RIFAMPIN injection, powder, lyophilized, for solution
Rifampin by
Drug Labeling and Warnings
Rifampin by is a Prescription medication manufactured, distributed, or labeled by AKORN, Inc., Patheon Italia S.p.A. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Rifampin for injection USP contains rifampin 600 mg, sodium formaldehyde sulfoxylate dihydrate 10 mg, and sodium hydroxide to adjust pH. Rifampin for injection is a sterile, lyophilized powder for intravenous infusion only.
Rifampin is a semisynthetic antibiotic derivative of rifamycin SV. Rifampin is a red-brown crystalline powder very slightly soluble in water at neutral pH, freely soluble in chloroform, soluble in ethyl acetate and in methanol. Its molecular weight is 822.95 and its chemical formula is C43H58N4O12. The chemical name for rifampin is either:
3-[[(4-Methyl-1-piperazinyl)imino]methyl]rifamycin
or
5,6,9,17,19,21-hexahydroxy-23-methoxy-2,4,12,16,20,22–heptamethyl-8-[N-(4-methyl-1-piperazinyl)formimidoyl]-2,7- (epoxypentadeca[1,11,13]trienimino)naphtho[2,1-b]furan-1,11(2H)-dione 21-acetate.
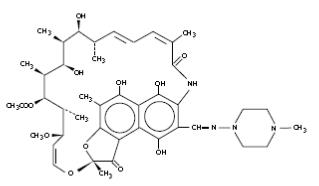
Its structural formula is:
-
CLINICAL PHARMACOLOGY
Intravenous Administration
After intravenous administration of a 300 or 600 mg dose of rifampin infused over 30 minutes to healthy male volunteers (n=12), mean peak plasma concentrations were 9.0 ± 3.0 and 17.5 ± 5.0 mcg/mL, respectively. Total body clearances after the 300 and 600 mg IV doses were 0.19 ± 0.06 and 0.14 ± 0.03 L/hr/kg, respectively. Volumes of distribution at steady state were 0.66 ± 0.14 and 0.64 ± 0.11 L/kg for the 300 and 600 mg IV doses, respectively. After intravenous administration of 300 or 600 mg doses, rifampin plasma concentrations in these volunteers remained detectable for 8 and 12 hours, respectively (see Table).
Plasma Concentrations (mean ± standard deviation, mcg/mL)
Rifampin
Dosage IV30 min
1 hr
2 hr
4 hr
8 hr
12 hr
300 mg
8.9±2.9
4.9±1.3
4.0±1.3
2.5±1.0
1.1±0.6
<0.4
600 mg
17.4±5.1
11.7±2.8
9.4±2.3
6.4±1.7
3.5±1.4
1.2±0.6
Plasma concentrations after the 600 mg dose, which were disproportionately higher (up to 30% greater than expected) than those found after the 300 mg dose, indicated that the elimination of larger doses was not as rapid.
After repeated once-a-day infusions (3 hr duration) of 600 mg in patients (n=5) for 7 days, concentrations of IV rifampin decreased from 5.81 ± 3.38 mcg/mL 8 hours after the infusion on day 1 to 2.6 ± 1.88 mcg/mL 8 hours after the infusion on day 7.
Rifampin is widely distributed throughout the body. It is present in effective concentrations in many organs and body fluids, including cerebrospinal fluid. Rifampin is about 80% protein bound. Most of the unbound fraction is not ionized and therefore diffuses freely into tissues.
Rifampin is rapidly eliminated in the bile and undergoes progressive enterohepatic circulation and deacetylation to the primary metabolite, 25-desacetyl-rifampin. This metabolite is microbiologically active. Less than 30% of the dose is excreted in the urine as rifampin or metabolites. Serum concentrations do not differ in patients with renal failure at a studied dose of 300 mg and consequently, no dosage adjustment is required.
Pediatrics
Intravenous Administration
In pediatric patients 0.25 to 12.8 years old (n=12), the mean peak serum concentration of rifampin at the end of a 30 minute infusion of approximately 300 mg/m2 was 25.9 ± 1.3 mcg/mL; individual peak concentrations 1 to 4 days after initiation of therapy ranged from 11.7 to 41.5 mcg/mL; individual peak concentrations 5 to 14 days after initiation of therapy were 13.6 to 37.4 mcg/mL. The individual serum half-life of rifampin changed from 1.04 to 3.81 hours early in therapy to 1.17 to 3.19 hours 5 to 14 days after therapy was initiated.
Microbiology
Mechanism of Action
Rifampin inhibits DNA-dependent RNA polymerase activity in susceptible Mycobacterium tuberculosis organisms. Specifically, it interacts with bacterial RNA polymerase but does not inhibit the mammalian enzyme.
Resistance
Organisms resistant to rifampin are likely to be resistant to other rifamycins. In addition, resistance to rifampin has been determined to occur as single-step mutations of the DNA-dependent RNA polymerase. Since resistance can emerge rapidly, appropriate susceptibility tests should be performed in the event of persistent positive cultures.
Activity in vitro and in vivo
Rifampin has bactericidal activity in vitro against slow and intermittently growing M tuberculosis organisms.
Rifampin has been shown to be active against most strains of the following microorganisms, both in vitro and in clinical infections as described in the INDICATIONS AND USAGE section.
Aerobic Gram-Negative Microorganisms:
Neisseria meningitidis"Other" Microorganisms:
Mycobacterium tuberculosisThe following in vitro data are available, but their clinical significance is unknown.
Rifampin exhibits in vitro activity against most strains of the following microorganisms; however, the safety and effectiveness of rifampin in treating clinical infections due to these microorganisms have not been established in adequate and well-controlled trials.
Aerobic Gram-Positive Microorganisms:
Staphylococcus aureus (including Methicillin-Resistant S. aureus/MRSA)
Staphylococcus epidermidisAerobic Gram-Negative Microorganisms:
Haemophilus influenzae"Other" Microorganisms:
Mycobacterium lepraeβ-lactamase production should have no effect on rifampin activity.
-
INDICATIONS AND USAGE
In the treatment of both tuberculosis and the meningococcal carrier state, the small number of resistant cells present within large populations of susceptible cells can rapidly become the predominant type. Bacteriologic cultures should be obtained before the start of therapy to confirm the susceptibility of the organism to rifampin and they should be repeated throughout therapy to monitor the response to treatment. Since resistance can emerge rapidly, susceptibility tests should be performed in the event of persistent positive cultures during the course of treatment. If test results show resistance to rifampin and the patient is not responding to therapy, the drug regimen should be modified.
Tuberculosis
Rifampin is indicated in the treatment of all forms of tuberculosis.
A three-drug regimen consisting of rifampin, isoniazid, and pyrazinamide [e.g., RIFATER®]1 is recommended in the initial phase of short-course therapy which is usually continued for 2 months. The Advisory Council for the Elimination of Tuberculosis, the American Thoracic Society, and Centers for Disease Control and Prevention recommend that either streptomycin or ethambutol be added as a fourth drug in a regimen containing isoniazid (INH), rifampin, and pyrazinamide for initial treatment of tuberculosis unless the likelihood of INH resistance is very low. The need for a fourth drug should be reassessed when the results of susceptibility testing are known. If community rates of INH resistance are currently less than 4%, an initial treatment regimen with less than four drugs may be considered.
Following the initial phase, treatment should be continued with rifampin and isoniazid [eg, RIFAMATE®]2 for at least 4 months. Treatment should be continued for longer if the patient is still sputum or culture positive, if resistant organisms are present, or if the patient is HIV positive.
Rifampin for injection is indicated for the initial treatment and retreatment of tuberculosis when the drug cannot be taken by mouth.
Meningococcal Carriers
Rifampin is indicated for the treatment of asymptomatic carriers of Neisseria meningitidis to eliminate meningococci from the nasopharynx. Rifampin is not indicated for the treatment of meningococcal infection because of the possibility of the rapid emergence of resistant organisms. (See WARNINGS).
Rifampin should not be used indiscriminately, and therefore, diagnostic laboratory procedures, including serotyping and susceptibility testing, should be performed for establishment of the carrier state and the correct treatment. So that the usefulness of rifampin in the treatment of asymptomatic meningococcal carriers is preserved, the drug should be used only when the risk of meningococcal disease is high.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of rifampin and other antibacterial drugs, rifampin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
CONTRAINDICATIONS
Rifampin for injection is contraindicated in patients with a history of hypersensitivity to rifampin or any of the components, or to any of the rifamycins. (See WARNINGS).
Rifampin is contraindicated in patients who are also receiving ritonavir-boosted saquinavir due to an increased risk of severe hepatocellular toxicity. (See PRECAUTIONS, Drug Interactions.)
Rifampin is contraindicated in patients who are also receiving atazanavir, darunavir, fosamprenavir, saquinavir, or tipranavir due to the potential of rifampin to substantially decrease plasma concentrations of these antiviral drugs, which may result in loss of antiviral efficacy and/or development of viral resistance.
-
WARNINGS
Rifampin has been shown to produce liver dysfunction. Fatalities associated with jaundice have occurred in patients with liver disease and in patients taking rifampin with other hepatotoxic agents. Patients with impaired liver function should be given rifampin only in cases of necessity and then with caution and under strict medical supervision. In these patients, careful monitoring of liver function, especially SGPT/ALT and SGOT/AST should be carried out prior to therapy and then every 2 to 4 weeks during therapy. If signs of hepatocellular damage occur, rifampin should be withdrawn.
In some cases, hyperbilirubinemia resulting from competition between rifampin and bilirubin for excretory pathways of the liver at the cell level can occur in the early days of treatment. An isolated report showing a moderate rise in bilirubin and/or transaminase level is not in itself an indication for interrupting treatment; rather, the decision should be made after repeating the tests, noting trends in the levels, and considering them in conjunction with the patient's clinical condition.
Rifampin has enzyme-inducing properties, including induction of delta amino levulinic acid synthetase. Isolated reports have associated porphyria exacerbation with rifampin administration.
The possibility of rapid emergence of resistant meningococci restricts the use of rifampin for injection to short-term treatment of the asymptomatic carrier state. Rifampin for injection is not to be used for the treatment of meningococcal disease.
Systemic hypersensitivity reactions, including Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) syndrome, may occur in patients receiving rifampin for injection (see ADVERSE REACTIONS). Signs and symptoms of hypersensitivity reactions may include fever, rash, urticaria, angioedema, hypotension, acute bronchospasm, conjunctivitis, thrombocytopenia, neutropenia, elevated liver transaminases or flu-like syndrome (weakness, fatigue, muscle pain, nausea, vomiting, headache, chills, aches, itching, sweats, dizziness, shortness of breath, chest pain, cough, syncope, palpitations). These reactions may be severe and DRESS may be fatal. Manifestations of hypersensitivity, such as fever, lymphadenopathy or laboratory abnormalities (including eosinophilia, liver abnormalities) may be present even though rash is not evident. Monitor patients receiving rifampin for injection for signs and/or symptoms of hypersensitivity reactions. If these signs or symptoms occur, discontinue rifampin for injection and administer supportive measures.
-
PRECAUTIONS
General
Rifampin for injection should be used with caution in patients with a history of diabetes mellitus, as diabetes management may be more difficult.
Prescribing rifampin in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
For the treatment of tuberculosis, rifampin is usually administered on a daily basis. Doses of rifampin greater than 600 mg given once or twice weekly have resulted in a higher incidence of adverse reactions, including the "flu syndrome" (fever, chills and malaise), hematopoietic reactions (leukopenia, thrombocytopenia, or acute hemolytic anemia), cutaneous, gastrointestinal, and hepatic reactions, shortness of breath, shock, anaphylaxis, and renal failure. Recent studies indicate that regimens using twice-weekly doses of rifampin 600 mg plus isoniazid 15 mg/kg are much better tolerated.
Rifampin is not recommended for intermittent therapy; the patient should be cautioned against intentional or accidental interruption of the daily dosage regimen since rare renal hypersensitivity reactions have been reported when therapy was resumed in such cases.
Rifampin has enzyme induction properties that can enhance the metabolism of endogenous substrates including adrenal hormones, thyroid hormones, and vitamin D. Rifampin and isoniazid have been reported to alter vitamin D metabolism. In some cases, reduced levels of circulating 25-hydroxy vitamin D and 1,25-dihydroxy vitamin D have been accompanied by reduced serum calcium and phosphate, and elevated parathyroid hormone.
Rifampin IV
For intravenous infusion only. Must not be administered by intramuscular or subcutaneous route. Avoid extravasation during injection: local irritation and inflammation due to extravascular infiltration of the infusion have been observed. If these occur, the infusion should be discontinued and restarted at another site.
Information for Patients
Patients should be counseled that antibacterial drugs including rifampin should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When rifampin is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by rifampin or other antibacterial drugs in the future.
The patient should be told that rifampin may produce a discoloration (yellow, orange, red, brown) of the teeth, urine, sweat, sputum, and tears, and the patient should be forewarned of this. Soft contact lenses may be permanently stained.
The patient should be advised that the reliability of oral or other systemic hormonal contraceptives may be affected; consideration should be given to using alternative contraceptive measures.
Patients should be instructed to notify their physicians promptly if they experience any of the following: fever, loss of appetite, malaise, nausea and vomiting, darkened urine, yellowish discoloration of the skin and eyes, and pain or swelling of the joints.
Compliance with the full course of therapy must be emphasized, and the importance of not missing any doses must be stressed.
Laboratory Tests
Adults treated for tuberculosis with rifampin should have baseline measurements of hepatic enzymes, bilirubin, serum creatinine, a complete blood count, and a platelet count (or estimate). Baseline tests are unnecessary in pediatric patients unless a complicating condition is known or clinically suspected.
Patients should be seen at least monthly during therapy and should be specifically questioned concerning symptoms associated with adverse reactions. All patients with abnormalities should have follow-up, including laboratory testing, if necessary. Routine laboratory monitoring for toxicity in people with normal baseline measurements is generally not necessary.
Drug Interactions
Healthy subjects who received rifampin 600 mg once daily concomitantly with saquinavir 1000 mg/ritonavir 100 mg twice daily (ritonavir-boosted saquinavir) developed severe hepatocellular toxicity. Therefore, concomitant use of these medications is contraindicated. (See CONTRAINDICATIONS.)
Enzyme Induction: Rifampin is known to induce certain cytochrome P-450 enzymes. Administration of rifampin with drugs that undergo biotransformation through these metabolic pathways may accelerate elimination of coadministered drugs. To maintain optimum therapeutic blood levels, dosages of drugs metabolized by these enzymes may require adjustment when starting or stopping concomitantly administered rifampin.
Rifampin has been reported to substantially decrease the plasma concentrations of the following antiviral drugs: atazanavir, darunavir, fosamprenavir, saquinavir, and tipranavir. These antiviral drugs must not be co-administered with rifampin. (See CONTRAINDICATIONS.)
Rifampin has been reported to accelerate the metabolism of the following drugs: anticonvulsants (e.g., phenytoin), digitoxin, antiarrhythmics (e.g., disopyramide, mexiletine, quinidine, tocainide), oral anticoagulants, antifungals (e.g., fluconazole, itraconazole, ketoconazole), barbiturates, beta-blockers, calcium channel blockers (e.g., diltiazem, nifedipine, verapamil), chloramphenicol, clarithromycin, corticosteroids, cyclosporine, cardiac glycoside preparations, clofibrate, oral or other systemic hormonal contraceptives, dapsone, diazepam, doxycycline, fluoroquinolones (e.g., ciprofloxacin), haloperidol, oral hypoglycemic agents (sulfonylureas), levothyroxine, methadone, narcotic analgesics, progestins, quinine, tacrolimus, theophylline, tricyclic antidepressants (e.g., amitriptyline, nortriptyline) and zidovudine. It may be necessary to adjust the dosages of these drugs if they are given concurrently with rifampin.
Patients using oral or other systemic hormonal contraceptives should be advised to change to nonhormonal methods of birth control during rifampin therapy.
Rifampin has been observed to increase the requirements for anticoagulant drugs of the coumarin type. In patients receiving anticoagulants and rifampin concurrently, it is recommended that the prothrombin time be performed daily or as frequently as necessary to establish and maintain the required dose of anticoagulant.
Other Interactions: When the two drugs were taken concomitantly, decreased concentrations of atovaquone and increased concentrations of rifampin were observed.
Concurrent use of ketoconazole and rifampin has resulted in decreased serum concentrations of both drugs. Concurrent use of rifampin and enalapril has resulted in decreased concentrations of enalaprilat, the active metabolite of enalapril. Dosage adjustments should be made if indicated by the patient's clinical condition.
Concomitant antacid administration may reduce the absorption of rifampin. Daily doses of rifampin should be given at least 1 hour before the ingestion of antacids.
Probenecid and cotrimoxazole have been reported to increase the blood level of rifampin.
When rifampin is given concomitantly with either halothane or isoniazid, the potential for hepatotoxicity is increased. The concomitant use of rifampin and halothane should be avoided. Patients receiving both rifampin and isoniazid should be monitored close for hepatotoxicity.
Plasma concentrations of sulfapyridine may be reduced following the concomitant administration of sulfasalazine and rifampin. This finding may be the result of alteration in the colonic bacteria responsible for the reduction of sulfasalazine to sulfapyridine and mesalamine.
Drug/Laboratory Interactions
Cross-reactivity and false-positive urine screening tests for opiates have been reported in patients receiving rifampin when using the KIMS (Kinetic Interaction of Microparticles in Solution) method (e.g., Abuscreen OnLine opiates assay; Roche Diagnostic Systems). Confirmatory tests, such as gas chromatography/mass spectrometry, will distinguish rifampin from opiates.
Therapeutic levels of rifampin have been shown to inhibit standard microbiological assays for serum folate and vitamin B12. Thus, alternate assay methods should be considered. Transient abnormalities in liver function tests (e.g., elevation in serum bilirubin, alkaline phosphatase, and serum transaminases) and reduced biliary excretion of contrast media used for visualization of the gallbladder have also been observed. Therefore, these tests should be performed before the morning dose of rifampin.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A few cases of accelerated growth of lung carcinoma have been reported in man, but a causal relationship with the drug has not been established. Hepatomas were increased in female (C3Hf/DP) mice dosed for 60 weeks with rifampicin followed by an observation period of 46 weeks, at 20 to 120 mg/kg (equivalent to 0.1 to 0.5 times the maximum dosage used clinically, based on body surface area comparisons). There was no evidence of tumorigenicity in male C3Hf/DP mice or in similar studies in BALB/c mice, or in two year studies in Wistar rats.
There was no evidence of mutagenicity in both prokaryotic (Salmonella typhi, Escherichia coli) and eukaryotic (Saccharomyces cerevisiae) bacteria, Drosophila melanogaster, or ICR/Ha Swiss mice. An increase in chromatid breaks was noted when whole blood cell cultures were treated with rifampin. Increased frequency of chromosomal aberrations was observed in vitro in lymphocytes obtained from patients treated with combinations of rifampin, isoniazid, and pyrazinamide and combinations of streptomycin, rifampin, isoniazid, and pyrazinamide.
Pregnancy –Teratogenic Effects
Category C. Rifampin has been shown to be teratogenic in rodents. Congenital malformations, primarily spina bifida were increased in the offspring of pregnant rats given rifampin during organogenesis at oral doses of 150 to 250 mg/kg/day (about 1 to 2 times the maximum recommended human dose based on body surface area comparisons). Cleft palate was increased in a dose-dependent fashion in fetuses of pregnant mice treated at oral doses of 50 to 200 mg/kg (about 0.2 to 0.8 times the maximum recommended human dose based on body surface area comparisons). Imperfect osteogenesis and embryotoxicity were also reported in pregnant rabbits given rifampin at oral doses up to 200 mg/kg/day (about 3 times the maximum recommended human dose based on body surface area comparisons). There are no adequate and well-controlled studies of rifampin in pregnant women. Rifampin has been reported to cross the placental barrier and appear in cord blood. Rifampin should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Pregnancy–Non-Teratogenic Effects
When administered during the last few weeks of pregnancy, rifampin can cause post-natal hemorrhages in the mother and infant for which treatment with vitamin K may be indicated.
Nursing Mothers
Because of the potential for tumorigenicity shown for rifampin in animal studies, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of rifampin did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. Caution should therefore be observed in using rifampin in elderly patients. (See WARNINGS).
-
ADVERSE REACTIONS
Gastrointestinal
Heartburn, epigastric distress, anorexia, nausea, vomiting, jaundice, flatulence, cramps, and diarrhea have been noted in some patients. Although Clostridium difficile has been shown in vitro to be sensitive to rifampin, pseudomembranous colitis has been reported with the use of rifampin (and other broad spectrum antibiotics). Therefore, it is important to consider this diagnosis in patients who develop diarrhea in association with antibiotic use. Tooth discoloration (which may be permanent) may occur.
Hepatic
Transient abnormalities in liver function tests (e.g., elevations in serum bilirubin, alkaline phosphatase, serum transaminases) have been observed. Rarely, hepatitis or a shock-like syndrome with hepatic involvement and abnormal liver function tests has been reported.
Hematologic
Thrombocytopenia has occurred primarily with high dose intermittent therapy, but has also been noted after resumption of interrupted treatment. It rarely occurs during well supervised daily therapy. This effect is reversible if the drug is discontinued as soon as purpura occurs. Cerebral hemorrhage and fatalities have been reported when rifampin administration has been continued or resumed after the appearance of purpura.
Rare reports of disseminated intravascular coagulation have been observed.
Leukopenia, hemolytic anemia, and decreased hemoglobin have been observed.Agranulocytosis has been reported very rarely.
Central Nervous System
Headache, fever, drowsiness, fatigue, ataxia, dizziness, inability to concentrate, mental confusion, behavioral changes, muscular weakness, pains in extremities, and generalized numbness have been observed.
Psychoses have been rarely reported.
Rare reports of myopathy have also been observed.
Endocrine
Menstrual disturbances have been observed.
Rare reports of adrenal insufficiency in patients with compromised adrenal function have been observed.
Renal
Elevations in BUN and serum uric acid have been reported. Rarely, hemolysis, hemoglobinuria, hematuria, interstitial nephritis, acute tubular necrosis, renal insufficiency, and acute renal failure have been noted. These are generally considered to be hypersensitivity reactions. They usually occur during intermittent therapy or when treatment is resumed following intentional or accidental interruption of a daily dosage regimen, and are reversible when rifampin is discontinued and appropriate therapy instituted.
Dermatologic
Cutaneous reactions are mild and self-limiting and do not appear to be hypersensitivity reactions. Typically, they consist of flushing and itching with or without a rash. More serious cutaneous reactions which may be due to hypersensitivity occur but are uncommon.
Hypersensitivity Reactions
Occasionally, pruritus, urticaria, rash, pemphigoid reaction, erythema multiforme including Stevens-Johnson Syndrome, toxic epidermal necrolysis, Drug Reaction with Eosinophilia and Systemic Symptoms syndrome (see WARNINGS), vasculitis, eosinophilia, sore mouth, sore tongue, and conjunctivitis have been observed.
Anaphylaxis has been reported rarely.
Miscellaneous
Edema of the face and extremities have been reported. Other reactions which have occurred with intermittent dosage regimens include "flu syndrome" (such as episodes of fever, chills, headache, dizziness, and bone pain), shortness of breath, wheezing, decrease in blood pressure and shock. The "flu syndrome" may also appear if rifampin is taken irregularly by the patient or if daily administration is resumed after a drug free interval.
To report SUSPECTED ADVERSE EVENTS, contact FDA at 1-800-FDA-1088 or www.fda.gov.
-
OVERDOSAGE
Signs and Symptoms
Nausea, vomiting, abdominal pain, pruritus, headache, and increasing lethargy will probably occur within a short time after ingestion; unconsciousness may occur when there is severe hepatic disease. Transient increases in liver enzymes and/or bilirubin may occur. Brownish-red or orange discoloration of the skin, urine, sweat, saliva, tears, and feces will occur, and its intensity is proportional to the amount ingested.
Liver enlargement, possibly with tenderness, can develop within a few hours after severe overdosage; bilirubin levels may increase and jaundice may develop rapidly. Hepatic involvement may be more marked in patients with prior impairment of hepatic function. Other physical findings remain essentially normal. A direct effect upon the hematopoietic system, electrolyte levels, or acid-base balance is unlikely.
Facial or periorbital edema has also been reported in pediatric patients. Hypotension, sinus tachycardia, ventricular arrhythmias, seizures and cardiac arrest were reported in some fatal cases.
Acute Toxicity
The minimum acute lethal or toxic dose is not well established. However, nonfatal acute overdoses in adults have been reported with doses ranging from 9 to 12 gm rifampin. Fatal acute overdoses in adults have been reported with doses ranging from 14 to 60 gm. Alcohol or a history of alcohol abuse was involved in some of the fatal and nonfatal reports. Nonfatal overdoses in pediatric patients ages 1 to 4 years old of 100 mg/kg for one to two doses has been reported.
Treatment
Intensive support measures should be instituted and individual symptoms treated as they arise. The airway should be secured and adequate respiratory exchange established. Since nausea and vomiting are likely to be present, gastric lavage within the first 2 to 3 hours after ingestion is probably preferable to induction of emesis. Following evacuation of the gastric contents, the instillation of activated charcoal slurry into the stomach may help absorb any remaining drug from the gastrointestinal tract. Antiemetic medication may be required to control severe nausea and vomiting.
Active diuresis (with measured intake and output) will help promote excretion of the drug. For severe cases, extracorporeal hemodialysis may be required. If this is not available, peritoneal dialysis can be used along with forced diuresis.
-
DOSAGE AND ADMINISTRATION
Rifampin for injection is administered by IV infusion (see INDICATIONS AND USAGE). IV doses are the same as those for oral.
See CLINICAL PHARMACOLOGY for dosing information in patients with renal failure.
Tuberculosis
Adults: 10 mg/kg, in a single daily administration, not to exceed 600 mg/day, IV.
Pediatric Patients: 10–20 mg/kg, not to exceed 600 mg/day, IV.
Rifampin is indicated in the treatment of all forms of tuberculosis. A three-drug regimen consisting of rifampin, isoniazid, and pyrazinamide [e.g., RIFATER®]1 is recommended in the initial phase of short-course therapy which is usually continued for 2 months. The Advisory Council for the Elimination of Tuberculosis, the American Thoracic Society, and the Centers for Disease Control and Prevention recommend that either streptomycin or ethambutol be added as a fourth drug in a regimen containing isoniazid (INH), rifampin and pyrazinamide for initial treatment of tuberculosis unless the likelihood of INH resistance is very low. The need for a fourth drug should be reassessed when the results of susceptibility testing are known. If community rates of INH resistance are currently less than 4%, an initial treatment regimen with less than four drugs may be considered.
Following the initial phase, treatment should be continued with rifampin and isoniazid [e.g., RIFAMATE®]2 for at least 4 months. Treatment should be continued for longer if the patient is still sputum or culture positive, if resistant organisms are present, or if the patient is HIV positive.
Preparation of Solution for IV Infusion: Reconstitute the lyophilized powder by transferring 10 mL of sterile water for injection to a vial containing 600 mg of rifampin for injection. Swirl vial gently to completely dissolve the antibiotic. The reconstituted solution contains 60 mg rifampin per mL and is stable at room temperature for 24 hours. Prior to administration, withdraw from the reconstituted solution a volume equivalent to the amount of rifampin calculated to be administered and add to 500 mL of infusion medium. Mix well and infuse at a rate allowing for complete infusion within 3 hours. Alternatively, the amount of rifampin calculated to be administered may be added to 100 mL of infusion medium and infused in 30 minutes.
Dilutions in dextrose 5% for injection (D5W) are stable at room temperature for up to 4 hours and should be prepared and used within this time. Precipitation of rifampin from the infusion solution may occur beyond this time. Dilutions in normal saline are stable at room temperature for up to 24 hours and should be prepared and used within this time. Other infusion solutions are not recommended.
Incompatibilities: Physical incompatibility (precipitate) was observed with undiluted (5 mg/mL) and diluted (1 mg/mL in normal saline) diltiazem hydrochloride and rifampin (6 mg/mL in normal saline) during simulated Y-site administration.
Meningococcal Carriers
Adults: For adults, it is recommended that 600 mg rifampin be administered twice daily for two days.
Pediatric Patients: Pediatric patients 1 month of age or older: 10 mg/kg (not to exceed 600 mg per dose) every 12 hours for two days.
Pediatric patients under 1 month of age: 5 mg/kg every 12 hours for two days.
-
HOW SUPPLIED
Rifampin for Injection USP, containing 600 mg of rifampin, is supplied in sterile glass vials, individually boxed (NDC # 17478-151-42).
Storage: Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature]. Avoid excessive heat (temperatures above 40°C or 104°F). Protect from light.
- REFERENCES
- Rx only
-
PRINCIPAL DISPLAY PANEL- 600 mg Vial Carton
NDC: 17478-151-42
RIFAMPIN
FOR INJECTION, USP
(Sterile)
600 mg*/vial
FOR IV INFUSION ONLY
Rx Only
One Sterile Vial
AKORN
-
INGREDIENTS AND APPEARANCE
RIFAMPIN
rifampin injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 17478-151 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIFAMPIN (UNII: VJT6J7R4TR) (RIFAMPIN - UNII:VJT6J7R4TR) RIFAMPIN 600 mg in 10 mL Inactive Ingredients Ingredient Name Strength SODIUM FORMALDEHYDE SULFOXYLATE DIHYDRATE (UNII: SQ4705447D) 10 mg in 10 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 17478-151-42 1 in 1 BOX, UNIT-DOSE 06/01/2012 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA065502 06/01/2012 Labeler - AKORN, Inc. (062649876) Establishment Name Address ID/FEI Business Operations Patheon Italia S.p.A 434078638 MANUFACTURE(17478-151) , PACK(17478-151) , ANALYSIS(17478-151)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.