DENGVAXIA- dengue tetravalent vaccine, live kit
DENGVAXIA by
Drug Labeling and Warnings
DENGVAXIA by is a Other medication manufactured, distributed, or labeled by Sanofi Pasteur Inc., Sanofi Pasteur VDR, Sanofi Pasteur NVL. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DENGVAXIA safely and effectively. See full prescribing information for DENGVAXIA.
DENGVAXIA (Dengue Tetravalent Vaccine, Live)
Suspension for Subcutaneous Injection
Initial U.S. Approval: 2019INDICATIONS AND USAGE
DENGVAXIA is a vaccine indicated for the prevention of dengue disease caused by dengue virus serotypes 1, 2, 3 and 4. DENGVAXIA is approved for use in individuals 9 through 16 years of age with laboratory-confirmed previous dengue infection and living in endemic areas.
Limitations of use:
- DENGVAXIA is not approved for use in individuals not previously infected by any dengue virus serotype or for whom this information is unknown. Those not previously infected are at increased risk for severe dengue disease when vaccinated and subsequently infected with dengue virus. (5.1) Previous dengue infection can be assessed through a medical record of a previous laboratory-confirmed dengue infection or through serological testing prior to vaccination. (1)
- The safety and effectiveness of DENGVAXIA have not been established in individuals living in dengue nonendemic areas who travel to dengue endemic areas. (1)
DOSAGE AND ADMINISTRATION
Three doses (0.5 mL each) 6 months apart (at month 0, 6, and 12). (2.1)
DOSAGE FORMS AND STRENGTHS
Suspension for injection (0.5 mL) supplied as a lyophilized powder to be reconstituted with the supplied diluent. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most frequently reported adverse reactions regardless of the dengue serostatus prior to vaccination were headache (40%), injection site pain (32%), malaise (25%), asthenia (25%), and myalgia (29%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pharmacovigilance Department, Sanofi Pasteur Inc., Discovery Drive, Swiftwater, PA 18370 at 1-800-822-2463 (1-800-VACCINE) or VAERS at 1-800-822-7967 or http://vaers.hhs.gov.
DRUG INTERACTIONS
False negative tuberculin purified protein derivative (PPD) test results may occur within 1 month following vaccination with DENGVAXIA. (7.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Preparation
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Hypersensitivity
4.2 Immunocompromised Individuals
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Severe Dengue Disease Following DENGVAXIA in Persons not Previously Infected with Dengue Virus
5.2 Management of Acute Allergic Reactions
5.3 Limitations of Vaccine Effectiveness
5.4 Syncope
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Data from Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Administration with Other Vaccines
7.2 Immunosuppressive Treatments
7.3 Drug/Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Efficacy
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
DENGVAXIA® (Dengue Tetravalent Vaccine, Live) is a vaccine indicated for the prevention of dengue disease caused by dengue virus serotypes 1, 2, 3, and 4. DENGVAXIA is approved for use in individuals 9 through 16 years of age with laboratory-confirmed previous dengue infection and living in endemic areas.
Limitations of use
- DENGVAXIA is not approved for use in individuals not previously infected by any dengue virus serotype or for whom this information is unknown. Those not previously infected are at increased risk for severe dengue disease when vaccinated and subsequently infected with dengue virus. [See Warnings and Precautions (5.1).] Previous dengue infection can be assessed through a medical record of a previous laboratory-confirmed dengue infection or through serological testing prior to vaccination.
- The safety and effectiveness of DENGVAXIA have not been established in individuals living in dengue nonendemic areas who travel to dengue endemic areas.
-
2 DOSAGE AND ADMINISTRATION
For subcutaneous use only.
2.2 Preparation
The package contains a vial of lyophilized vaccine antigen and a vial of saline diluent (0.4% NaCl).
After removing the "flip-off" caps, cleanse the lyophilized vaccine antigen and diluent vial stoppers with a suitable germicide. Do not remove the vial stoppers or metal seals holding them in place.
To reconstitute DENGVAXIA, use a sterile needle and syringe to withdraw 0.6 mL from the diluent vial and inject it into the vial of the lyophilized vaccine antigen. Swirl the vial gently.
Changing needles between withdrawing the vaccine from the vial and injecting it into a recipient is not necessary unless the needle has been damaged or contaminated.
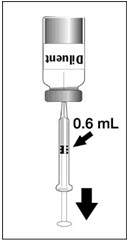

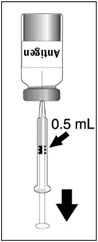
Figure 1 Figure 2 Figure 3 Figure 4 Insert the syringe needle through the stopper of the vial of diluent and withdraw 0.6 mL liquid content. Insert the syringe needle through the stopper of the vial of lyophilized vaccine antigen and inject the liquid into the vial. Swirl vial gently. The syringe needle is not removed while swirling the vial. After reconstitution, withdraw 0.5 mL.
DENGVAXIA should be used immediately after reconstitution.After reconstitution, the suspension is colorless and may develop trace amounts of white to translucent endogenous proteinaceous particles. [See Description (11).]
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard the vial if the solution is cloudy or contains particles other than trace amounts of white to translucent particles.
Discard reconstituted vaccine if not used within 30 minutes. [See How Supplied/Storage and Handling (16.2).]
DENGVAXIA should not be mixed in the same syringe with other parenteral products.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Hypersensitivity
Do not administer DENGVAXIA to individuals with a history of severe allergic reaction to a previous dose of DENGVAXIA or to any component of DENGVAXIA. [See Description (11).]
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Severe Dengue Disease Following DENGVAXIA in Persons not Previously Infected with Dengue Virus
In unvaccinated individuals, first dengue infections rarely cause severe dengue, while second dengue infections with a different serotype are associated with an increased risk of severe dengue. DENGVAXIA administration to individuals not previously infected by dengue virus is associated with an increased risk of severe dengue disease when the vaccinated individual is subsequently infected with any dengue virus serotype. Therefore, healthcare professionals must evaluate individuals for prior dengue infection to avoid vaccinating individuals who have not been previously infected by dengue virus.
Previous infection by dengue virus can be evaluated through a medical record of previous laboratory-confirmed dengue infection or through serotesting prior to vaccination.
There is no FDA cleared test available to determine a previous dengue infection. Available non-FDA cleared tests may yield false positive results (e.g., due to cross-reactivity with other flaviviruses).
5.2 Management of Acute Allergic Reactions
DENGVAXIA may cause hypersensitivity reactions, including anaphylaxis. Appropriate medical treatment and supervision must be available following administration of DENGVAXIA.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
The safety of DENGVAXIA in subjects 9 through 16 years of age was evaluated in 9 randomized, placebo-controlled, multicenter clinical trials. In these studies, a total of 19,102 subjects 9 through 16 years of age received at least one dose of DENGVAXIA and 9,484 received placebo (0.9% sodium chloride). Overall, 50.9% of trial participants who received DENGVAXIA or placebo were female. Racial groups were reported as 18.9% Asian, 13% American Indian or Alaska native, 6.4% Caucasian, 2.6% black, and 59.1% as other. In the largest study (Study 1, NCT01374516; N = 20,869) conducted in four Latin American countries and Puerto Rico, most subjects (99.9%) reported Hispanic ethnicity. All studies enrolled subjects irrespective of evidence of previous dengue infection.
Solicited Adverse Reactions
In a multi-center, observer-blind, randomized (2:1), placebo-controlled trial conducted in four Latin American countries and Puerto Rico (Study 1, NCT01374516), 2,000 subjects (out of a total of 20,869 subjects) were recruited during the first 2 months of enrollment for inclusion in the reactogenicity subset. Solicited adverse reactions were recorded daily for 14 days following each vaccination.
Table 1 presents the frequency and severity of solicited injection site reactions reported within 7 days and systemic adverse reactions reported within 14 days following receipt of DENGVAXIA or placebo.
Table 1: Percentages of Subjects with Solicited Injection Site Reactions within 7 Days and Systemic Adverse Reactions within 14 Days Following Receipt of Each Dose of DENGVAXIA or Placebo in Children and Adolescents 9 through 16 Years of Age in Study 1 Dose 1 Dose 2 Dose 3 DENGVAXIA
%
N = 1,264–1,326Placebo
%
N = 635–657DENGVAXIA
%
N = 1,228–1,298Placebo
%
N = 594–639DENGVAXIA
%
N = 1,215–1,279Placebo
%
N = 597 –631N: range number of subjects with available data for the specified endpoints
Study 1, NCT01374516
Placebo: 0.9% sodium chloride- * For subjects 9 through 11 years of age – Grade 3: Incapacitating, unable to perform usual activities. For subjects 12 through 16 years of age – Grade 3: Significant; prevents daily activity.
- † For subjects 9 through 11 years of age – Grade 3: ≥50 mm. For subjects 12 through 16 years of age – Grade 3: >100 mm.
- ‡ For all subjects – Grade 3: Significant; prevents daily activity.
- § For all subjects – Any Fever: ≥38.0°C. Grade 3: ≥39.0°C.
Injection Site Reactions Pain* Any
Grade 332.4
0.826.3
0.925.6
0.516.4
0.022.5
0.916.5
0.3Erythema† Any
Grade 34.1
0.04.7
0.21.9
<0.11.7
0.01.5
0.01.6
0.0Swelling† Any
Grade 33.5
0.02.7
0.21.9
0.00.9
0.01.6
0.01.3
0.0Systemic Adverse Reactions Asthenia‡ Any
Grade 324.6
2.722.5
2.617.8
1.816.4
1.116.3
1.317.4
1.3Fever§ Any
Grade 36.8
1.76.6
1.15.9
0.87.1
1.27.3
1.18.7
0.8Headache‡ Any
Grade 339.9
5.141.6
4.129.8
2.128.5
2.329.6
2.625.0
1.9Malaise‡ Any
Grade 324.5
2.425.9
2.320.8
1.316.6
1.319.3
1.415.2
1.1Myalgia‡ Any
Grade 329.2
2.227.4
1.521.0
1.615.8
0.820.0
1.518.4
0.8Unsolicited Non-serious Adverse Reactions
In Study 1, 1.2% of subjects in the DENGVAXIA group (16/1,333) and 0.8% of subjects in the placebo group (5/664) reported at least 1 unsolicited non-serious adverse reaction within 28 days following any dose.
In this study, 0.7% of the subjects in the DENGVAXIA group and 0.5% in the placebo group reported at least one unsolicited non-serious injection site adverse reaction. The unsolicited non-serious adverse reactions were injection site pain, hematoma, pruritus and anesthesia in the vaccine group and pain and induration in the control group.
In this study, 0.5% of the subjects in the DENGVAXIA group and 0.3% in the placebo group reported at least one unsolicited non-serious systemic adverse reaction. The unsolicited non-serious systemic adverse reactions were malaise, abdominal pain, vomiting, dyspnea, generalized erythema, vertigo, asthma crisis and urticaria in the vaccine group and pruritus and lymphadenitis in the control group.
Most unsolicited non-serious adverse reactions started within 3 days of any injection and resolved within 3 days or less.
A total of 2 subjects (one subject with asthma crisis and urticaria occurring the day of the first dose, and one subject with malaise occurring 20 days after the first dose) in the DENGVAXIA group (0.2%) and none in the placebo group reported unsolicited non-serious Grade 3 (significant; prevents daily activity) adverse reactions.
Severe Dengue Following Vaccination with DENGVAXIA and Subsequent Dengue Infection
Subjects were monitored for severe dengue from Day 0 (day of first study vaccination) to Month 60–72 (after first study vaccination) in three multi-center, observer-blind, randomized (2:1), placebo-controlled trials conducted in Latin America and Puerto Rico (Study 1, NCT01374516) and the Asia-Pacific region (Study 2, NCT01373281; Study 3, NCT00842530). A subset of 3,203 subjects (80.1%) enrolled in Study 3 were re-consented to participate in an extension study to evaluate safety of DENGVAXIA for 72 months (Study 4, NCT01983553). A total of 18,265 children and adolescents 9 through 16 years of age enrolled in these trials received at least one dose of DENGVAXIA. Table 2 presents the incidences and hazard ratios of severe dengue from Month 13 to Month 60–72 post vaccination with DENGVAXIA or placebo in children and adolescents 9 through 16 years of age, by dengue baseline serostatus. An increased rate of severe dengue was observed following vaccination with DENGVAXIA and subsequent infection with any dengue virus serotype in persons not previously infected by dengue virus. [See Warnings and Precautions (5.1).]
Table 2: Number of Events and Incidence of Severe Dengue* From Month 13 to Month 60-72† in Children 9 through 16 Years of Age, by Previous Dengue Infection Status, in Studies 1, 2, 3, and 4 Dengue Infection Status at Month 13‡ DENGVAXIA
n
(Incidence§, %)Placebo
n
(Incidence§, %)Hazard Ratio of Severe Dengue
(95% CI)n: number of subject with severe dengue cases
CI: confidence interval
Study 1, NCT01374516; Study 2, NCT01373281; Study 3, NCT00842530; Study 4, NCT01983553- * Severe Dengue according to IDMC (Independent Data Monitoring Committee) definition: Proven dengue fever (2 days fever + virological confirmation) plus one of the following: (a) Platelet count ≤100,000/μL and bleeding plus plasma leakage (effusion on chest x-ray [CXR] or clinically apparent ascites including imaging procedures or hematocrit ≥20% above baseline recovery level or standard for age if only one reading); (b) shock; (c) bleeding (requiring blood transfusion); (d) encephalopathy; (e) liver impairment; (f) impaired kidney function; (g) myocarditis, pericarditis or clinical heart failure.
- † The follow-up period corresponded to a minimum of 60 months for Study 1, a minimum of 63 months for Study 2 and 72 months for the combination of Study 3 and its extension, Study 4.
- ‡ Based on measured Dengue anti-NS1 IgG ELISA at Month 13 from first vaccination (Dengue Seropositive= ≥9EU/mL).
- § Cumulative incidence over 4 years from 13 months after the first vaccination.
Previous Dengue Infection
(Dengue seropositive at Month 13)10
(0.068)27
(0.401)0.18 (0.09; 0.37) No Previous Dengue Infection
(Dengue seronegative at Month 13)12
(0.380)1
(0.069)6.25 (0.81; 48.32) Non-fatal Serious Adverse Events
In the 9 studies conducted among subjects 9 through 16 years of age (NCT 01374516, NCT01373281, NCT00842530, NCT00993447, NCT00875524, NCT00788151, NCT00880893, NCT01187433, NCT01254422), subjects were monitored for serious adverse events (SAEs) for at least 6 months after the last dose of DENGVAXIA.
The proportions of subjects who reported at least 1 non-fatal SAE within 28 days following any dose were 0.6% (123/19,102) in the DENGVAXIA group and 0.8% (73/9,484) in the placebo group. The following events were considered related to DENGVAXIA: asthma attack (day of Dose 1), urticaria (day of Dose 2) and convulsion (day of Dose 1).
The proportions of subjects who reported at least 1 non-fatal SAE after 28 days and up to 6 months after any dose were similar in the 2 groups: 2.8% in the DENGVAXIA group (534/19,102) and 3.2% in the placebo group (307/9,484). None of these SAEs were considered related to the vaccination.
Deaths
From the first administered dose up to Month 72, 51 deaths (0.3%) for subjects who received DENGVAXIA and 26 deaths (0.3 %) for subjects who received placebo were reported in the 9 studies conducted among subjects 9 though 16 years of age. None of the deaths were considered related to vaccination. Causes of death among subjects were consistent with those generally reported in children and adolescent populations.
6.2 Data from Postmarketing Experience
In addition to events reported in clinical trials for DENGVAXIA, the following adverse events have been spontaneously reported during postapproval use. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to the vaccine.
The following adverse events were included based on one or more of the following factors: severity, frequency of reporting, or strength of evidence for a causal relationship to DENGVAXIA.
-
7 DRUG INTERACTIONS
7.1 Concomitant Administration with Other Vaccines
Data are not available to establish the safety and immunogenicity of concomitant administration of DENGVAXIA with recommended adolescent vaccines.
7.2 Immunosuppressive Treatments
Immunosuppressive therapies, including irradiation, antimetabolites, alkylating agents, cytotoxic drugs and corticosteroids (used in greater than physiologic doses), may reduce the immune response to DENGVAXIA.
7.3 Drug/Laboratory Test Interactions
DENGVAXIA may cause temporary depression of tuberculin purified protein derivative (PPD) test sensitivity, leading to false negative results. Tuberculin testing should be performed before DENGVAXIA is administered or at least 1 month following vaccination with DENGVAXIA.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to DENGVAXIA during pregnancy. Women who receive DENGVAXIA during pregnancy are encouraged to contact directly, or have their healthcare professional contact, Sanofi Pasteur Inc. at 1-800-822-2463 (1-800-VACCINE) to enroll in or obtain information about the registry.
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
No specific studies of DENGVAXIA have been performed among pregnant women. A limited number of cases of inadvertent exposure during pregnancy were reported during clinical studies. Isolated adverse pregnancy outcomes (e.g., stillbirth, intrauterine death, spontaneous abortion, blighted ovum) have been observed for these exposed pregnancies, with similar frequency and nature in the vaccinated individuals compared to the control group, and with risk factors identified for all cases. Available data in pregnant women are not sufficient to determine the effects of DENGVAXIA on pregnancy, embryo-fetal development, parturition and postnatal development.
In two developmental toxicity studies, the effect of DENGVAXIA on embryo-fetal and postnatal development was evaluated in pregnant rabbits and mice. A developmental toxicity study was performed in female rabbits given a 5 log10 50% cell culture infectious dose (CCID50) of DENGVAXIA (full human dose ranging from 4.5 log10 to 6.0 log10 CCID50) by intravenous injection prior to mating and during gestation. The study revealed no evidence of harm to the fetus due to DENGVAXIA. In another study, female mice were administered a single dose of 5 log10 CCID50, 6.5 log10 CCID50 (about 3 times the highest human dose) or 8 log10 CCID50 (about 100 times the highest human dose) of DENGVAXIA by intravenous injection during gestation. Fetal toxicities were observed at maternally toxic doses. [See Animal Data (8.1).]
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Pregnant women are at increased risk of complications associated with dengue infection compared to non-pregnant women. Pregnant women with dengue may be at increased risk for adverse pregnancy outcomes, including preterm labor and delivery. Vertical transmission of dengue virus from mothers with viremia at delivery to their infants has been reported.
Fetal/neonatal adverse reactions
Vaccine viremia can occur 7 to 14 days after vaccination with a duration of <7 days [See Pharmacokinetics (12.3).]. The potential for transmission of the vaccine virus from mother to infant is unknown.
Animal Data
In two developmental toxicity studies, the effect of DENGVAXIA on embryo-fetal and postnatal development was evaluated in pregnant rabbits and mice.
Rabbits were administered a full human dose [0.5 mL (5 log10 CCID50/animal/occasion)] of DENGVAXIA by intravenous injection 30 and 10 days before mating and on Days 6, 12 and 27 during gestation. No vaccine-related fetal malformation or variations and adverse effects on female fertility or preweaning development were reported in this study. Pregnant mice were administered a single dose of either 5 log10 CCID50 (full human dose ranging from 4.5 log10 to 6.0 log10 CCID50), 6.5 log10 CCID50 (about 3 times the highest human dose) or 8 log10 CCID50 (about 100 times the highest human dose) of DENGVAXIA by intravenous injection on Day 6, 9 or 12 of gestation. At doses of 6.5 log10 CCID50 or 8 log10 CCID50 of DENGVAXIA, maternal toxicity was observed which was associated with increased postimplantation loss and at doses of 8 log10 CCID50 with reduced fetal body weight. The significance of this observation for humans is unknown, especially considering the different route of administration (the human route of administration is subcutaneous) and dose levels which exceeded the intended human dose. There were no vaccine related fetal malformations or other evidence of teratogenesis noted in this study.
8.2 Lactation
Risk Summary
Human data are not available to assess the impact of DENGVAXIA on milk production, its presence in breast milk, or its effects on the breastfed child. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for DENGVAXIA and any potential adverse effects on the breastfed child from DENGVAXIA or from the underlying maternal condition. For preventive vaccines, the underlying condition is susceptibility to disease prevented by the vaccine. A lactation study in which female mice were administered a single dose of DENGVAXIA on day 14 of lactation did not show the presence of DENGVAXIA in breast milk.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Vertical transmission of dengue virus, including potentially through breastmilk, has been reported.
Fetal/neonatal adverse reactions
Vaccine viremia can occur 7 to 14 days after vaccination with a duration of <7 days. [See Pharmacokinetics (12.3).] The potential for transmission of the vaccine virus from mother to infant through breastmilk is unknown.
Animal Data
A developmental toxicity study in which female mice were administered a single injection of 5 log10 CCID50 (full human dose ranging from 4.5 log10 to 6.0 log10 CCID50), 6.5 log10 CCID50 or 8 log10 CCID50 of DENGVAXIA by intravenous injection on Day 14 of lactation did not show the presence of DENGVAXIA in breast milk in mice when measured 24 hours after vaccine administration.
-
11 DESCRIPTION
DENGVAXIA (Dengue Tetravalent Vaccine, Live) is a sterile suspension for subcutaneous injection. DENGVAXIA is supplied as a vial of lyophilized vaccine antigen, which must be reconstituted at the time of use with 0.6 mL from the accompanying vial of diluent (0.4% sodium chloride). After reconstitution, DENGVAXIA is a clear, colorless suspension (trace amounts of white to translucent proteinaceous particles may be present). [See Dosage and Administration (2.3).]
After reconstitution, each 0.5 mL dose of DENGVAXIA contains 4.5 – 6.0 log10 CCID50 of each of the chimeric yellow fever dengue (CYD) virus serotypes 1, 2, 3, and 4. Each 0.5 mL dose is formulated to contain 2 mg sodium chloride and the following ingredients as stabilizers: 0.56 mg essential amino acids (including L-phenylalanine), 0.2 mg non-essential amino acids, 2.5 mg L-arginine hydrochloride, 18.75 mg sucrose, 13.75 mg D-trehalose dihydrate, 9.38 mg D-sorbitol, 0.18 mg trometamol, and 0.63 mg urea.
Each of the four CYD viruses (CYD-1, CYD-2, CYD-3, and CYD-4) in DENGVAXIA was constructed using recombinant DNA technology by replacing the sequences encoding the pre-membrane (prM) and envelope (E) proteins in the yellow fever (YF) 17D204 vaccine virus genome with those encoding for the homologous sequences of dengue virus serotypes 1, 2, 3, and 4, respectively. Each CYD virus is cultured separately in Vero cells (African Green Monkey kidney) under serum-free conditions, harvested from the supernatant of the Vero cells and purified by membrane chromatography and ultrafiltration. The purified and concentrated harvest of each CYD virus is then diluted in a stabilizer solution to produce the four monovalent drug substances. The final bulk product is a mixture of the four monovalent drug substances diluted in the stabilizer solution. The final bulk product is sterilized by filtration at 0.22 µm, filled into vials and freeze-dried.
DENGVAXIA does not contain preservative.
The vial stoppers for the Lyophilized Vaccine Antigen and Diluent vials of DENGVAXIA are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Following administration, DENGVAXIA elicits dengue-specific immune responses against the four dengue virus serotypes. The exact mechanism of protection has not been determined.
12.3 Pharmacokinetics
Viremia
In studies that evaluated the occurrence of vaccine viremia systematically at pre-specified timepoints, vaccine viremia (measured by genomic amplification methods) was observed following vaccination with DENGVAXIA in 5.6% of subjects, with 90% of these occurrences documented after the first injection. Vaccine viremia was observed 7 to 14 days after DENGVAXIA vaccination with a duration of <7 days.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
DENGVAXIA has not been evaluated for carcinogenic or mutagenic potential or impairment of male fertility. Exposure of female rabbits to DENGVAXIA prior to and during gestation did not impair fertility. [See Use in Specific Populations (8.1).]
-
14 CLINICAL STUDIES
14.1 Efficacy
The efficacy of DENGVAXIA was evaluated in two randomized, observer-blind, placebo-controlled, multi-center studies. Study 1 (N=20,869) was conducted in individuals 9 through 16 years of age in four Latin American countries and Puerto Rico; and Study 2 (N=10,275) was conducted in individuals 2 through 14 years of age in five Asia-Pacific countries. A subset of subjects in each study (10% in Study 1; 20% in Study 2) was evaluated for antibodies to dengue virus at the time of enrollment and at later time points. Both studies enrolled subjects irrespective of evidence of previous dengue infection. Subjects were randomized 2:1 to receive either DENGVAXIA or saline placebo and were monitored for symptomatic virologically confirmed dengue (VCD) starting at Day 0. Per protocol vaccine efficacy was assessed beginning 28 days after the third vaccination for 12 months. VCD was defined as an acute febrile illness (temperature ≥38°C on at least 2 consecutive days) virologically confirmed by dengue RT-PCR and/or dengue non-structural protein 1 (NS1) ELISA Antigen Test. For each study, in pre-specified vaccine efficacy analyses including the full age range of subjects enrolled, the pre-specified criterion for demonstrating efficacy of DENGVAXIA against VCD due to any dengue virus serotype and irrespective of previous dengue virus infection, was met (lower bound of 95% CI for vaccine efficacy >25%). These studies were not designed to demonstrate efficacy of DENGVAXIA against individual dengue serotypes.
Given the identification of the increased risk for severe dengue following vaccination with DENGVAXIA and subsequent infection with dengue virus in persons not previously infected with dengue virus [see Adverse Reactions (6.1)], Table 3 presents analyses of vaccine efficacy against VCD due to any dengue virus serotype, limited to subjects who had baseline sera evaluated and who were dengue seropositive at baseline. These analyses include subjects 9 through 16 years of age from Study 1 and subjects 9 through 14 years of age from Study 2.
Table 3: Efficacy of DENGVAXIA against Symptomatic VCD in Subjects Seropositive for Dengue at Baseline from 28 Days Post Dose 3 for a Period of 12 months – Study 1 (Ages 9 through 16 Years) and Study 2 (Ages 9 through 14 Years) – mFASE* – Subjects Included in the Immunogenicity Subset DENGVAXIA group
Cases (Subjects)Placebo group
Cases (Subjects)VE % (95% CI)† - * mFASE (Modified Full Analysis Set): Set of the subjects who received 3 injections as per randomization including those with protocol deviations.
- † VE is calculated as 1- ratio of density incidence of dengue between DENGVAXIA and Placebo groups.
Study 1
(Subjects 9 through 16 years of age)7 (1034) 17 (492) 80.6 (50.7;93.2) Study 2
(Subjects 9 through 14 years of age)4 (483) 9 (250) 77.2 (18.3;94.9) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
An outer package of 1 dose (NDC: 49281-605-01) contains 1 single dose vial of Lyophilized Vaccine Antigen (NDC: 49281-606-58) and 1 single dose vial of Saline Diluent (NDC: 49281-549-58).
The vial stoppers for the Lyophilized Vaccine Antigen vials and the Saline Diluent vials of DENGVAXIA are not made with natural rubber latex.
16.2 Storage and Handling
Store Lyophilized Vaccine Antigen and Saline Diluent in a refrigerator at 2°C to 8°C (36°F to 46°F). Do not freeze. Protect from light.
Do not use after the expiration date shown on the vial labels of the Lyophilized Vaccine Antigen and Saline Diluent.
After reconstitution, administer DENGVAXIA immediately or store refrigerated at 2°C to 8°C (36°F to 46°F) and use within 30 minutes. Discard reconstituted vaccine if not used within 30 minutes.
-
17 PATIENT COUNSELING INFORMATION
Educate vaccine recipients regarding the most common adverse reactions that occur within 14 days following administration of DENGVAXIA (headache, injection site pain, malaise, asthenia, and myalgia).
Inform individuals to seek medical care if they develop signs and symptoms of dengue fever with particular attention to severe dengue warning signs (e.g., high fever, severe abdominal pain or tenderness, persistent vomiting, mucosal bleeding, somnolence and hyperactivity).
Register women who receive DENGVAXIA during pregnancy in the Pregnancy Registry by calling 1-800-822- 2463 (1-800-VACCINE). [See Pregnancy (8.1).]
Instruct vaccine recipients to report adverse reactions to their healthcare provider.
- SPL UNCLASSIFIED SECTION
-
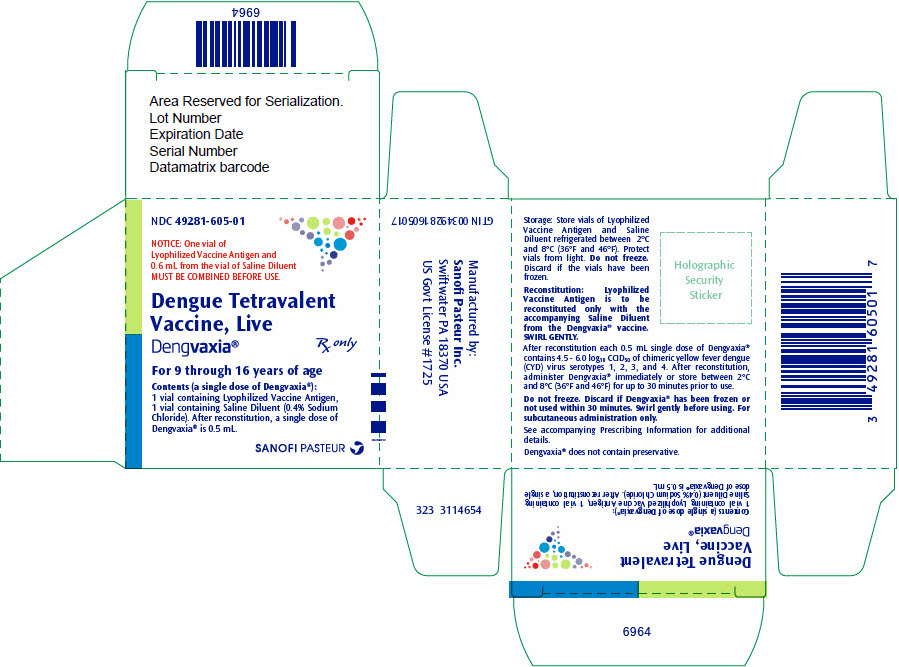
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC: 49281-605-01
NOTICE: One vial of
Lyophilized Vaccine Antigen and
0.6 mL from the vial of Saline Diluent
MUST BE COMBINED BEFORE USE.Dengue Tetravalent
Vaccine, Live
Dengvaxia®Rx only
For 9 through 16 years of age
Contents (a single dose of Dengvaxia®):
1 vial containing Lyophilized Vaccine Antigen,
1 vial containing Saline Diluent (0.4% Sodium
Chloride). After reconstitution, a single dose of
Dengvaxia® is 0.5 mL.SANOFI PASTEUR
- PRINCIPAL DISPLAY PANEL - 0.5 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 0.6 mL Vial Label
-
INGREDIENTS AND APPEARANCE
DENGVAXIA
dengue tetravalent vaccine, live kitProduct Information Product Type VACCINE Item Code (Source) NDC: 49281-605 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49281-605-01 1 in 1 CARTON Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-DOSE 0.5 mL Part 2 1 VIAL, SINGLE-DOSE 0.6 mL Part 1 of 2 DENGVAXIA
dengue tetravalent vaccine, live injection, powder, lyophilized, for suspensionProduct Information Item Code (Source) NDC: 49281-606 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CYD DENGUE VIRUS SEROTYPE 1 LIVE (ATTENUATED) ANTIGEN (UNII: 75KB2HPX5H) (CYD DENGUE VIRUS SEROTYPE 1 LIVE (ATTENUATED) ANTIGEN - UNII:75KB2HPX5H) CYD DENGUE VIRUS SEROTYPE 1 LIVE (ATTENUATED) ANTIGEN 100000 [CCID_50] in 0.5 mL CYD DENGUE VIRUS SEROTYPE 2 LIVE (ATTENUATED) ANTIGEN (UNII: FH5SVG7GLC) (CYD DENGUE VIRUS SEROTYPE 2 LIVE (ATTENUATED) ANTIGEN - UNII:FH5SVG7GLC) CYD DENGUE VIRUS SEROTYPE 2 LIVE (ATTENUATED) ANTIGEN 100000 [CCID_50] in 0.5 mL CYD DENGUE VIRUS SEROTYPE 3 LIVE (ATTENUATED) ANTIGEN (UNII: RHT2Q37FYG) (CYD DENGUE VIRUS SEROTYPE 3 LIVE (ATTENUATED) ANTIGEN - UNII:RHT2Q37FYG) CYD DENGUE VIRUS SEROTYPE 3 LIVE (ATTENUATED) ANTIGEN 100000 [CCID_50] in 0.5 mL CYD DENGUE VIRUS SEROTYPE 4 LIVE (ATTENUATED) ANTIGEN (UNII: RS26HP5ND2) (CYD DENGUE VIRUS SEROTYPE 4 LIVE (ATTENUATED) ANTIGEN - UNII:RS26HP5ND2) CYD DENGUE VIRUS SEROTYPE 4 LIVE (ATTENUATED) ANTIGEN 100000 [CCID_50] in 0.5 mL Inactive Ingredients Ingredient Name Strength AMINO ACIDS, ESSENTIAL (UNII: N7U7BXP2OI) 0.56 mg in 0.5 mL AMINO ACIDS (UNII: 0O72R8RF8A) 0.2 mg in 0.5 mL ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 2.5 mg in 0.5 mL PHENYLALANINE (UNII: 47E5O17Y3R) 0.56 mg in 0.5 mL SUCROSE (UNII: C151H8M554) 18.75 mg in 0.5 mL TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 13.75 mg in 0.5 mL SORBITOL (UNII: 506T60A25R) 9.38 mg in 0.5 mL TROMETHAMINE (UNII: 023C2WHX2V) 0.18 mg in 0.5 mL UREA (UNII: 8W8T17847W) 0.63 mg in 0.5 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 2 mg in 0.5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49281-606-58 0.5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125682 05/01/2019 Part 2 of 2 SALINE DILUENT
sodium chloride injectionProduct Information Item Code (Source) NDC: 49281-549 Route of Administration SUBCUTANEOUS Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49281-549-58 0.6 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125682 05/01/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125682 05/01/2019 Labeler - Sanofi Pasteur Inc. (086723285) Establishment Name Address ID/FEI Business Operations Sanofi Pasteur Inc. 086723285 LABEL, MANUFACTURE Establishment Name Address ID/FEI Business Operations Sanofi Pasteur VDR 766693436 MANUFACTURE Establishment Name Address ID/FEI Business Operations Sanofi Pasteur NVL 260680254 MANUFACTURE


Trademark Results [DENGVAXIA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 DENGVAXIA 86633354 4877362 Live/Registered |
Sanofi Pasteur 2015-05-18 |
 DENGVAXIA 85499167 4891090 Live/Registered |
Sanofi Pasteur 2011-12-19 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.