatocyte UDS test and the mouse micronucleus test.
In rats, budesonide had no effect on fertility at subcutaneous doses up to 80 mcg/kg (approximately 0.07 times the maximum recommended human dose on a body surface area basis). However, it caused a decrease in prenatal viability and viability in pups at birth and during lactation, along with a decrease in maternal body-weight gain, at subcutaneous doses of 20 mcg/kg (approximately 0.02 times the maximum recommended human dose on a body surface area basis) and above. No such effects were noted at 5 mcg/kg (approximately 0.005 times the maximum recommended human dose on a body surface area basis).
14 CLINICAL STUDIES
The safety and efficacy of ORTIKOS have been established based on adequate and well-controlled adult studies of another oral budesonide product in patients with Crohn’s Disease. Below is a display of the results of these adequate and well-controlled studies of budesonide in these conditions.
14.1 Treatment of Mild to Moderate Active Crohn’s Disease
Adults
The efficacy of oral budesonide were evaluated in 994 patients with mild to moderate active Crohn’s disease of the ileum and/or ascending colon in 5 randomized and double-blind studies of 8 weeks duration. The study patients ranged in age from 17 to 85 (mean 35), 40% were male and 97% were white. The Crohn’s Disease Activity Index (CDAI) was the main clinical assessment used for determining efficacy in these 5 studies.1 The CDAI is a validated index based on subjective aspects rated by the patient (frequency of liquid or very soft stools, abdominal pain rating and general well-being) and objective observations (number of extraintestinal symptoms, need for antidiarrheal drugs, presence of abdominal mass, body weight and hematocrit). Clinical improvement, defined as a CDAI score of less than or equal to 150 assessed after 8 weeks of treatment, was the primary efficacy variable in these 5 comparative efficacy studies of oral budesonide. Safety assessments in these studies included monitoring of adverse reactions. A checklist of potential symptoms of hypercorticism was used.
One study (Study 1) compared the efficacy of budesonide 9 mg daily in the morning to a comparator. At baseline, the median CDAI was 272. Budesonide 9 mg daily resulted in a significantly higher clinical improvement rate at Week 8 than the comparator. See Table 5.
Table 5: Clinical Improvement Rates (CDAI less than or equal to 150) After 8 weeks of Treatment
|
|
|||||
| Clinical
Study | Budesonide
| Comparator*
| Placebo
| Prednisolone
|
|
| 9 mg Daily
| 4.5 mg Twice Daily
|
| |||
| 1 | 62/91 (69%)†
| | 37/83 (45%) | | |
| 2 | | 31/61 (51%)‡
| | 13/64 (20%) | |
| 3 | 38/79 (48%) | 41/78 (53%) | | 13/40 (33%) | |
| 4 | 35/58 (60%) | 25/60 (42%) | | | 35/58 (60%) |
| 5 | 45/86 (52%) | | | | 56/85 (65%) |
Two placebo-controlled clinical trials (Studies 2 and 3) were conducted. Study 2 involved 258 patients and tested the effects of graded doses of budesonide (1.5 mg twice daily, 4.5 mg twice daily, or 7.5 mg twice daily) versus placebo. At baseline, the median CDAI was 290. The 1.5 mg twice daily arm (data not shown) could not be differentiated from placebo. The 4.5 mg twice daily arm was statistically different from placebo (Table 5), while no additional benefit was seen when the daily budesonide dose was increased to 15 mg per day (data not shown). Study 3 was a 3-armed parallel group study. The groups were treated with budesonide 9 mg once daily, budesonide 4.5 mg twice daily and placebo for 8 weeks, followed by a 2-week double-blind taper phase. The median CDAI at baseline was 263. Neither 9 mg daily nor 4.5 mg twice daily budesonide dose levels were statistically different from placebo (Table 5). The recommended dosage of budesonide for the treatment of mild to moderate active Crohn’s disease involving the ileum and/or the ascending colon in adults is 9 mg once daily in the morning for up to 8 weeks [see Dosage and Administration (2.1)].
Two clinical trials (Studies 4 and 5) compared oral budesonide with oral prednisolone (initial dose 40 mg per day). Study 4 was a 3-armed parallel group study. The groups were treated with budesonide 9 mg once daily, budesonide 4.5 mg twice daily and prednisolone 40 mg (tapered dose) for 8 weeks, followed by a 4-week double blind taper phase. At baseline, the median CDAI was 277. Equal clinical improvement rates (60%) were seen in the budesonide 9 mg daily and the prednisolone groups in Study 4. In Study 5, 13% fewer patients in the budesonide group experienced clinical improvement than in the prednisolone group (no statistical difference) (Table 5).
The proportion of patients with normal plasma cortisol values (greater than 150 nmol/L) was significantly higher in the budesonide groups in both trials (60% to 66%) than in the prednisolone groups (26% to 28%) at Week 8.
Pediatric Patients (8 to 17 Years of Age)
The effectiveness of oral budesonide, in pediatric patients aged 8 to 17 years, who weigh more than 25 kg with mild to moderate active Crohn’s disease (defined as Crohn's Disease Activity Index (CDAI) ≥ 200) involving the ileum and/or the ascending colon, was assessed in one randomized, double-blind, active control study. This study compared budesonide 9 mg once daily, with prednisolone, administered at tapering doses starting from 1 mg/kg. Twenty-two (22) patients were treated with budesonide and 24 patients were treated with prednisolone. After 8 weeks of treatment, 55% (95% CI: 32%, 77%) of patients treated with budesonide reached the endpoint (CDAI ≤150), as compared to 68% (95% CI: 47%, 89%) of patients treated with prednisolone. The average number of liquid or very soft stools per day (assessed over 7 days) decreased from 1.49 at baseline to 0.96 after treatment with budesonide and 2.00 at baseline to 0.52 after treatment with prednisolone. The average daily abdominal pain rating (where 0=none, 1=mild, 2=moderate, and 3=severe) decreased from 1.49 at baseline to 0.54 after treatment with budesonide and 1.64 at baseline to 0.38 after 8 weeks of treatment with prednisolone.
Use of budesonide in this age group is supported by evidence from adequate and well-controlled studies of budesonide in adults, and by safety and pharmacokinetic studies performed in pediatric patients.
14.2 Maintenance of Clinical Remission of Mild to Moderate Crohn’s Disease
Adults
The efficacy of oral budesonide for maintenance of clinical remission were evaluated in four double-blind, placebo-controlled, 12-month trials in which 380 patients were randomized and treated once daily with 3 mg or 6 mg budesonide or placebo. Patients ranged in age from 18 to 73 (mean 37) years. Sixty percent of the patients were female and 99% were Caucasian. The mean CDAI at entry was 96. Among the four clinical trials, approximately 75% of the patients enrolled had exclusively ileal disease. Colonoscopy was not performed following treatment. Budesonide 6 mg per day prolonged the time to relapse, defined as an increase in CDAI of at least 60 units to a total score greater than 150 or withdrawal due to disease deterioration. The median time to relapse in the pooled population of the 4 studies was 154 days for patients taking placebo, and 268 days for patients taking budesonide 6 mg per day. Budesonide 6 mg per day reduced the proportion of patients with loss of symptom control relative to placebo in the pooled population for the 4 studies at 3 months (28% versus 45% for placebo).
15 REFERENCES
- Best WR, Becktel JM, Singleton JW, Kern F: Development of a Crohn’s Disease Activity Index, National Cooperative Crohn’s Disease Study. Gastroenterology 1976; 70(3): 439-444.
16 HOW SUPPLIED/STORAGE AND HANDLING
- ORTIKOS 6 mg are hard gelatin capsules with light grey colored cap and pink colored body imprinted with “061” on cap and body in black ink containing white to off-white pellets.
Bottles of 30's with Child Resistant Cap.................................................................NDC: 55566-1002-1
- ORTIKOS 9 mg are hard gelatin capsules with pink colored cap and pink colored body imprinted with “062” on cap and body in black ink containing white to off-white pellets.
Bottles of 30's with Child Resistant Cap.................................................................NDC: 55566-1020-1
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Keep container tightly closed.
17 PATIENT COUNSELING INFORMATION
Advise Patients to read the FDA-Approved patient labeling (Patient Information).
Hypercorticism and Adrenal Axis Suppression
Advise patients that ORTIKOS may cause hypercorticism and adrenal axis suppression and to follow a taper schedule, as instructed by their healthcare provider if transferring to ORTIKOS from systemic corticosteroids [see Warnings and Precautions (5.1), (5.2)]. Advise patients that replacement of systemic corticosteroids with ORTIKOS may unmask allergies (e.g., rhinitis and eczema), which were previously controlled by the systemic drug.
Increased Risk of Infection
Advise patients to avoid exposure to people with chicken pox or measles and, if exposed, to consult their healthcare provider immediately. Inform patients that they are at increased risk of developing a variety of infections; including worsening of existing tuberculosis, fungal, bacterial, viral or parasitic infections or ocular herpes simplex and to contact their healthcare provider if they develop any symptoms of infection [see Warnings and Precautions (5.3)].
Pregnancy
Advise female patients that ORTIKOS may cause fetal harm and to inform their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Administration
Advise patients to:
- Take ORTIKOS once daily in the morning.
- Swallow ORTIKOS capsules whole. Do not chew or crush.
Avoid consumption of grapefruit juice for the duration of therapy with ORTIKOS [see Drug Interactions (7.1)].
Distributor:
Ferring Pharmaceuticals Inc.
Parsippany, NJ 07054
Manufactured by:
Sun Pharmaceutical Industries Ltd.
Halol-Baroda Highway,
Halol-389 350, Gujarat, India
|
Patient Information ORTIKOS™ (or-TEE-kos) (budesonide) extended-release capsules, for oral use |
| Read this Patient Information before you start taking ORTIKOS and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment. |
| What is ORTIKOS?
ORTIKOS is a prescription corticosteroid medicine used to treat mild to moderate Crohn’s disease that affects part of the small intestine (ileum) and part of the large intestine (ascending colon):
It is not known if ORTIKOS is safe and effective in children to help keep symptoms of mild to moderate Crohn’s disease that affects part of the small intestine and part of the large intestine from coming back. |
Do not take ORTIKOS if:
|
Before you take ORTIKOS tell your healthcare provider if you have any other medical conditions including if you:
|
How should I take ORTIKOS?
|
What should I avoid while taking ORTIKOS?
|
| What are the possible side effects of ORTIKOS?
ORTIKOS may cause serious side effects, including:
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of ORTIKOS. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
How should I store ORTIKOS?
|
| General information about the safe and effective use of ORTIKOS.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ORTIKOS for a condition for which it was not prescribed. Do not give ORTIKOS to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ORTIKOS that is written for health professionals. |
| What are the ingredients in ORTIKOS?
Active ingredient: budesonide Inactive ingredients: acetyl tributyl citrate, corn starch, ethylcellulose aqueous dispersion, methacrylic acid and ethyl acrylate copolymer dispersion, polysorbate 80, simethicone emulsion, sucrose, talc, and triethyl citrate. Capsule shell contains gelatin, iron oxide black (for 6 mg), iron oxide red, iron oxide yellow, sodium lauryl sulphate and titanium dioxide. The imprinting ink contains, black iron oxide, potassium hydroxide and shellac. Distributor: Ferring Pharmaceuticals Inc. Parsippany, NJ 07054 Manufactured by: Sun Pharmaceutical Industries Ltd. Halol-Baroda Highway, Halol-389 350, Gujarat, India For more information, call 1-800-818-4555. PJPI0648A 6794-02 Issued: October 2019 |
This Patient Information has been approved by the U.S. Food and Drug Administration
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 6 mg
NDC: 55566-1002-1
OrtikosTM
(budesonide) Extended-Release Capsules
6 mg
Rx only
30 Capsules
FERRING PHARMACEUTICALS
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - 9 mg
NDC: 55566-1020-1
OrtikosTM
(budesonide) Extended-Release Capsules
9 mg
Rx only
30 Capsules
FERRING PHARMACEUTICALS

| ORTIKOS
budesonide capsule |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| ORTIKOS
budesonide capsule |
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||
| Labeler - FERRING PHARMACEUTICALS INC. (103722955) |
| Registrant - Sun Pharmaceutical Industries, Inc. (146974886) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sun Pharmaceutical Industries Limited | 725959238 | ANALYSIS(55566-1002, 55566-1020) , MANUFACTURE(55566-1002, 55566-1020) | |
Trademark Results [Ortikos]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ORTIKOS 88284058 not registered Live/Pending |
Sun Pharma Global FZE 2019-01-31 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.