TABRECTA- capmatinib tablet, film coated
TABRECTA by
Drug Labeling and Warnings
TABRECTA by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TABRECTA safely and effectively. See full prescribing information for TABRECTA.
TABRECTA™ (capmatinib) tablets, for oral use
Initial U.S. Approval: 2020
INDICATIONS AND USAGE
TABRECTA is a kinase inhibitor indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have a mutation that leads to mesenchymal-epithelial transition (MET) exon 14 skipping as detected by an FDA-approved test.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 150 mg and 200 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD)/Pneumonitis: Monitor for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Permanently discontinue TABRECTA in patients with ILD/pneumonitis. (2.3, 5.1)
- Hepatotoxicity: Monitor liver function tests. Withhold, dose reduce, or permanently discontinue TABRECTA based on severity. (2.3, 5.2)
- Risk of Photosensitivity: May cause photosensitivity reactions. Advise patients to limit direct ultraviolet exposure. (5.3)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.4, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (≥ 20%) are peripheral edema, nausea, fatigue, vomiting, dyspnea, and decreased appetite. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
Strong and Moderate CYP3A Inducers: Avoid concomitant use. (7.1)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
5.2 Hepatotoxicity
5.3 Risk of Photosensitivity
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TABRECTA
7.2 Effect of TABRECTA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
TABRECTA is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors have a mutation that leads to mesenchymal-epithelial transition (MET) exon 14 skipping as detected by an FDA-approved test.
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment with TABRECTA based on the presence of a mutation that leads to MET exon 14 skipping in tumor specimens [see Clinical Studies (14)]. Information on FDA-approved tests is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of TABRECTA is 400 mg orally twice daily with or without food.
Swallow TABRECTA tablets whole. Do not break, crush or chew the tablets.
If a patient misses or vomits a dose, instruct the patient not to make up the dose, but to take the next dose at its scheduled time.
2.3 Dosage Modifications for Adverse Reactions
The recommended dose reductions for the management of adverse reactions are listed in Table 1.
Table 1: Recommended TABRECTA Dose Reductions for Adverse Reactions Dose Reduction Dose and Schedule First 300 mg orally twice daily Second 200 mg orally twice daily Permanently discontinue TABRECTA in patients who are unable to tolerate 200 mg orally twice daily.
The recommended dosage modifications of TABRECTA for adverse reactions are provided in Table 2.
Table 2: Recommended TABRECTA Dosage Modifications for Adverse Reactions Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ILD, interstitial lung disease; ULN, upper limit of normal.
Grading according to CTCAE Version 4.03 (CTCAE = Common Terminology Criteria for Adverse Events).Adverse Reaction Severity Dosage Modification Interstitial Lung Disease (ILD)/Pneumonitis
[see Warnings and Precautions (5.1)]Any grade Permanently discontinue TABRECTA. Increased ALT and/or AST without increased total bilirubin
[see Warnings and Precautions (5.2)]Grade 3 Withhold TABRECTA until recovery to baseline ALT/AST.
If recovered to baseline within 7 days, then resume TABRECTA at the same dose; otherwise resume TABRECTA at a reduced dose.Grade 4 Permanently discontinue TABRECTA. Increased ALT and/or AST with increased total bilirubin in the absence of cholestasis or hemolysis
[see Warnings and Precautions (5.2)]ALT and/or AST greater than 3 times ULN with total bilirubin greater than 2 times ULN Permanently discontinue TABRECTA. Increased total bilirubin without concurrent increased ALT and/or AST
[see Warnings and Precautions (5.2)]Grade 2 Withhold TABRECTA until recovery to baseline bilirubin.
If recovered to baseline within 7 days, then resume TABRECTA at the same dose; otherwise resume TABRECTA at a reduced dose.Grade 3 Withhold TABRECTA until recovery to baseline bilirubin.
If recovered to baseline within 7 days, then resume TABRECTA at a reduced dose; otherwise permanently discontinue TABRECTA.Grade 4 Permanently discontinue TABRECTA. Other Adverse Reactions
[see Adverse Reactions (6.1)]Grade 2 Maintain dose level. If intolerable, consider withholding TABRECTA until resolved, then resume TABRECTA at a reduced dose. Grade 3 Withhold TABRECTA until resolved, then resume TABRECTA at a reduced dose. Grade 4 Permanently discontinue TABRECTA. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
ILD/pneumonitis, which can be fatal, occurred in patients treated with TABRECTA [see Adverse Reactions (6.1)]. ILD/pneumonitis occurred in 4.5% of patients treated with TABRECTA in GEOMETRY mono-1, with 1.8% of patients experiencing Grade 3 ILD/pneumonitis and one patient experiencing death (0.3%). Eight patients (2.4%) discontinued TABRECTA due to ILD/pneumonitis. The median time-to-onset of Grade 3 or higher ILD/pneumonitis was 1.4 months (range: 0.2 months to 1.2 years).
Monitor for new or worsening pulmonary symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Immediately withhold TABRECTA in patients with suspected ILD/pneumonitis and permanently discontinue if no other potential causes of ILD/pneumonitis are identified [see Dosage and Administration (2.3)].
5.2 Hepatotoxicity
Hepatotoxicity occurred in patients treated with TABRECTA [see Adverse Reactions (6.1)]. Increased alanine aminotransferase (ALT)/aspartate aminotransferase (AST) occurred in 13% of patients treated with TABRECTA in GEOMETRY mono-1. Grade 3 or 4 increased ALT/AST occurred in 6% of patients. Three patients (0.9%) discontinued TABRECTA due to increased ALT/AST. The median time-to-onset of Grade 3 or higher increased ALT/AST was 1.4 months (range: 0.5 to 4.1 months).
Monitor liver function tests (including ALT, AST, and total bilirubin) prior to the start of TABRECTA, every 2 weeks during the first 3 months of treatment, then once a month or as clinically indicated, with more frequent testing in patients who develop increased transaminases or bilirubin. Based on the severity of the adverse reaction, withhold, dose reduce, or permanently discontinue TABRECTA [see Dosage and Administration (2.3)].
5.3 Risk of Photosensitivity
Based on findings from animal studies, there is a potential risk of photosensitivity reactions with TABRECTA [see Nonclinical Toxicology (13.2)]. In GEOMETRY mono-1, it was recommended that patients use precautionary measures against ultraviolet exposure such as use of sunscreen or protective clothing during treatment with TABRECTA. Advise patients to limit direct ultraviolet exposure during treatment with TABRECTA.
5.4 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TABRECTA can cause fetal harm when administered to a pregnant woman. Oral administration of capmatinib to pregnant rats and rabbits during the period of organogenesis resulted in malformations at exposures less than the human exposure based on area under the curve (AUC) at the 400 mg twice daily clinical dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- ILD/Pneumonitis [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Metastatic Non-Small Cell Lung Cancer
The safety of TABRECTA was evaluated in GEOMETRY mono-1 [see Clinical Studies (14)]. Patients received TABRECTA 400 mg orally twice daily until disease progression or unacceptable toxicity (N=334). Among patients who received TABRECTA, 31% were exposed for at least 6 months and 16% were exposed for at least one year.
Serious adverse reactions occurred in 51% of patients who received TABRECTA. Serious adverse reactions in ≥ 2% of patients included dyspnea (7%), pneumonia (4.8%), pleural effusion (3.6%), general physical health deterioration (3%), vomiting (2.4%), and nausea (2.1%). A fatal adverse reaction occurred in one patient (0.3%) due to pneumonitis.
Permanent discontinuation of TABRECTA due to an adverse reaction occurred in 16% of patients. The most frequent adverse reactions (≥ 1%) leading to permanent discontinuation of TABRECTA were peripheral edema (1.8%), pneumonitis (1.8%), and fatigue (1.5%).
Dose interruptions due to an adverse reaction occurred in 54% of patients who received TABRECTA. Adverse reactions requiring dosage interruption in > 2% of patients who received TABRECTA included peripheral edema, increased blood creatinine, nausea, vomiting, increased lipase, increased ALT, dyspnea, increased amylase, increased AST, increased blood bilirubin, fatigue, and pneumonia.
Dose reductions due to an adverse reaction occurred in 23% of patients who received TABRECTA. Adverse reactions requiring dosage reductions in > 2% of patients who received TABRECTA included peripheral edema, increased ALT, increased blood creatinine, and nausea.
The most common adverse reactions (≥ 20%) in patients who received TABRECTA were peripheral edema, nausea, fatigue, vomiting, dyspnea, and decreased appetite.
Table 3 summarizes the adverse reactions in GEOMETRY mono-1.
Table 3: Adverse Reactions (≥ 10%) in Patients Who Received TABRECTA in GEOMETRY mono-1 aOnly includes Grade 3 adverse reactions with exception of dyspnea. Grade 4 dyspnea was reported in 0.6% of patients.
bPeripheral edema includes peripheral swelling, peripheral edema, and fluid overload.
cFatigue includes fatigue and asthenia.
dNon-cardiac chest pain includes chest discomfort, musculoskeletal chest pain, non-cardiac chest pain, and chest pain.
ePyrexia includes pyrexia and body temperature increased.Adverse Reactions TABRECTA
(N = 334)Grades 1 to 4
(%)Grades 3 to 4a
(%)General disorders and administration-site conditions Peripheral edemab 52 9 Fatiguec 32 8 Non-cardiac chest paind 15 2.1 Back pain 14 0.9 Pyrexiae 14 0.6 Weight decreased 10 0.6 Gastrointestinal disorders Nausea 44 2.7 Vomiting 28 2.4 Constipation 18 0.9 Diarrhea 18 0.3 Respiratory, thoracic, and mediastinal disorders Dyspnea 24 7a Cough 16 0.6 Metabolism and nutrition disorders Decreased appetite 21 0.9 Clinically relevant adverse reactions occurring in < 10% of patients treated with TABRECTA included pruritus (allergic and generalized), ILD/pneumonitis, cellulitis, acute kidney injury (including renal failure), urticaria, and acute pancreatitis.
Table 4 summarizes the laboratory abnormalities in GEOMETRY mono-1.
Table 4: Select Laboratory Abnormalities (≥ 20%) Worsening from Baseline in Patients Who Received TABRECTA in GEOMETRY mono-1 aThe denominator used to calculate the rate varied from 320 to 325 based on the number of patients with a baseline value and at least one post-treatment value. Laboratory Abnormalities TABRECTAa Grades 1 to 4
(%)Grades 3 to 4
(%)Chemistry Decreased albumin 68 1.8 Increased creatinine 62 0.3 Increased alanine aminotransferase 37 8 Increased alkaline phosphatase 32 0.3 Increased amylase 31 4.4 Increased gamma-glutamyltransferase 29 7 Increased lipase 26 7 Increased aspartate aminotransferase 25 4.9 Decreased sodium 23 6 Decreased phosphate 23 4.6 Increased potassium 23 3.1 Decreased glucose 21 0.3 Hematology Decreased lymphocytes 44 14 Decreased hemoglobin 24 2.8 Decreased leukocytes 23 0.9 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TABRECTA
Strong CYP3A Inhibitors
Coadministration of TABRECTA with a strong CYP3A inhibitor increased capmatinib exposure, which may increase the incidence and severity of adverse reactions of TABRECTA [see Clinical Pharmacology (12.3)]. Closely monitor patients for adverse reactions during coadministration of TABRECTA with strong CYP3A inhibitors.
Strong and Moderate CYP3A Inducers
Coadministration of TABRECTA with a strong CYP3A inducer decreased capmatinib exposure. Coadministration of TABRECTA with a moderate CYP3A inducer may also decrease capmatinib exposure. Decreases in capmatinib exposure may decrease TABRECTA anti-tumor activity [see Clinical Pharmacology (12.3)]. Avoid coadministration of TABRECTA with strong and moderate CYP3A inducers.
7.2 Effect of TABRECTA on Other Drugs
CYP1A2 Substrates
Coadministration of TABRECTA increased the exposure of a CYP1A2 substrate, which may increase the adverse reactions of these substrates [see Clinical Pharmacology (12.3)]. If coadministration is unavoidable between TABRECTA and CYP1A2 substrates where minimal concentration changes may lead to serious adverse reactions, decrease the CYP1A2 substrate dosage in accordance with the approved prescribing information.
P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP) Substrates
Coadministration of TABRECTA increased the exposure of a P-gp substrate and a BCRP substrate, which may increase the adverse reactions of these substrates [see Clinical Pharmacology (12.3)]. If coadministration is unavoidable between TABRECTA and P-gp or BCRP substrates where minimal concentration changes may lead to serious adverse reactions, decrease the P-gp or BCRP substrate dosage in accordance with the approved prescribing information.
MATE1 and MATE2K Substrates
Coadministration of TABRECTA may increase the exposure of MATE1 and MATE2K substrates, which may increase the adverse reactions of these substrates [see Clinical Pharmacology (12.3)]. If coadministration is unavoidable between TABRECTA and MATE1 or MATE2K substrates where minimal concentration changes may lead to serious adverse reactions, decrease the MATE1 or MATE2K substrate dosage in accordance with the approved prescribing information.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], TABRECTA can cause fetal harm when administered to a pregnant woman. There are no available data on TABRECTA use in pregnant women. Oral administration of capmatinib to pregnant rats and rabbits during the period of organogenesis resulted in malformations at maternal exposures less than the human exposure based on AUC at the 400 mg twice daily clinical dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In rats, maternal toxicity (reduced body weight gain and food consumption) occurred at 30 mg/kg/day (approximately 1.4 times the human exposure based on AUC at the 400 mg twice daily clinical dose). Fetal effects included reduced fetal weights, irregular/incomplete ossification, and increased incidences of fetal malformations (e.g., abnormal flexure/inward malrotation of hindpaws/forepaws, thinness of forelimbs, lack of/reduced flexion at the humerus/ulna joints, and narrowed or small tongue) at doses of ≥ 10 mg/kg/day (approximately 0.6 times the human exposure based on AUC at the 400 mg twice daily clinical dose).
In rabbits, no maternal effects were detected at doses up to 60 mg/kg/day (approximately 1.5 times the human exposure based on AUC at the 400 mg twice daily clinical dose). Fetal effects included small lung lobe at ≥ 5 mg/kg/day (approximately 0.016 times the human exposure based on AUC at the 400 mg twice daily clinical dose), and reduced fetal weights, irregular/incomplete ossification and increased incidences of fetal malformations (e.g., abnormal flexure/malrotation of hindpaws/forepaws, thinness of forelimbs/hindlimbs, lack of/reduced flexion at the humerus/ulna joints, small lung lobes, narrowed or small tongue) at the dose of 60 mg/kg/day.
8.2 Lactation
Risk Summary
There are no data on the presence of capmatinib or its metabolites in either human or animal milk or its effects on the breastfed child or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with TABRECTA and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, TABRECTA can cause malformations at doses less than the human exposure based on AUC at the 400 mg twice daily clinical dose [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status for females of reproductive potential prior to starting treatment with TABRECTA.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose.
Males
Advise males with female partners of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose.
8.4 Pediatric Use
Safety and effectiveness of TABRECTA in pediatric patients have not been established.
8.5 Geriatric Use
In GEOMETRY mono-1, 57% of the 334 patients were 65 years or older and 16% were 75 years or older. No overall differences in the safety or effectiveness were observed between these patients and younger patients.
8.6 Renal Impairment
No dosage adjustment is recommended in patients with mild (baseline creatinine clearance [CLcr] 60 to 89 mL/min by Cockcroft-Gault) or moderate renal impairment (CLcr 30 to 59 mL/min) [see Clinical Pharmacology (12.3)]. TABRECTA has not been studied in patients with severe renal impairment (CLcr 15 to 29 mL/min).
-
11 DESCRIPTION
Capmatinib is a kinase inhibitor. The chemical name is 2-Fluoro-N-methyl-4-[7-(quinolin-6-ylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide—hydrogen chloride—water (1/2/1). The molecular formula for capmatinib hydrochloride is C23H21Cl2FN6O2. The relative molecular mass is 503.36 g/mol for the hydrochloride salt and 412.43 g/mol for the free base. The chemical structure for capmatinib hydrochloride is shown below:
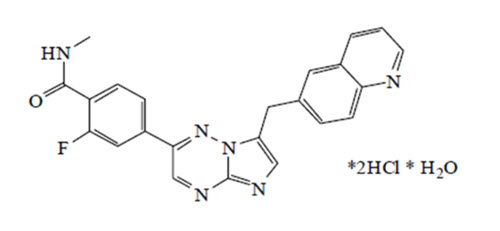
Capmatinib hydrochloride is a yellow powder with a pKa1 of 0.9 (calculated) and pKa2 of 4.5 (experimentally). Capmatinib hydrochloride is slightly soluble in acidic aqueous solutions at pH 1 and 2 and of further decreasing solubility towards neutral condition. The log of the distribution coefficient (n-octanol/acetate buffer pH 4.0) is 1.2.
TABRECTA is supplied for oral use as ovaloid, curved film-coated tablets with beveled edges, unscored containing 150 mg (pale orange brown color) or 200 mg (yellow color) capmatinib (equivalent to 176.55 mg or 235.40 mg respectively of capmatinib hydrochloride anhydrous). Each tablet strength contains colloidal silicon dioxide; crospovidone; magnesium stearate; mannitol; microcrystalline cellulose; povidone; and sodium lauryl sulfate as inactive ingredients.
The 150 mg tablet coating contains ferric oxide, red; ferric oxide, yellow; ferrosoferric oxide; hypromellose; polyethylene glycol (PEG) 4000; talc; and titanium dioxide. The 200 mg tablet coating contains ferric oxide, yellow; hypromellose; polyethylene glycol (PEG) 4000; talc; and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Capmatinib is a kinase inhibitor that targets MET, including the mutant variant produced by exon 14 skipping. MET exon 14 skipping results in a protein with a missing regulatory domain that reduces its negative regulation leading to increased downstream MET signaling. Capmatinib inhibited cancer cell growth driven by a mutant MET variant lacking exon 14 at clinically achievable concentrations and demonstrated anti-tumor activity in murine tumor xenograft models derived from human lung tumors with either a mutation leading to MET exon 14 skipping or MET amplification. Capmatinib inhibited the phosphorylation of MET triggered by binding of hepatocyte growth factor or by MET amplification, as well as MET-mediated phosphorylation of downstream signaling proteins and proliferation and survival of MET-dependent cancer cells.
12.2 Pharmacodynamics
Exposure-Response
Capmatinib exposure-response relationships and the time course of pharmacodynamics response are unknown.
Cardiac Electrophysiology
No large mean increase in QTc (i.e. > 20 ms) was detected following treatment with TABRECTA at the recommended dosage of 400 mg orally twice daily.
12.3 Pharmacokinetics
Capmatinib exposure (AUC0-12h and Cmax) increased approximately proportionally over a dose range of 200 mg (0.5 times the recommended dosage) to 400 mg. Capmatinib reached steady-state by day 3 following twice daily dosing, with a mean (% coefficient of variation [%CV]) accumulation ratio of 1.5 (41%).
Absorption
After administration of TABRECTA 400 mg orally in patients with cancer, capmatinib peak plasma concentrations (Cmax) were reached in approximately 1 to 2 hours (Tmax). The absorption of capmatinib after oral administration is estimated to be greater than 70%.
Effect of Food
A high-fat meal (containing approximately 1000 calories and 50% fat) in healthy subjects increased capmatinib AUC0-INF by 46% with no change in Cmax compared to under fasted conditions. A low-fat meal (containing approximately 300 calories and 20% fat) in healthy subjects had no clinically meaningful effect on capmatinib exposure. When capmatinib was administered at 400 mg orally twice daily in cancer patients, exposure (AUC0-12h) was similar after administration of capmatinib with food and under fasted conditions.
Distribution
Capmatinib plasma protein binding is 96%, independent of capmatinib concentration. The apparent mean volume of distribution at steady-state is 164 L.
The blood-to-plasma ratio was 1.5, but decreased at higher concentrations to 0.9.
Elimination
The effective elimination half-life of capmatinib is 6.5 hours. The mean (%CV) steady-state apparent clearance of capmatinib is 24 L/hr (82%).
Metabolism
Capmatinib is primarily metabolized by CYP3A4 and aldehyde oxidase.
Excretion
Following a single oral administration of radiolabeled-capmatinib to healthy subjects, 78% of the total radioactivity was recovered in feces with 42% as unchanged and 22% was recovered in urine with negligible as unchanged.
Specific Populations
No clinically significant effects on the pharmacokinetic parameters of capmatinib were identified for the following covariates assessed: age (26 to 90 years), sex, race (White, Asian, Native American, Black, unknown), body weight (35 to 131 kg), mild to moderate renal impairment (baseline CLcr 30 to 89 mL/min by Cockcroft-Gault) and mild, moderate or severe hepatic impairment (Child-Pugh classification). The effect of severe renal impairment (baseline CLcr 15 to 29 mL/min) on capmatinib pharmacokinetics has not been studied.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A Inhibitors: Coadministration with itraconazole (a strong CYP3A inhibitor) increased capmatinib AUC0-INF by 42% with no change in capmatinib Cmax.
Strong CYP3A Inducers: Coadministration with rifampicin (a strong CYP3A inducer) decreased capmatinib AUC0-INF by 67% and decreased Cmax by 56%.
Moderate CYP3A Inducers: Coadministration with efavirenz (a moderate CYP3A inducer) was predicted to decrease capmatinib AUC0-12h by 44% and decrease Cmax by 34%.
Proton Pump Inhibitors: Coadministration with rabeprazole (a proton pump inhibitor) decreased capmatinib AUC0-INF by 25% and decreased Cmax by 38%.
Substrates of CYP Enzymes: Coadministration of capmatinib increased caffeine (a CYP1A2 substrate) AUC0-INF by 134% with no change in its Cmax. Coadministration of capmatinib had no clinically meaningful effect on exposure of midazolam (a CYP3A substrate).
P-gp Substrates: Coadministration of capmatinib increased digoxin (a P-gp substrate) AUC0-INF by 47% and increased Cmax by 74%.
BCRP Substrates: Coadministration of capmatinib increased rosuvastatin (a BCRP substrate) AUC0-INF by 108% and increased Cmax by 204%.
In Vitro Studies
Transporter Systems: Capmatinib is a substrate of P-gp, but not a substrate of BCRP or MRP2. Capmatinib reversibly inhibits MATE1 and MATE2K, but does not inhibit OATP1B1, OATP1B3, OCT1, OAT1, OAT3, or MRP2.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were not conducted with capmatinib. Capmatinib was not mutagenic in an in vitro bacterial reverse mutation assay and did not cause chromosomal aberrations in an in vitro chromosome aberration assay in human peripheral blood lymphocytes. Capmatinib was not clastogenic in an in vivo bone marrow micronucleus test in rats.
Dedicated fertility studies were not conducted with capmatinib. No effects on male and female reproductive organs occurred in general toxicology studies conducted in rats and monkeys at doses resulting in exposures of up to approximately 3.6 times the human exposure based on AUC at the 400 mg twice daily clinical dose.
13.2 Animal Toxicology and/or Pharmacology
In rats, capmatinib administration resulted in vacuolation of white matter of the brain in both 4- and 13-week studies at doses ≥ 2.2 times the human exposure (AUC) at the 400 mg twice daily clinical dose. In some cases, the brain lesions were associated with early death and/or convulsions or tremors. Concentrations of capmatinib in the brain tissue of rats was approximately 9% of the corresponding concentrations in plasma.
In vitro and in vivo assays demonstrated that capmatinib has some potential for photosensitization; however, the no-observed-adverse-effect level for in vivo photosensitization was 30 mg/kg/day (Cmax of 14000 ng/mL), about 2.9 times the human Cmax at the 400 mg twice daily clinical dose.
-
14 CLINICAL STUDIES
Metastatic NSCLC with a Mutation that Leads to MET Exon 14 Skipping
The efficacy of TABRECTA was evaluated in GEOMETRY mono-1, a multicenter, non-randomized, open-label, multi-cohort study (NCT02414139). Eligible patients were required to have NSCLC with a mutation that leads to MET exon 14 skipping, epidermal growth factor receptor (EGFR) wild-type and anaplastic lymphoma kinase (ALK) negative status, and at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Patients with symptomatic CNS metastases, clinically significant uncontrolled cardiac disease, or who received treatment with any MET or hepatocyte growth factor (HGF) inhibitor were not eligible for the study.
Out of the 97 patients enrolled in GEOMETRY mono-1 following the central confirmation of MET exon 14 skipping by a RNA-based clinical trial assay, 78 patient samples were retested with the FDA-approved FoundationOne® CDx (22 treatment-naïve and 56 previously treated patients) to detect mutations that lead to MET exon 14 skipping. Out of 78 samples retested with FoundationOne® CDx, 73 samples were evaluable (20 treatment-naïve and 53 previously treated patients), 72 (20 treatment-naïve and 52 previously treated patients) of which were confirmed to have a mutation that leads to MET exon 14 skipping, demonstrating an estimated positive percentage agreement of 99% (72/73) between the clinical trial assay and the FDA-approved assay.
Patients received TABRECTA 400 mg orally twice daily until disease progression or unacceptable toxicity. The major efficacy outcome measure was overall response rate (ORR) as determined by a Blinded Independent Review Committee (BIRC) according to RECIST 1.1. An additional efficacy outcome measure was duration of response (DOR) by BIRC.
The efficacy population included 28 treatment-naïve patients and 69 previously treated patients. The median age was 71 years (range: 49 to 90 years); 60% female; 75% White; 24% had Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) 0 and 75% had ECOG PS 1; 60% never smoked; 80% had adenocarcinoma; and 12% had CNS metastases. Amongst previously treated patients, 88% received prior platinum-based chemotherapy.
Efficacy results are presented in Table 5.
Table 5: Efficacy Results in GEOMETRY mono-1 Abbreviations: CI = Confidence Interval
aBlinded Independent Review Committee (BIRC) review.
bConfirmed response.
cClopper and Pearson exact binomial 95% CI.
dBased on Kaplan-Meier estimate.Efficacy Parameters Treatment-Naïve
N = 28Previously Treated
N = 69Overall Response Ratea,b (95% CI)c 68% (48, 84) 41% (29, 53) Complete Response 4% 0 Partial Response 64% 41% Duration of Response (DOR)a Median (months) (95% CI)d 12.6 (5.5, 25.3) 9.7 (5.5, 13.0) Patients with DOR ≥ 12 months 47% 32% -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
TABRECTA (capmatinib) 150 mg and 200 mg tablets Strength Description Tablets per Bottle NDC Number 150 mg Pale orange brown, ovaloid, curved film-coated tablet with beveled edges, unscored, debossed with ‘DU’ on one side and ‘NVR’ on the other side. 56 0078-0709-56 200 mg Yellow, ovaloid, curved film-coated tablet with beveled edges, unscored, debossed with ‘LO’ on one side and ‘NVR’ on the other side. 56 0078-0716-56 Storage
Dispense in the original package with the desiccant cartridge. Store at 20˚C to 25˚C (68˚F to 77˚F), excursions permitted between 15˚C and 30˚C (59˚F and 86˚F) [see USP Controlled Room Temperature]. Protect from moisture.
Discard any unused TABRECTA remaining after 6 weeks of first opening the bottle.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the risks of severe or fatal ILD/pneumonitis. Advise patients to immediately contact their healthcare provider for new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Hepatotoxicity
Inform patients that they will need to undergo lab tests to monitor liver function. Advise patients to immediately contact their healthcare provider for signs and symptoms of liver dysfunction [see Warnings and Precautions (5.2)].
Risk of Photosensitivity
Inform patients that there is a potential risk of photosensitivity reactions with TABRECTA. Advise patients to limit direct ultraviolet exposure by using sunscreen or protective clothing during treatment with TABRECTA [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with TABRECTA and for 1 week after the last dose [see Use in Specific Populations (8.3)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
Lactation
Advise women not to breastfeed during treatment with TABRECTA and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936© Novartis
T2020-47
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 5/2020 PATIENT INFORMATION
TABRECTA™ (ta brek tah)
(capmatinib) tabletsWhat is TABRECTA?
TABRECTA is a prescription medicine used to treat adults with a kind of lung cancer called non-small cell lung cancer (NSCLC) that:- has spread to other parts of the body or cannot be removed by surgery (metastatic), and
- whose tumors have an abnormal mesenchymal epithelial transition (MET) gene
Before taking TABRECTA, tell your healthcare provider about all of your medical conditions, including if you: - have or have had lung or breathing problems other than your lung cancer
- have or have had liver problems
- are pregnant or plan to become pregnant. TABRECTA can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider should do a pregnancy test before you start your treatment with TABRECTA.
- You should use effective birth control during treatment and for 1 week after your last dose of TABRECTA. Talk to your healthcare provider about birth control choices that might be right for you during this time.
- Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with TABRECTA.
Males who have female partners who can become pregnant:
- You should use effective birth control during treatment and for 1 week after your last dose of TABRECTA.
- are breastfeeding or plan to breastfeed. It is not known if TABRECTA passes into your breast milk. Do not breastfeed during treatment and for 1 week after your last dose of TABRECTA.
How should I take TABRECTA? - Take TABRECTA exactly as your healthcare provider tells you.
- Take TABRECTA 2 times a day with or without food.
- Swallow TABRECTA tablets whole. Do not break, chew, or crush TABRECTA tablets.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with TABRECTA if you have certain side effects.
- Do not change your dose or stop taking TABRECTA unless your healthcare provider tells you to.
- If you miss or vomit a dose of TABRECTA, do not make up the dose. Take your next dose at your regular scheduled time.
What should I avoid while taking TABRECTA? - Your skin may be sensitive to the sun (photosensitivity) during treatment with TABRECTA. Use sunscreen or wear clothes that cover your skin during your treatment with TABRECTA to limit direct sunlight exposure.
What are the possible side effects of TABRECTA?
TABRECTA may cause serious side effects, including:- lung or breathing problems. TABRECTA may cause inflammation of the lungs that can cause death. Tell your healthcare provider right away if you develop any new or worsening symptoms, including:
◦ cough ◦ fever ◦ trouble breathing or shortness of breath Your healthcare provider may temporarily stop or permanently stop treatment with TABRECTA if you develop lung or breathing problems during treatment.
- liver problems. TABRECTA may cause abnormal liver blood test results. Your healthcare provider will do blood tests to check your liver function before you start treatment and during treatment with TABRECTA. Tell your healthcare provider right away if you develop any signs and symptoms of liver problems, including:
◦ your skin or the white part of your eyes turns yellow (jaundice)
◦ dark or “tea-colored” urine
◦ light-colored stools (bowel movements)
◦ confusion
◦ tiredness◦ loss of appetite for several days or longer
◦ nausea and vomiting
◦ pain, aching, or tenderness on the right side of your stomach-area (abdomen)
◦ weakness
◦ swelling in your stomach-areaYour healthcare provider may change your dose, temporarily stop, or permanently stop treatment with TABRECTA if you develop liver problems during treatment.
- risk of sensitivity to sunlight (photosensitivity). See “What should I avoid while taking TABRECTA?”
The most common side effects of TABRECTA include: swelling of your hands or feet
nausea
tiredness and weaknessvomiting
loss of appetite
changes in certain blood testsThese are not all of the possible side effects of TABRECTA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store TABRECTA? - Store TABRECTA at room temperature between 68°F to 77°F (20°C to 25°C).
- Store TABRECTA in the original package with the drying agent (desiccant) cartridge.
- Protect TABRECTA from moisture.
- Throw away (discard) any unused TABRECTA you have left after 6 weeks of first opening the bottle.
General information about the safe and effective use of TABRECTA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TABRECTA for a condition for which it was not prescribed. Do not give TABRECTA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TABRECTA that is written for health professionals.What are the ingredients in TABRECTA?
Active ingredient: capmatinib
Inactive ingredients: Tablet core: colloidal silicon dioxide; crospovidone; magnesium stearate; mannitol; microcrystalline cellulose; povidone; and sodium lauryl sulfate.
Tablet coating (150 mg): ferric oxide, red; ferric oxide, yellow; ferrosoferric oxide; hypromellose; polyethylene glycol (PEG) 4000; talc; and titanium dioxide.
Tablet coating (200 mg): ferric oxide, yellow; hypromellose; polyethylene glycol (PEG) 4000; talc; and titanium dioxide.
Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936
For more information, go to www.TABRECTA.com or call 1-844-638-5864.
© Novartis
T2020-48
-
PRINCIPAL DISPLAY PANEL
NDC: 0078-0709-56
TABRECTA™
(capmatinib) tablets
150 mg*
Attention: Dispense and store Tabrecta
in original container with the desiccant
to protect from moisture.Rx only
56 Film-coated tablets
NOVARTIS

-
PRINCIPAL DISPLAY PANEL
NDC: 0078-0716-56
TABRECTA™
(capmatinib) tablets
200 mg*
Attention: Dispense and store Tabrecta
in original container with the desiccant
to protect from moisture.Rx only
56 Film-coated tablets
NOVARTIS

-
INGREDIENTS AND APPEARANCE
TABRECTA
capmatinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0709 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CAPMATINIB (UNII: TY34L4F9OZ) (CAPMATINIB - UNII:TY34L4F9OZ) CAPMATINIB 150 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POVIDONE (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) HYPROMELLOSES (UNII: 3NXW29V3WO) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color ORANGE (Pale orange brown) Score no score Shape OVAL (ovaloid, curved) Size 18mm Flavor Imprint Code DU;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0709-56 56 in 1 BOTTLE; Type 0: Not a Combination Product 05/06/2020 2 NDC: 0078-0709-94 56 in 1 BOTTLE; Type 0: Not a Combination Product 05/06/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213591 05/06/2020 TABRECTA
capmatinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0716 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CAPMATINIB (UNII: TY34L4F9OZ) (CAPMATINIB - UNII:TY34L4F9OZ) CAPMATINIB 200 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POVIDONE (UNII: FZ989GH94E) SODIUM LAURYL SULFATE (UNII: 368GB5141J) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) HYPROMELLOSES (UNII: 3NXW29V3WO) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color YELLOW Score no score Shape OVAL (ovaloid, curved) Size 20mm Flavor Imprint Code LO;NVR Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0716-56 56 in 1 BOTTLE; Type 0: Not a Combination Product 05/06/2020 2 NDC: 0078-0716-94 56 in 1 BOTTLE; Type 0: Not a Combination Product 05/06/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA213591 05/06/2020 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [TABRECTA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TABRECTA 79281021 not registered Live/Pending |
NOVARTIS AG 2020-01-22 |
 TABRECTA 79148570 4630489 Live/Registered |
NOVARTIS AG 2014-03-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.