HEPCLUDEX- bulevirtide injection, powder, lyophilized, for solution
Drug Labeling and Warnings
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HEPCLUDEX safely and effectively. See full prescribing information for HEPCLUDEX.
HEPCLUDEX® (bulevirtide-gmod) for injection, for subcutaneous use
Initial U.S. Approval: 2026WARNING: POSTTREATMENT SEVERE ACUTE EXACERBATION OF HEPATITIS D and B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis D and hepatitis B may occur after HEPCLUDEX is discontinued, especially in patients with cirrhosis, who may be at increased risk of more severe flares or progression to hepatic decompensation. Monitor hepatic function closely with both clinical and laboratory follow-up, including hepatitis B virus (HBV) DNA and hepatitis delta virus (HDV) RNA viral load, for at least six months in patients who discontinue HEPCLUDEX. Resumption of antiviral therapy may be warranted. (5.1)
INDICATIONS AND USAGE
HEPCLUDEX is a sodium taurocholate co-transporting polypeptide (NTCP)-directed HDV attachment inhibitor indicated for the treatment of chronic HDV infection in adults without cirrhosis or with compensated cirrhosis. This indication is approved under accelerated approval based on participants who achieved a decrease in HDV RNA and alanine aminotransferase (ALT) normalization. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1, 14)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
For injection: 8.5 mg as a lyophilized powder or cake, in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Hypersensitivity Reactions Including Anaphylaxis: Hypersensitivity reactions have been reported with HEPCLUDEX. If signs or symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue HEPCLUDEX and initiate appropriate treatment. (5.2)
ADVERSE REACTIONS
The most common adverse reactions (incidence greater than or equal to 10%, all grades) observed with treatment with HEPCLUDEX are injection site reactions, headache, abdominal pain, fatigue, and pruritus. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: POSTTREATMENT SEVERE ACUTE EXACERBATION OF HEPATITIS D and B
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adults
2.2 Dose Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Exacerbation of Hepatitis D and B After Discontinuation of Treatment
5.2 Hypersensitivity Reactions Including Anaphylaxis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of HEPCLUDEX on Other Drugs
7.2 Effects of Other Drugs on HEPCLUDEX
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
14 CLINICAL STUDIES
14.1 Clinical Trials in Adults with Chronic HDV Infection Without Cirrhosis or With Compensated Cirrhosis
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: POSTTREATMENT SEVERE ACUTE EXACERBATION OF HEPATITIS D and B
Severe acute exacerbations of hepatitis D and hepatitis B may occur after HEPCLUDEX is discontinued, especially in patients with cirrhosis, who may be at increased risk of more severe flares or progression to hepatic decompensation. Monitor hepatic function closely with both clinical and laboratory follow-up, including hepatitis B virus (HBV) DNA and hepatitis delta virus (HDV) RNA viral load, for at least six months in patients who discontinue HEPCLUDEX. Resumption of antiviral therapy may be warranted [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
HEPCLUDEX is indicated for the treatment of chronic hepatitis delta virus (HDV) infection in adults without cirrhosis or with compensated cirrhosis.
This indication is approved under accelerated approval based on a decrease in HDV RNA and alanine aminotransferase (ALT) normalization [see Clinical Studies (14)]. An improvement in disease-related clinical outcomes has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adults
The recommended dosage in adults is HEPCLUDEX 8.5 mg once daily administered by subcutaneous injection.
- HEPCLUDEX should be continued as long as it is associated with a response to treatment. The optimal treatment duration is unknown.
- In all patients, manage the underlying hepatitis B virus (HBV) infection as clinically appropriate.
- If a dose is missed, that dose should be taken as soon as possible. However, if it is almost time for the next dose, skip the missed dose and resume the original schedule.
2.2 Dose Preparation and Administration
- See the HEPCLUDEX full Instructions for Use for details on the preparation and administration of HEPCLUDEX.
- Healthcare professionals should train patients or caregivers on the proper reconstitution and administration of HEPCLUDEX, and subcutaneous injection techniques. Consider preparation and administration of the first dose under healthcare professional supervision.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Aseptically reconstitute HEPCLUDEX lyophilized powder or cake by adding 1 mL of Sterile Water for Injection to the HEPCLUDEX vial.
- Administer entire contents of vial by subcutaneous injection into the upper thigh, lower abdomen, or back of the upper arm (only if administered by a caregiver).
- Use reconstituted product immediately. Do not store for later use.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Exacerbation of Hepatitis D and B After Discontinuation of Treatment
Severe acute exacerbations of HDV and HBV infection may occur after HEPCLUDEX is discontinued, especially in patients with cirrhosis, who may be at increased risk of more severe flares or progression to hepatic decompensation. Monitor hepatic function closely with both clinical and laboratory follow-up, including monitoring HBV DNA and HDV RNA viral load, for at least six months in patients who discontinue HEPCLUDEX. Resumption of antiviral therapy may be warranted.
5.2 Hypersensitivity Reactions Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported with HEPCLUDEX. If signs or symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue HEPCLUDEX and initiate appropriate treatment [see Adverse Reactions (6.2)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Exacerbation of Hepatitis D and B After Discontinuation of Treatment [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The overall safety profile of HEPCLUDEX is based on Phase 2 and Phase 3 data from 165 adults with chronic HDV infection without cirrhosis or with compensated cirrhosis who received at least 48 weeks of HEPCLUDEX 8.5 mg subcutaneous injection once daily. Trial MYR301 was a Phase 3 randomized, multi-center, open-label, parallel-arm trial in 101 adults. In this trial, 50 adults received 8.5 mg HEPCLUDEX daily for 144 weeks and 51 adults who were in the Delayed Treatment group received no HDV treatment for the first 48 weeks; 50 adults in the Delayed Treatment group then received HEPCLUDEX 8.5 mg once daily for 96 weeks [see Clinical Studies (14)].
Table 1 displays the frequency of the adverse reactions (all grades) ≥ 10% in the HEPCLUDEX group at Week 48. No participant discontinued treatment with HEPCLUDEX due to an adverse reaction through Week 48.
Table 1 Adverse Reactions* (All Grades) Reported in ≥ 10% of Participants with Chronic HDV Infection Without Cirrhosis or With Compensated Cirrhosis Receiving HEPCLUDEX in Trial MYR301 (Week 48 Analysis) Adverse Reaction HEPCLUDEX
(N=50)Delayed Treatment†
(N=51)- * Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug.
- † Participants who received no HDV treatment in Trial MYR301 for the first 48 weeks.
- ‡ Grouped term includes injection site abscess, injection site erythema, injection site reaction, injection site pruritus, injection site swelling, injection site hematoma, injection site rash, injection site dermatitis, and injection site pain.
- § Grouped term includes abdominal pain, abdominal pain lower, and abdominal pain upper.
Injection site reactions‡ 30% 0 Headache 20% 0 Abdominal pain§ 18% 2% Fatigue 14% 2% Pruritus 14% 0 A similar safety profile was observed through Week 144 in Trial MYR301 and for participants in the Delayed Treatment group who switched to treatment with HEPCLUDEX at Week 48 through to Week 144. Additionally, a similar safety profile was observed through Week 96 in Phase 2b Trial MYR204.
Laboratory Abnormalities
Eosinophil Count Increased: In MYR301, increases in eosinophil counts were reported in 33% of participants (all Grade 1) receiving HEPCLUDEX; there were no associated clinical sequelae, hepatic adverse reactions, or significant liver-related laboratory abnormalities.
Total Bile Salts Increased: HEPCLUDEX inhibits sodium taurocholate co-transporting polypeptide (NTCP)-mediated bile acid transport. Consistent with this, elevations in total serum bile salt levels were observed in clinical trials of HEPCLUDEX. In MYR301, all participants who received HEPCLUDEX had elevated serum bile salts. Bile salt levels showed visit-to-visit variability and peaked by Week 8 of treatment in both participants without cirrhosis and those with compensated cirrhosis, although median levels trended higher in the latter group. Bile salt elevations resolved upon discontinuation of HEPCLUDEX.
In MYR301, 14% of HEPCLUDEX recipients experienced Grade 1 or 2 pruritus that was self-limited. The magnitude of total serum bile salt elevations did not correlate with the severity of pruritus.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of HEPCLUDEX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: hypersensitivity, including anaphylactic reaction
-
7 DRUG INTERACTIONS
7.1 Effects of HEPCLUDEX on Other Drugs
No CYP enzyme or transporter mediated inhibition or induction by bulevirtide is anticipated at clinically relevant concentrations [see Clinical Pharmacology (12.3)].
7.2 Effects of Other Drugs on HEPCLUDEX
Due to peptide catabolism of bulevirtide, the drug-drug interaction potential of other drugs to impact bulevirtide pharmacokinetics, via CYP enzymes, is low [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are insufficient human data on the use of HEPCLUDEX during pregnancy to inform a drug-associated risk of birth defects and miscarriage. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
In nonclinical reproductive toxicity studies, bulevirtide demonstrated no adverse effect on embryofetal development when administered to pregnant rats and rabbits at systemic exposures (AUC) 4- and 37-fold relative to exposure in humans at the recommended human dose (RHD).
Data
Animal Data
Bulevirtide was administered via subcutaneous injection to pregnant rats and rabbits (2.5 mg/kg/day) on Gestation Days 6 through 17 and 6 through 20, respectively, and also to rats from Gestation Day 6 to Lactation/Postpartum Day 20. There were no adverse effects on embryofetal development in rats and rabbits. During organogenesis, exposure in rats and rabbits was 4 and 37 times higher, respectively, than the exposure in humans at the RHD. In a pre/postnatal development study in rats, bulevirtide (2.5 mg/kg/day) was administered via subcutaneous injection from Gestation Day 6 to Lactation Day 21. No effects were observed in the offspring at maternal exposures 3 times the exposure at the RHD.
8.2 Lactation
Risk Summary
It is not known whether bulevirtide is present in human breast milk, affects human milk production, or has effects on the breastfed infant. In nonclinical pre- and postnatal developmental rat studies, bulevirtide was not measured in the plasma of pups or in the milk of nursing animals. However, due to its high protein binding, liver tropism, and high specificity for NTCP, bulevirtide is not likely to be secreted in milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for HEPCLUDEX and any potential adverse effects on the breastfed child from HEPCLUDEX or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of HEPCLUDEX in pediatric patients less than 18 years of age have not been established.
8.5 Geriatric Use
Clinical trials of HEPCLUDEX did not include participants aged 65 and over to determine whether they respond differently from younger participants [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of HEPCLUDEX is recommended in patients with mild, moderate, or severe renal impairment (creatinine clearance [CrCl] greater than or equal to 15 mL per minute) [see Clinical Pharmacology (12.3)]. HEPCLUDEX has not been studied in patients with end-stage renal disease (CrCl less than 15 mL per minute).
8.7 Hepatic Impairment
No dosage adjustment of HEPCLUDEX is recommended in patients with mild hepatic impairment (Child-Pugh Class A) [see Clinical Pharmacology (12.3)]. The safety and efficacy of HEPCLUDEX have not been studied in patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment.
-
10 OVERDOSAGE
No data are available on overdose of HEPCLUDEX in patients. Treatment of overdose with HEPCLUDEX should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with HEPCLUDEX. Hemodialysis is unlikely to result in significant removal of bulevirtide since bulevirtide is highly bound to plasma protein.
-
11 DESCRIPTION
Bulevirtide-gmod is an NTCP-directed HDV attachment inhibitor. Bulevirtide as an acetate salt, is a 47-amino acid protein with a fatty acid myristoyl residue at the N-terminus and an amidated C-terminus. All chiral amino acids are in the L-configuration. The counter ion acetate is bound in ionic form to basic groups of the peptide molecule in a nonstoichiometric ratio. Bulevirtide acetate has a molecular formula of C248H355N65O72 (net) and a molecular weight of 5398.9 Da (average mass, net), and has the following structural formula:
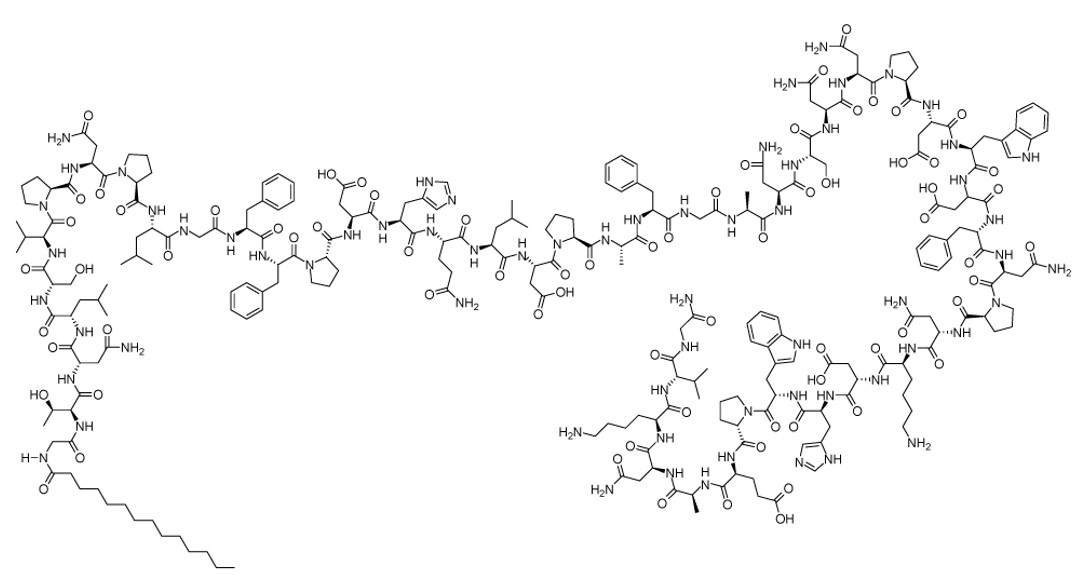
HEPCLUDEX (bulevirtide-gmod) for injection is a sterile, preservative-free, white to off-white lyophilized powder or cake for subcutaneous injection after reconstitution. Each single-dose vial delivers 8.5 mg of bulevirtide-gmod (equivalent to approximately 8.6 mg of bulevirtide acetate). The inactive ingredients are histidine (3.3 mg), mannitol (51 mg), and sucrose (8.5 mg), and may include hydrochloric acid and/or sodium hydroxide to adjust the pH to 8.5.
HEPCLUDEX requires reconstitution prior to administration by subcutaneous injection [see Dosage and Administration (2.2)].
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
HEPCLUDEX 8.5 mg once daily was associated with a higher percentage of trial participants with undetectable HDV RNA at Week 144 compared to a lower once daily dose.
12.3 Pharmacokinetics
The pharmacokinetic properties of bulevirtide were characterized after intravenous administration in healthy participants, and after subcutaneous administration in healthy participants and participants with chronic HDV infection. The systemic exposure of bulevirtide increased in a more than proportional manner with increasing doses. At steady state, AUC and Cmax increased by approximately 2-fold compared to the AUC and Cmax after the first dose.
The pharmacokinetic parameters of bulevirtide are provided in Table 2. The steady-state PK parameters of bulevirtide (based on population PK analysis of participants with chronic HDV infection) are provided in Table 3.
Table 2 Pharmacokinetic Parameters of Bulevirtide - * t1/2 values refer to mean terminal plasma half-life.
- † Bulevirtide, a linear peptide consisting of L-amino acids, is expected to be degraded to smaller peptides and individual amino acids. No active metabolites are expected.
Absorption % absolute bioavailability 57 Tmax (h) (range) 3.00 (1.00 - 4.00) Distribution % bound to human plasma proteins > 99 Elimination t1/2 (h)* (range) 3 (2 - 6) Metabolism Metabolic pathway† Catabolized by peptidases to amino acids Excretion Major route of elimination Excreted as smaller peptides and amino acids Table 3 Steady-State Pharmacokinetic Parameters of Bulevirtide Following Subcutaneous Administration of HEPCLUDEX in Adults with HDV Infection* Parameter Geometric Mean (90% CI) CI=Confidence Interval - * Empirical Bayesian post hoc exposure estimates from Population pharmacokinetic analysis in MYR301 trial, N=100.
Cmax (ng/mL) 184 (160 - 211) AUC0-24h (ngh/mL) 1935 (1680 - 2230) Specific Populations
Age (18 to 65 years), sex, race (87.5% White, 1.9% Black, 10.3% Asian, 0.2% Other), or body weight (39.7 to 110 kg) did not have a clinically relevant impact on the systemic exposure of bulevirtide.
Geriatric Patients
The pharmacokinetics of bulevirtide have not been evaluated in elderly participants with HDV infection (65 years of age and older) [see Use in Specific Populations (8.5)].
Patients with Renal Impairment
In a Phase 1, open-label study in participants without HDV infection, the steady state pharmacokinetics of bulevirtide were similar among participants with normal renal function and participants with severe renal impairment (CrCl 15 to less than 30 mL per minute), and no clinically relevant differences in total bile acid elevations were observed between the two groups. The pharmacokinetics of bulevirtide have not been evaluated in participants with end-stage renal disease (CrCl less than 15 mL per minute), including those on dialysis. As bulevirtide is greater than 99% protein bound, dialysis is not expected to alter exposures of bulevirtide [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
In a Phase 1, open-label study in participants without HDV infection, the steady-state pharmacokinetics of bulevirtide were approximately 27% lower in participants with moderate hepatic impairment (Child-Pugh Class B) than participants with normal hepatic function. The steady state pharmacokinetics of bulevirtide were similar among participants with severe hepatic impairment (Child-Pugh Class C) and participants with normal hepatic function [see Use in Specific Populations (8.7)].
Drug Interaction Studies
Effect of Bulevirtide on Other Drugs
Cytochrome P450 (CYP) Enzymes: In vitro studies have shown, bulevirtide is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Bulevirtide is not an inducer of CYP1A2, CYP2B6, or CYP3A4. Consistent with in vitro results, bulevirtide at steady state did not impact the pharmacokinetics of the CYP3A4 probe substrate midazolam, administered as an oral 30 μg microdose, in clinical drug-interaction studies.
Transporter Systems: In vitro studies have shown that no clinically relevant interactions are expected for efflux transporters including MDR1, BCRP, BSEP, MATE1, and MATE2K and uptake transporters including OATP2B1, OAT1, OAT3, OCT1, and OCT2. In vitro studies have shown that bulevirtide inhibits the organic anion transporting polypeptides, OATP1B1 and OATP1B3, with IC50 values of 0.5 and 8.7 μM, respectively, and taurocholate uptake via NTCP receptors with an IC50 value of 0.320 μM; however, no clinical drug interaction is expected for OATP1B or NTCP substrates at clinically relevant concentrations of bulevirtide.
Steady state exposures of bulevirtide administered once daily did not impact the pharmacokinetics of TDF (at 300 mg) in a dedicated clinical drug-interaction study.
12.4 Microbiology
Mechanism of Action
Bulevirtide is a synthetic 47-amino acid lipopeptide with a myristoylated N-terminus and an amidated C-terminus derived from amino acids 13-59 of the L-HBsAg preS1 domain from an HBV genotype (GT)-C consensus sequence (corresponding to GT-D preS1 amino acids 2-48). Bulevirtide inhibits HDV infection by binding to the HDV receptor NTCP on the plasma membrane of hepatocytes, blocking HDV attachment to NTCP.
Antiviral Activity in Cell Culture
In primary human hepatocytes (PHH), bulevirtide inhibited infection of lab-generated HDV GTs 1-8 carrying envelopes from HBV GTs A-H with a median EC50 value of 0.52 nM (range: 0.23-0.93 nM) overall and median EC50 values of 0.32-0.72 nM across HBV GTs. In addition, bulevirtide inhibited infection of PHH with lab-generated HDV GT-1 carrying 24 different HBV envelopes (GT-A: 2, GT-B: 10, GT-C: 10, GT-D: 2) with a median EC50 value of 0.47 nM (range: 0.17-0.93 nM) overall and median EC50 values of 0.29-0.65 nM across HBV GTs. Lastly, bulevirtide inhibited infection of 264 HDV clinical isolates (mostly HBV GT-D [n=209] or HBV GT-A [n=34]) in PHH with median EC50 values of 0.40 nM (range: 0.10-1.27 nM) overall, 0.37 nM (range: 0.10-1.27 nM) against GT-D, and 0.65 nM (range: 0.27-1.08 nM) against GT-A. In the U.S., a surveillance study of individuals with HBV/HDV co-infection found that GT-D is the most prevalent HBV genotype (41%), followed by GT-A (33%).
Antiviral Resistance
In Cell Culture
HBV or HDV viruses resistant to bulevirtide in cell culture have not been identified to date. It is not possible to select for HBV or HDV resistance to antivirals using current cell culture systems. As described above, bulevirtide maintained activity against lab-generated HDV carrying envelopes from HBV GTs A-H, lab-generated HDV carrying 24 different envelope variants from HBV GTs A-D, and HDV clinical isolates in PHH. In addition, NTCP polymorphisms that disrupt bulevirtide activity while permitting HDV infection have not been identified to date.
In Clinical Trials
The antiviral activity of HEPCLUDEX 8.5 mg against different HBV and HDV genotypes was evaluated in trials MYR301 and MYR204. HBV GT-D was the most prevalent in these trials, in 129/150 (86%) participants, followed by GT-A in 12/150 (8%) participants. For participants treated with HEPCLUDEX 8.5 mg for 96 weeks, a virologic response (HDV RNA declining ≥ 2.0 log10 IU/mL or becoming undetectable) was achieved by 6/12 (50%) participants with GT-A and 112/129 (87%) participants with GT-D, which included 1/12 (8.3%) participants with GT-A who achieved undetectable HDV RNA compared with 52/129 (40%) participants with GT-D.
Resistance analysis was performed for participants who had virologic non-response (HDV RNA decline < 1 log10 IU/mL from baseline) or who experienced virologic breakthrough (2 consecutive increases in HDV RNA of ≥ 1 log10 IU/mL from nadir or 2 consecutive HDV RNA values ≥ lower limit of quantification [LLOQ] if previously < LLOQ during treatment with HEPCLUDEX).
In Trials MYR301 and MYR204, resistance analysis was performed for 13/150 participants at Week 48, 20/150 participants at Week 96, and 5/50 participants at Week 144 on HEPCLUDEX treatment (n=24 unique participants). Amino acid sequences for the bulevirtide region of HBsAg were determined at baseline for 17/24 participants with virologic non-response or breakthrough at any time point, and paired baseline and post-baseline sequence data were determined for 10/24 participants. For HDV, baseline sequence data for the HDAg region were determined for 23/24 participants, and paired baseline and post-baseline sequence data were determined for 21/24 participants.
No baseline polymorphisms identified in the bulevirtide region of HBsAg or in HDAg were associated with virologic non-response or breakthrough. Similarly, no post-baseline substitutions in the bulevirtide region or in HDAg showed an association with virologic breakthrough. All identified baseline and post-baseline variants retained susceptibility to bulevirtide in cell culture assays. A positive control for resistance was not available for these experiments.
Cross-Resistance
Cross-resistance is not expected between bulevirtide and nucleos(t)ide analog reverse transcriptase inhibitors approved for the treatment of chronic HBV infection given their different mechanisms of action.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADAs across trials.
In Trials MYR301 and MYR204, during the first 96 weeks of HEPCLUDEX treatment, the incidence of ADA was 66% (97/148) and the incidence of neutralizing antibodies (NAbs) in ADA-positive participants was 79% (77/97). The development of these antibodies did not appear to be associated with a reduced treatment response or differences in the safety profile.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Clinical Trials in Adults with Chronic HDV Infection Without Cirrhosis or With Compensated Cirrhosis
Trial MYR301
The efficacy of HEPCLUDEX once daily in the treatment of adults with chronic HDV infection without cirrhosis or with compensated cirrhosis is based on data through Week 144 from a multi-center, randomized, open-label, parallel-arm Phase 3 trial, Trial MYR301(NCT03852719), in which 100 participants received HEPCLUDEX 8.5 mg once daily. The MYR301 protocol specified the HEPCLUDEX dose as 10 mg; however, a dose recovery study later showed that the delivered dose was 8.5 mg.
In Trial MYR301, participants with chronic HDV infection without cirrhosis or with compensated cirrhosis were randomized to immediate treatment with HEPCLUDEX 8.5 mg once daily by subcutaneous injection for 144 weeks or to delayed treatment with an observational period of 48 weeks followed by HEPCLUDEX 8.5 mg once daily by subcutaneous injection for 96 weeks. Randomization was stratified by the presence or absence of compensated cirrhosis. The groups were followed for 96 weeks after treatment ended.
Demographic and clinical characteristics at baseline were balanced between treatment groups. The mean age was 41 years; 55% were male, 82% were White, 17% were Asian, and 1% were Black or African American. Forty-eight percent had compensated liver cirrhosis, 98% had HDV genotype 1, and 87% had HBV genotype D. Fifty-seven percent of participants had received previous interferon therapy and 59% were receiving nucleos(t)ide analog reverse transcriptase inhibitors for chronic hepatitis B.
The primary efficacy endpoint was combined response, defined as undetectable HDV RNA or ≥ 2 log10 IU/mL decline from baseline and ALT normalization, at Week 48.
Table 4 presents efficacy outcomes at Week 48 from Trial MYR301.
Table 4 Trial MYR301: Efficacy Outcomes of HEPCLUDEX versus Delayed Treatment at Week 48 HEPCLUDEX
(Immediate Treatment)
(N=50)Delayed Treatment
(N=51)Rate Difference
96% CI (%)CI=Confidence Interval, NA=Not applicable - * Defined as virologic response (HDV RNA undetectable or ≥ 2 log10 IU/mL decline) and ALT normalization.
- † p < 0.0001 (by Fisher’s exact test) for HEPCLUDEX vs. Delayed Treatment. The 96% confidence interval was calculated using score statistics with unconditional confidence limits method. A two-sided significance level of 0.04 was used to control the overall Type I error rate of 0.05 following a prespecified interim analysis conducted at the 0.01 level.
- ‡ Defined as HDV RNA below lower limit of quantification (LLOQ) (50 IU/mL) with target not detected or ≥ 2 log10 IU/mL decline from baseline.
- § Defined as an ALT value within the normal range: Russian sites, ≤ 31 U/L for females and ≤ 41 U/L for males; all other sites, ≤ 34 U/L for females and ≤ 49 U/L for males.
Combined Response* 48% 2% 46%†
(96% CI: 31% to 61%)Virologic Response‡ 76% 4% NA ALT Normalization§ 56% 12% NA At Week 48, the rate of undetectable HDV RNA (defined as less than the lower limit of quantification [LLOQ] [50 IU/mL] with target not detected) was 20% in the HEPCLUDEX group compared with 0% in the Delayed Treatment group. At Weeks 96 and 144, these rates increased to 36% and 50%, respectively, in the HEPCLUDEX group.
At Week 96, the HEPCLUDEX group demonstrated a 56% combined response rate, 82% virologic response rate (defined as undetectable HDV RNA or ≥ 2 log10 IU/mL decline from baseline), and 64% ALT normalization rate. At Week 144, these rates were 54%, 76%, and 60%, respectively. Participants in the Delayed Treatment group switched to HEPCLUDEX 8.5 mg once daily at Week 48. At Week 144 (after 96 weeks of treatment), the combined response rate in the Delayed Treatment group was 56%, the rate of undetectable HDV RNA was 52%, virologic response rate was 92%, and ALT normalization rate was 58%.
At posttreatment Week 24, 32% and 20% of participants in the HEPCLUDEX group and the Delayed Treatment group, respectively, had combined response, and 26% and 18% of participants, respectively, had undetectable HDV RNA. At posttreatment Week 96, 24% of participants in both the HEPCLUDEX group and the Delayed Treatment group had combined response and 22% and 20% of participants, respectively, had undetectable HDV RNA.
Phase 2b Trial MYR204
Further supportive data from an additional 50 adult participants with chronic HDV infection without cirrhosis or with compensated cirrhosis receiving HEPCLUDEX 8.5 mg subcutaneously once daily for 96 weeks is available from a randomized, open-label, exploratory Phase 2b trial, Trial MYR204 (NCT03852433). At Week 96, 48% and 22% of participants achieved combined response and undetectable HDV RNA, respectively. At 24 weeks posttreatment, response rates were 26% and 12%, respectively.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
HEPCLUDEX (bulevirtide-gmod) for injection 8.5 mg is supplied in a carton (NDC: 61958-3104-1) of 30 single-dose vials.
Each single-dose vial contains a sterile, preservative-free, white to off-white lyophilized powder or cake. It requires reconstitution prior to administration by subcutaneous injection [see Dosage and Administration (2.2)]. The container closure is not made with natural rubber latex.
Store HEPCLUDEX vials at room temperature between 68 °F to 77 °F (20 °C to 25 °C), excursions permitted from 59 °F to 86 °F (15 °C to 30 °C).
After reconstitution, use vials immediately. Discard unused portion.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use) for proper preparation and administration instructions.
Important Preparation and Administration Considerations
Healthcare professionals should train patients or caregivers in the proper technique for reconstituting HEPCLUDEX with Sterile Water for Injection and administering subcutaneous injections using a syringe and consider preparation and administration of the first dose under the supervision of a healthcare provider.
Inform patients that the Sterile Water for Injection, syringes, and needles needed for preparation and injection of HEPCLUDEX are obtained separately from the pharmacy.
Exacerbation of Hepatitis D and B after Discontinuation of Treatment
Inform patients that discontinuation of HEPCLUDEX may result in severe acute exacerbations of hepatitis D and B. Advise the patient to inform their healthcare provider before they discontinue HEPCLUDEX [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions Including Anaphylaxis
Advise patients that hypersensitivity reactions, including anaphylaxis, have been reported with HEPCLUDEX. Advise patients to immediately discontinue HEPCLUDEX and alert their healthcare provider if signs or symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur [see Warnings and Precautions (5.2)].
Missed Dosage
Inform patients that it is important to take HEPCLUDEX on a regular dosing schedule and to avoid missing doses. If a dose is missed, that dose should be taken as soon as possible. However, if it is almost time for the next dose, skip the missed dose and resume the original schedule [see Dosage and Administration (2.1)].
Treatment Duration
Advise patients that in the treatment of chronic hepatitis D, the optimal duration of treatment is unknown [see Dosage and Administration (2.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 05/2026 PATIENT INFORMATION HEPCLUDEX® (hep-CLUE-decks)
(bulevirtide-gmod)
for injection, for subcutaneous useWhat is the most important information I should know about HEPCLUDEX?
HEPCLUDEX can cause serious side effects, including:-
Worsening of hepatitis delta virus (HDV) and hepatitis B virus (HBV) infection. Your HDV or HBV infection may get worse (flare-up) if you stop taking HEPCLUDEX, especially if you have cirrhosis. A "flare-up" is when your HDV or HBV infection suddenly returns in a worse way than before.
- Do not run out of HEPCLUDEX. Refill your prescription or talk to your healthcare provider before your HEPCLUDEX is all gone.
- Do not stop taking HEPCLUDEX without first talking to your healthcare provider.
- If you stop taking HEPCLUDEX, your healthcare provider will need to check your health often and do blood tests regularly for at least 6 months to check your liver, and may give you medicine to treat your virus infection. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking HEPCLUDEX.
What is HEPCLUDEX?
HEPCLUDEX is a prescription medicine used to treat chronic (long-lasting) hepatitis delta virus (HDV) infection in adults without cirrhosis or with compensated cirrhosis.
It is not known if HEPCLUDEX is safe and effective in children less than 18 years of age.Before taking HEPCLUDEX, tell your healthcare provider about all your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if HEPCLUDEX can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if HEPCLUDEX can pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with HEPCLUDEX.
How should I take HEPCLUDEX?
See the Instructions for Use for instructions on how to prepare and inject your prescribed dose of HEPCLUDEX.
- Take HEPCLUDEX exactly as your healthcare provider tells you to take it. Do not change your dose unless your healthcare provider tells you to.
- HEPCLUDEX is given as an injection under your skin (subcutaneous) by you or a caregiver.
- Your healthcare provider should show you or your caregiver how to prepare and inject HEPCLUDEX before you use it for the first time.
- Take HEPCLUDEX 1 time each day.
- Do not change your dose or stop taking HEPCLUDEX without first talking with your healthcare provider. Stay under a healthcare provider's care when taking HEPCLUDEX.
- Do not miss a dose of HEPCLUDEX.
- If you miss a dose, take that dose as soon as possible on that day. However, if it is almost time for your next dose, skip the missed dose and go back to the regular dosing schedule.
- If you take too much HEPCLUDEX, call your healthcare provider or go to the nearest hospital emergency room right away.
- When your HEPCLUDEX supply starts to run low, get more from your healthcare provider or pharmacy. This is very important because your HDV and HBV infection may get worse (flare-up) if you stop taking HEPCLUDEX.
What are the possible side effects of HEPCLUDEX?
HEPCLUDEX can cause serious side effects, including:- See “What is the most important information I should know about HEPCLUDEX?”
-
Allergic reactions, including severe allergic reactions (anaphylaxis). Stop taking HEPCLUDEX and tell your healthcare provider or get emergency medical help right away if you develop any signs or symptoms of a severe allergic reaction, including:
- trouble swallowing or breathing
- throat tightness
- severe rash, hives, or itching
- swelling of the mouth, lips, tongue, or face
- dizziness or fainting
- stomach area pain, nausea, diarrhea, or vomiting
The most common side effects of HEPCLUDEX include: - injection site reactions, including infection, redness, itching, swelling, bruising, rash, and pain at the injection site
- headache
- stomach area pain
- feeling tired
- itchy skin
These are not all of the possible side effects of HEPCLUDEX.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store HEPCLUDEX?
- Store HEPCLUDEX vials at room temperature between 68 °F to 77 °F (20 °C to 25 °C).
- After mixing, use HEPCLUDEX vial right away. Throw away (discard) unused portion.
General information about the safe and effective use of HEPCLUDEX.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use HEPCLUDEX for a condition for which it was not prescribed. Do not give HEPCLUDEX to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about HEPCLUDEX that is written for health professionals.What are the ingredients in HEPCLUDEX?
Active ingredient: bulevirtide-gmod
Inactive ingredients: histidine, mannitol, and sucrose, and may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
Manufactured by: Gilead Sciences, Inc., Foster City, CA 94404
U.S. License No. 2258
HEPCLUDEX is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2026 Gilead Sciences, Inc. All rights reserved.
761468-GS-000
For more information, call 1-800-445-3235 or go to www.HEPCLUDEX.com. -
Worsening of hepatitis delta virus (HDV) and hepatitis B virus (HBV) infection. Your HDV or HBV infection may get worse (flare-up) if you stop taking HEPCLUDEX, especially if you have cirrhosis. A "flare-up" is when your HDV or HBV infection suddenly returns in a worse way than before.
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
HEPCLUDEX® [hep-CLUE-decks]
(bulevirtide-gmod) for injection,
for subcutaneous use
single-dose vial
This Instructions for Use contains information on how to prepare and inject HEPCLUDEX.
Read this Instructions for Use before you start using HEPCLUDEX and each time you get a refill. There may be new information.Do not use HEPCLUDEX unless you have been trained by your healthcare provider. Your healthcare provider should show you or your caregiver how to prepare and inject HEPCLUDEX before you use it for the first time. If you have questions or do not understand the instructions, talk to your healthcare provider.
- Read the Patient Information that comes with HEPCLUDEX for more information.
- If you have any problems using HEPCLUDEX, including if you do not give a full dose of medicine, contact your healthcare provider.
Important Information You Need to Know Before Injecting HEPCLUDEX:
- Your carton of HEPCLUDEX contains 30 vials. This is a 30-day supply of medicine.
- HEPCLUDEX comes as a powder or cake in a vial. HEPCLUDEX must be mixed and dissolved with Sterile Water for Injection before use.
- Mixed HEPCLUDEX must be used right away. Do not reuse or save mixed HEPCLUDEX for later use.
- Your pharmacy will provide you with the following supplies for mixing and injecting HEPCLUDEX:
- Transfer Needles
- Empty Syringes
- Sterile Water for Injection Vials
- Injection Needles
- Note: The supplies not included in the HEPCLUDEX carton may slightly differ in size, shape and color from the figures in this Instructions for Use. These instructions provide the steps to mix and inject HEPCLUDEX using an example set of supplies. If you have any questions, contact your healthcare provider or pharmacist.
- Only use the Sterile Water for Injection Vial provided by your pharmacy to mix HEPCLUDEX. Sterile Water for Injection is different from bottled water or water from a faucet. Sterile Water for Injection Vials are for one-time use only and must be thrown away (disposed of) after that use, even if there is water remaining in the vial. Reusing Sterile Water for Injection Vials may lead to infection.
- Do not touch the needle or syringe tip or allow the needle or syringe tip to touch any other surface. If you touch the needle or syringe tip or allow the needle or syringe tip to touch any other surface, throw them away in a sharps disposal container (see “Disposing of Used Needles”) and use a new needle or syringe. Touching the needle or syringe tip would make it non-sterile and could lead to an infection.
- Do not reuse any of the supplies. Throw away any used Transfer Needles and Injection Needles in a sharps disposal container. See “Disposing of Used Needles”.
How should I store HEPCLUDEX?
- Store HEPCLUDEX Vials at room temperature between 68 °F to 77 °F (20 °C to 25 °C).
- After mixing, use the HEPCLUDEX Vial right away. Throw away (discard) unused portion.
- Store all other supplies according to the manufacturer instructions.
- Keep HEPCLUDEX Vials, all supplies, and all medicines out of the reach of children.
Guide to Parts
HEPCLUDEX Vial
Supplies Needed to Mix HEPCLUDEX
(not included in the HEPCLUDEX carton; supplies from the pharmacy may look different than supplies pictured here)
Sterile Water for Injection Vial
(used to mix HEPCLUDEX)
Sterile Transfer Needles
(used to transfer Sterile Water for Injection to HEPCLUDEX Vial and to withdraw mixed HEPCLUDEX)

Supplies Needed to Inject HEPCLUDEX After Mixing
(not included in the HEPCLUDEX carton; supplies from the pharmacy may look different than supplies pictured here)Sterile Injection Needle with Safety Shield
(used to inject after HEPCLUDEX has been mixed)
Do not insert the Injection Needle into the vials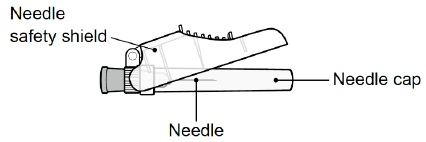
Note: When the needle safety shield is closed over the Injection Needle, the needle is locked and cannot be used.
Additional Supplies Needed (may be provided by pharmacy)
Gather HEPCLUDEX Vial and Supplies
Step 1.
- Take 1 vial from the HEPCLUDEX Vial carton.
- Gather the supplies for mixing and injecting (supplies provided by pharmacy):
- 2 Sterile Transfer Needles (21G, 1 inch)
- 1 Sterile Water for Injection Vial (5 mL or 10 mL)
- 1 Sterile Syringe (3 mL)
- 1 Sterile Injection Needle with safety shield (27G, ½ inch)
- Gather additional supplies needed (may be provided by pharmacy):
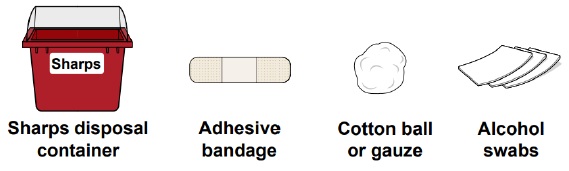
- 1 sharps disposal container
- 1 adhesive bandage
- 1 cotton ball or gauze
- 3 alcohol swabs
- Place the HEPCLUDEX Vial and all supplies on a clean flat surface.
Step 2.
- Check the expiration dates (YYYY/MM) on the HEPCLUDEX Vial (see Figure A) and on the supplies for mixing and injecting HEPCLUDEX.
- Do not use if any of the expiration dates have passed.
- Check the HEPCLUDEX Vial and the supplies packaging for damage.
- Do not use if the vial or supplies packaging has been dropped or appears damaged.
Step 3.
- Wash your hands well with soap and water (see Figure B).
Prepare HEPCLUDEX Vial,
Sterile Water for Injection Vial, and SyringeStep 4.
- Check the HEPCLUDEX Vial, Sterile Water for Injection Vial, Transfer Needles, Syringe, and Injection Needle (supplies) for damage.
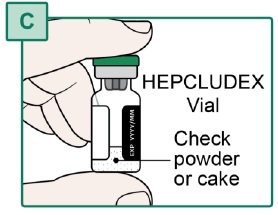
- Check the powder or cake in the HEPCLUDEX Vial. The powder or cake should be a white to off-white color (see Figure C).
-
Do not use if:
- the vials or supplies look damaged or have dropped onto a hard surface.
- the vial caps are missing, loose, or damaged.
- the powder or cake in the HEPCLUDEX Vial is discolored.
Step 5.
Step 6.
Step 7.
- Remove a Transfer Needle and Syringe from their packaging.
- Hold the Syringe and the Transfer Needle firmly.
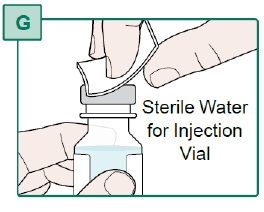
- Turn the Transfer Needle to the right (clockwise) onto the Syringe until it is fully connected (see Figure H).
- Remove the needle cap from the Transfer Needle by pulling it straight off (see Figure I). Keep the needle cap for later use.
- Do not use the Transfer Needle if it is bent or appears damaged.
Mix HEPCLUDEX
Step 8.
- Hold the Sterile Water for Injection Vial on a flat surface.
- Insert the Transfer Needle all the way into the Sterile Water for Injection Vial by pushing it straight through the center of the rubber stopper (see Figure J).
- Turn the Sterile Water for Injection Vial and Syringe upside down so the vial is pointing upwards.
- Be sure that the tip of the Transfer Needle is in the liquid.
- With one hand, keep the Syringe and Transfer Needle in place.
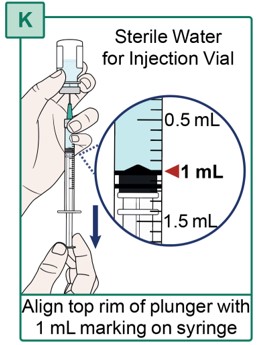
- With the other hand, carefully pull the plunger down until the top rim of the plunger lines up with the 1 mL marking on the Syringe (see Figure K).
- If you see large air gaps in the Syringe, push the plunger all the way up and repeat Step 9.
Step 10.
- Hold the plunger in place and turn the Sterile Water for Injection Vial and Syringe upright. Hold the vial on a flat surface.
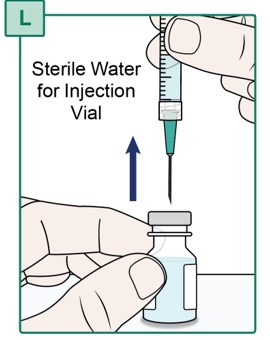
- Remove the water-filled Syringe with the Transfer Needle attached from the Sterile Water for Injection Vial by holding the Syringe body and pulling it straight up (see Figure L).
- Do not touch the Transfer Needle or let it touch any surface. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
- Throw away the Sterile Water for Injection Vial into the household trash. Sterile Water for Injection Vials are for one-time use only and must be thrown away (disposed of) after that use, even if there is water remaining in the vial. Reusing Sterile Water for Injection Vials may lead to infection.
Step 11.
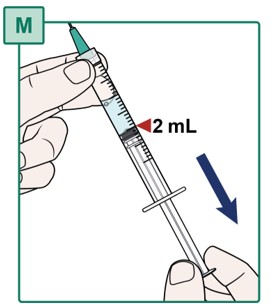
- Pull the plunger all the way down to the 2 mL marking of the Syringe to add air to the Syringe (see Figure M).
- Do not pull the plunger out of the Syringe.
- Get the HEPCLUDEX Vial and hold the vial on a flat surface.
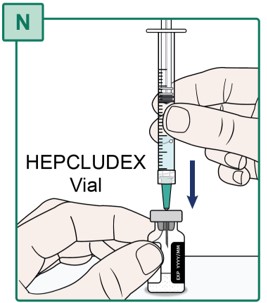
- Insert the Transfer Needle all the way into the HEPCLUDEX Vial by pushing it straight through the center of the rubber stopper (see Figure N).
Step 12.
- Turn the HEPCLUDEX Vial and Syringe upside down so the vial is pointing upwards.
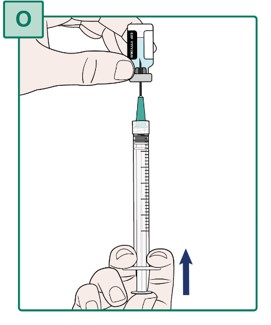
- Slowly push the plunger all the way up until the Syringe is empty (see Figure O).
- Pushing the plunger all the way up fills the HEPCLUDEX Vial with air and the Sterile Water for Injection.
Step 13.
- Hold the plunger in place and turn the HEPCLUDEX Vial and Syringe upright.
- Hold the vial on a flat surface.
- Remove the empty Syringe with the Transfer Needle attached from the HEPCLUDEX Vial by pulling it straight up (see Figure P).
Step 14.
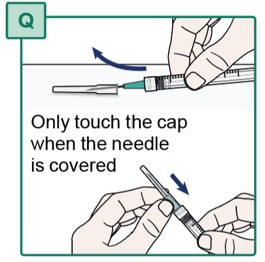
- Using one hand, slide the Transfer Needle into its cap and scoop upwards to cover the needle (see Figure Q).
- When the Transfer Needle is covered, carefully push the needle cap towards the Syringe to fully attach it.
- Do not touch the Transfer Needle. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
- Keep the Syringe with Transfer Needle attached for later use in Step 18.
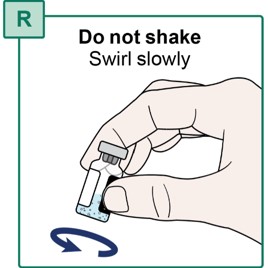
- Slowly swirl the HEPCLUDEX Vial for a few seconds to mix the powder with the Sterile Water for Injection (see Figure R).
- If there is powder stuck to the vial wall or rubber stopper, continue to swirl the vial slowly until it is removed.
- Do not shake the vial.
- Place the HEPCLUDEX Vial upright on a flat surface.
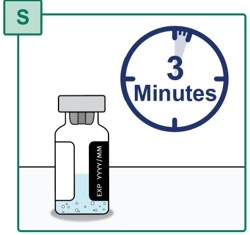
- Wait for the powder to fully dissolve (see Figure S). This may take up to 3 minutes.
Step 17.
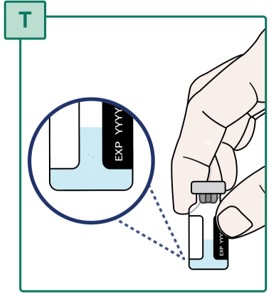
- Check that the powder is fully dissolved. The medicine should look clear without any clumps or particles (see Figure T).
- It is normal to see foam in the medicine.
- Do not use the medicine if you see particles or clumps that have not completely dissolved, including powder stuck to the vial wall or rubber stopper.
- Repeat Steps 15 and 16 if the powder is not fully dissolved.
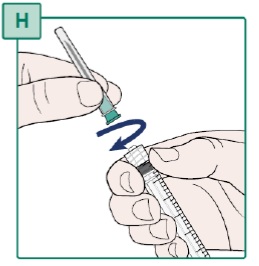
- Hold the Syringe firmly (see Figure U).
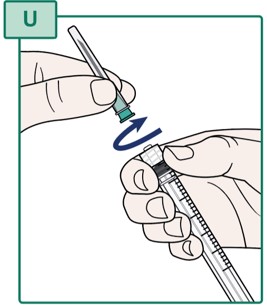
- Turn the capped Transfer Needle to the left (counterclockwise) to remove it from the Syringe.
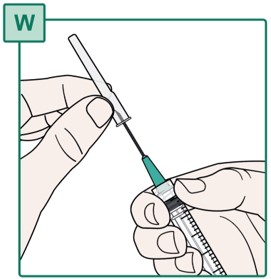
- Throw away (dispose of) the Transfer Needle in the sharps disposal container. See “Disposing of Used Needles”.
- Do not touch the Syringe tip or allow it to touch any surface. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
Step 19.
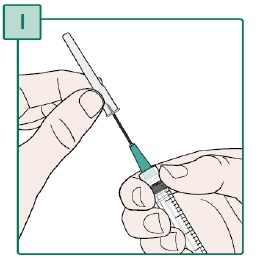
- Remove the second Transfer Needle from its packaging.
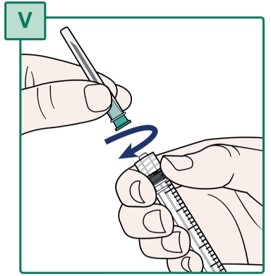
- Hold the Syringe and the Transfer Needle firmly.
- Turn the Transfer Needle to the right (clockwise) onto the Syringe until it is fully connected (see Figure V).
- Remove the needle cap from the Transfer Needle by pulling it straight off (see Figure W). Keep the needle cap for later use.
- Do not use the Transfer Needle if it is bent or appears damaged.
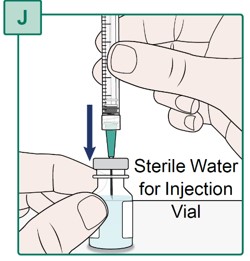
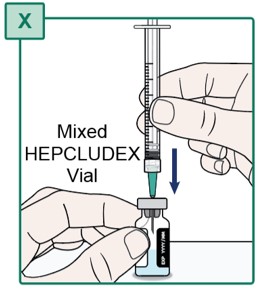
Step 20.
- Hold the mixed HEPCLUDEX Vial on a flat surface.
- Insert the Transfer Needle all the way into the HEPCLUDEX Vial by pushing it straight through the center of the rubber stopper (see Figure X).
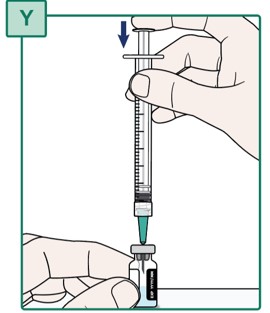
- Check the plunger is pushed all the way down. If the plunger has risen, push the plunger all the way down (see Figure Y). All air should be pushed out of the Syringe.
- Keep the plunger fully pushed down.
Step 22.
Step 23.
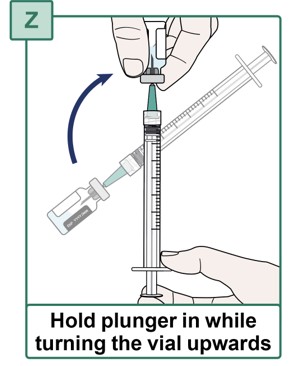
- Hold the HEPCLUDEX Vial pointing upwards.
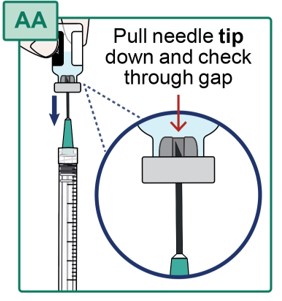
- Pull the Transfer Needle down until the needle tip is near the bottom of the rubber stopper in the vial. Check the position through the gap in the rubber stopper (see Figure AA).
- Do not completely remove the needle from the vial.
Step 24.
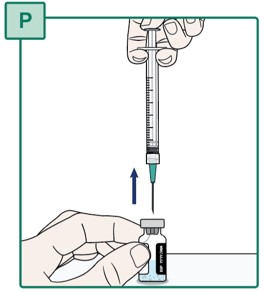
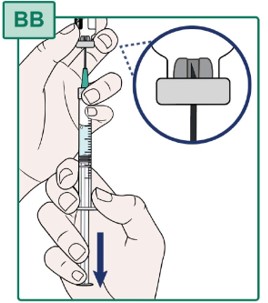
- Keep the Transfer Needle tip in place inside the HEPCLUDEX Vial (see Figure BB).
- Carefully pull the plunger until all the medicine is removed from the HEPCLUDEX Vial (see Figure BB).
- Hold the plunger in place to keep all the medicine in the Syringe.
- An air gap above the medicine is normal at this step and will be removed before injection. Small air bubbles in the medicine are normal.
- Leftover foam in the vial is normal.
- Do not pull the plunger out of the Syringe.
Step 25.
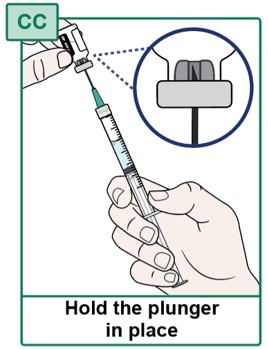
- Before continuing, check that all the liquid medicine is removed from the HEPCLUDEX Vial (see Figure CC).
- Hold the plunger in place and turn the HEPCLUDEX Vial and Syringe upright.
- Hold the vial on a flat surface.
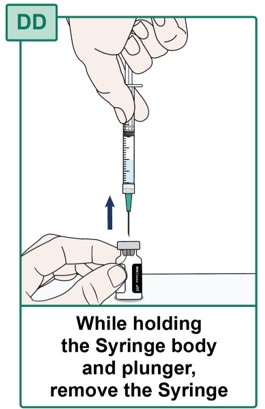
- While holding the Syringe body and plunger in place, remove the Syringe with the Transfer Needle attached from the HEPCLUDEX Vial (see Figure DD).
- Do not push the plunger until ready to inject. Pushing the plunger will cause medicine to come out of the Syringe.
- Throw away the HEPCLUDEX Vial into the household trash.
Step 26.
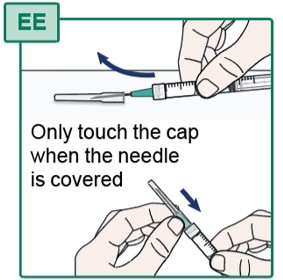
- Using one hand, slide the Transfer Needle into its cap and scoop upwards to cover the needle (see Figure EE).
- When the Transfer Needle is covered, carefully push the needle cap towards the Syringe to fully attach it.
- Do not touch the Transfer Needle. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
Step 27.
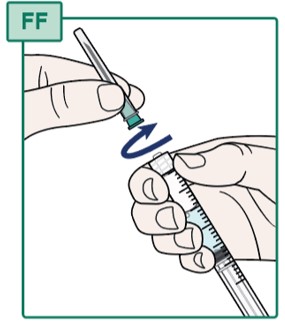
- Hold the Syringe firmly (see Figure FF).
- Turn the capped Transfer Needle to the left (counterclockwise) to remove it from the Syringe.
- Throw away (dispose of) the Transfer Needle in the sharps disposal container. See “Disposing of Used Needles”.
- Do not touch the Syringe tip or allow it to touch any surface. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
Inject HEPCLUDEX
Step 28.
- Remove the Injection Needle from the packaging.
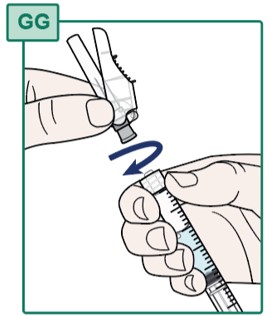
- Hold the Syringe firmly (see Figure GG).
- Turn the Injection Needle to the right (clockwise) onto the Syringe until it is fully connected (see Figure GG).
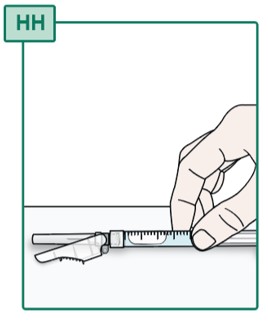
- Place the Syringe with the Injection Needle attached on a flat surface (see Figure HH).
- Do not remove the needle cap until you are ready to inject the medicine.
- Do not push the Syringe plunger until you are ready to inject the medicine, this may cause liquid to come out.
Step 29.
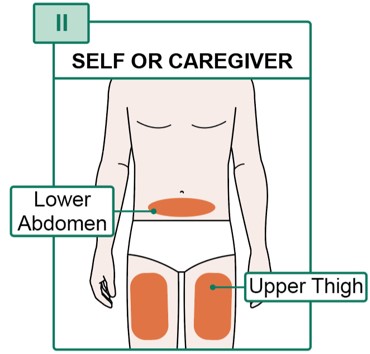
- To inject yourself, choose a site on your lower abdomen (stomach) or upper thighs (see Figure II).
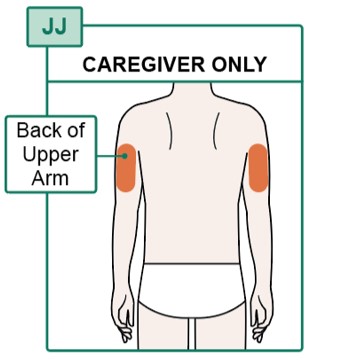
- If someone else is injecting the medicine for you, they can also choose the back of the upper arms (see Figure JJ).
- Do not inject into the same site on consecutive days.
- Do not inject into moles, scars, bruises, or tattoos or into areas where the skin is red, hard, tender, or injured.
- Do not inject into the 1-inch (2.5-cm) area around the belly button (navel).
Step 30.
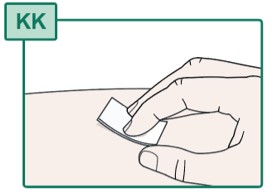
- Clean the injection site with a new alcohol swab (see Figure KK).
- Allow the skin to air dry.
- Do not touch or blow on the injection site after it has been cleaned.
Step 31.
- Hold the Syringe firmly.
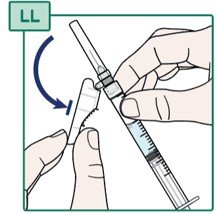
- Gently pull back the Injection Needle safety shield (see Figure LL).
- Do not remove the needle safety shield from the Injection Needle.
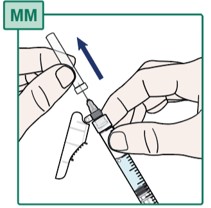
- Carefully pull off the Injection Needle cap (see Figure MM) and throw it away into the household trash.
- Do not touch the Injection Needle or allow it to touch any other surface. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
- Do not recap the needle.
Step 33.
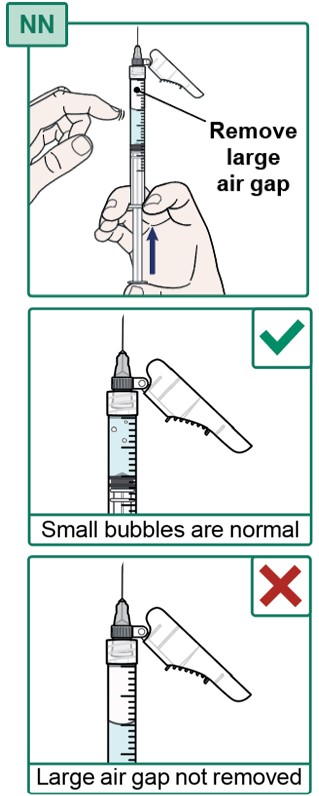
- Make sure the Injection Needle is pointing up and the air gap in the Syringe is at the top.
- If there are large air bubbles, tap the Syringe with your finger until the air bubbles move to the top.
- If there is a large air gap, carefully push the plunger up to remove most of the air gap at the top (see Figure NN).
- Small air bubbles are normal and do not need to be removed.
- It is okay for a few drops of medicine to come out of the Syringe.
- Do not touch the Injection Needle or allow it to touch any other surface. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
Step 34.
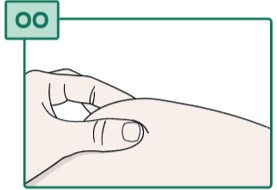
- Pinch the cleaned injection site (see Figure OO).
Step 35.
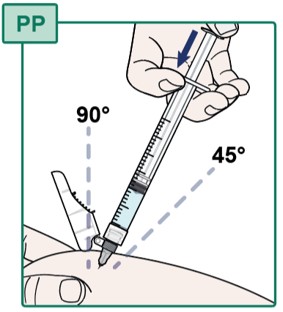
- With your other hand, insert the Injection Needle all the way into the pinched skin at a 45 to 90 degree angle (see Figure PP).
- Slowly push down on the plunger until all of the medicine is injected into the skin.
- Carefully pull out the needle at the same angle as you inserted it.
After Injecting HEPCLUDEX
Step 36.
- Right after the injection, place the Injection Needle safety shield on the work surface (see Figure QQ).
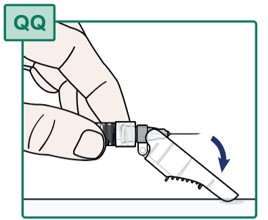
- Press the needle toward the needle safety shield to cover the needle and lock the safety shield (see Figure RR).
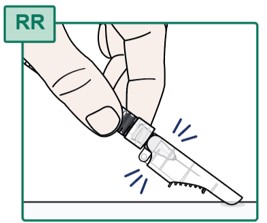
- Do not touch the Injection Needle. See “Important Information You Need to Know Before Injecting HEPCLUDEX”.
- Do not remove the needle from the syringe.
Step 37.
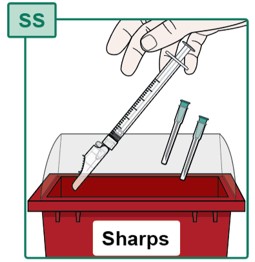
- Throw away (dispose of) the used Syringe with Injection Needle attached in the sharps disposal container (see Figure SS). See “Disposing of Used Needles” below.
- Dispose of any remaining waste in the household trash.
Step 38.
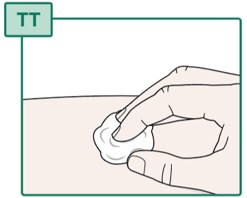
- If there is bleeding at the injection site, gently press a cotton ball or gauze over the injection site for several seconds (see Figure TT).
- Cover the injection site with an adhesive bandage if needed.
- Do not rub the injection site.
- Throw away (dispose of) the used Syringe with Injection Needle attached and the used Transfer Needles in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles with your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic;
- able to be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out;
- upright and stable during use;
- leak-resistant; and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your filled, used sharps disposal container.
- Keep the sharps disposal container out of the reach of children.
For more information, call 1-800-445-3235 or go to www.HEPCLUDEX.com.
Manufactured by: Gilead Sciences, Inc., Foster City, CA 94404
HEPCLUDEX is a trademark of Gilead Sciences, Inc., or its related companies.
© 2026 Gilead Sciences, Inc. All rights reserved.
761468-GS-IFU-V-000This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: May/2026
- PRINCIPAL DISPLAY PANEL - 8.5 mg Vial Label
- PRINCIPAL DISPLAY PANEL - 8.5 mg Vial Carton
-
INGREDIENTS AND APPEARANCE
HEPCLUDEX
bulevirtide injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-3104 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BULEVIRTIDE ACETATE (UNII: LLM855Y3S3) (BULEVIRTIDE - UNII:WKM56H3TLB) BULEVIRTIDE ACETATE 8.5 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) SUCROSE (UNII: C151H8M554) HISTIDINE (UNII: 4QD397987E) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) NITROGEN (UNII: N762921K75) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-3104-1 30 in 1 CARTON 05/22/2026 1 NDC: 61958-3104-2 1 in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761468 05/22/2026 Labeler - Gilead Sciences, Inc. (185049848)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.