These highlights do not include all the information needed to use PROPAFENONE HYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for PROPAFENONE HYDROCHLORIDE TABLETS. PROPAFENONE HYDROCHLORIDE tablets, for oral useInitial U.S. Approval: 1989
Propafenone Hydrochloride by
Drug Labeling and Warnings
Propafenone Hydrochloride by is a Prescription medication manufactured, distributed, or labeled by Bryant Ranch Prepack. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
PROPAFENONE HYDROCHLORIDE- propafenone hydrochloride tablet, film coated
Bryant Ranch Prepack
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PROPAFENONE HYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for PROPAFENONE HYDROCHLORIDE TABLETS.
PROPAFENONE HYDROCHLORIDE tablets, for oral use Initial U.S. Approval: 1989 WARNING: MORTALITYSee full prescribing information for complete boxed warning.
RECENT MAJOR CHANGESINDICATIONS AND USAGEPropafenone hydrochloride tablets are an antiarrhythmic indicated to:
Usage Considerations:
DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSTablets: 150 mg, 225 mg, 300 mg (3) CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most commonly reported adverse events with propafenone (greater than 5%) included: unusual taste, nausea and/or vomiting, dizziness, constipation, headache, fatigue, first-degree AV block, and intraventricular conduction delay. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Par Pharmaceutical at 1-800-828-9393 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 4/2022 |
FULL PRESCRIBING INFORMATION
WARNING: MORTALITY
- In the National Heart, Lung, and Blood Institute’s Cardiac Arrhythmia Suppression Trial (CAST), a long-term, multicenter, randomized, double-blind trial in subjects with asymptomatic non-life-threatening ventricular arrhythmias who had a myocardial infarction more than 6 days but less than 2 years previously, an increased rate of death or reversed cardiac arrest rate (7.7%; 56/730) was seen in subjects treated with encainide or flecainide (Class IC antiarrhythmics) compared with that seen in subjects assigned to placebo (3.0%; 22/725). The average duration of treatment with encainide or flecainide in this trial was 10 months.
- The applicability of the CAST results to other populations (e.g., those without recent myocardial infarction) or other antiarrhythmic drugs is uncertain, but at present, it is prudent to consider any IC antiarrhythmic to have a significant proarrhythmic risk in patients with structural heart disease. Given the lack of any evidence that these drugs improve survival, antiarrhythmic agents should generally be avoided in patients with non-life-threatening ventricular arrhythmias, even if the patients are experiencing unpleasant, but not life-threatening, symptoms or signs.
1 INDICATIONS AND USAGE
Propafenone hydrochloride tablets are indicated to:
-
prolong the time to recurrence of paroxysmal atrial fibrillation/flutter (PAF) associated with disabling symptoms in patients without structural heart disease.
-
prolong the time to recurrence of paroxysmal supraventricular tachycardia (PSVT) associated with disabling symptoms in patients without structural heart disease.
-
treat documented ventricular arrhythmias, such as sustained ventricular tachycardia that, in the judgment of the physician, are life-threatening. Initiate treatment in the hospital.
Usage Considerations:
-
The use of propafenone hydrochloride tablets in patients with permanent atrial fibrillation (AF) or in patients exclusively with atrial flutter or PSVT has not been evaluated. Do not use propafenone hydrochloride tablets to control ventricular rate during AF.
-
Some patients with atrial flutter treated with propafenone have developed 1:1 conduction, producing an increase in ventricular rate. Concomitant treatment with drugs that increase the functional atrioventricular (AV) nodal refractory period is recommended.
-
The use of propafenone hydrochloride tablets in patients with chronic atrial fibrillation has not been evaluated.
-
Because of the proarrhythmic effects of propafenone hydrochloride tablets, its use with lesser ventricular arrhythmias is not recommended, even if patients are symptomatic, and any use of the drug should be reserved for patients in whom, in the opinion of the physician, the potential benefits outweigh the risks.
-
The effect of propafenone on mortality has not been determined [see Boxed Warning].
2 DOSAGE AND ADMINISTRATION
The dose of propafenone hydrochloride tablets must be individually titrated on the basis of response and tolerance. Initiate therapy with propafenone hydrochloride tablets 150 mg given every 8 hours (450 mg/day). Dosage may be increased at a minimum of 3 to 4 day intervals to 225 mg every 8 hours (675 mg/day). If additional therapeutic effect is needed, the dose of propafenone hydrochloride tablets may be increased to 300 mg every 8 hours (900 mg/day). The usefulness and safety of dosages exceeding 900 mg per day have not been established.
In patients with hepatic impairment or those with significant widening of the QRS complex or second- or third-degree AV block, consider reducing the dose.
As with other antiarrhythmic agents, in the elderly or in ventricular arrhythmia patients with marked previous myocardial damage, the dose of propafenone hydrochloride tablets should be increased more gradually during the initial phase of treatment.
The combination of cytochrome P450 3A4 (CYP3A4) inhibition and either cytochrome P450 2D6 (CYP2D6) deficiency or CYP2D6 inhibition with the simultaneous administration of propafenone may significantly increase the concentration of propafenone and thereby increase the risk of proarrhythmia and other adverse events. Therefore, avoid simultaneous use of propafenone hydrochloride tablets with both a CYP2D6 inhibitor and a CYP3A4 inhibitor see Warnings and Precautions (5.4) and Drug Interactions (7.1)].
4 CONTRAINDICATIONS
Propafenone hydrochloride is contraindicated in the following circumstances:
-
Heart failure
-
Cardiogenic shock
-
Sinoatrial, atrioventricular and intraventricular disorders of impulse generation or conduction (e.g., sick sinus node syndrome, AV block) in the absence of an artificial pacemaker
-
Known Brugada Syndrome
-
Bradycardia
-
Marked hypotension
-
Bronchospastic disorders or severe obstructive pulmonary disease
-
Marked electrolyte imbalance
5 WARNINGS AND PRECAUTIONS
5.1 Proarrhythmic Effects
Propafenone has caused new or worsened arrhythmias. Such proarrhythmic effects include sudden death and life-threatening ventricular arrhythmias such as ventricular fibrillation, ventricular tachycardia, asystole and torsade de pointes. It may also worsen premature ventricular contractions or supraventricular arrhythmias, and it may prolong the QT interval. It is therefore essential that each patient given propafenone hydrochloride be evaluated electrocardiographically prior to and during therapy to determine whether the response to propafenone hydrochloride supports continued treatment. Because propafenone prolongs the QRS interval in the electrocardiogram, changes in the QT interval are difficult to interpret [see Clinical Pharmacology (12.2)].
In a U.S. uncontrolled, open label, multicenter trial in subjects with symptomatic supraventricular tachycardia (SVT), 1.9% (9/474) of these subjects experienced ventricular tachycardia (VT) or ventricular fibrillation (VF) during the trial. However, in 4 of the 9 subjects, the ventricular tachycardia was of atrial origin. Six of the nine subjects that developed ventricular arrhythmias did so within 14 days of onset of therapy. About 2.3% (11/474) of all subjects had a recurrence of SVT during the trial which could have been a change in the subjects’ arrhythmia behavior or could represent a proarrhythmic event. Case reports in patients treated with propafenone for atrial fibrillation/flutter have included increased premature ventricular contractions (PVCs), VT, VF, torsade de pointes, asystole, and death.
Overall in clinical trials with propafenone hydrochloride (which included subjects treated for ventricular arrhythmias, atrial fibrillation/flutter, and PSVT), 4.7% of all subjects had new or worsened ventricular arrhythmia possibly representing a proarrhythmic event (0.7% was an increase in PVCs; 4.0% a worsening, or new appearance of VT or VF). Of the subjects who had worsening of VT (4%), 92% had a history of VT and/or VT/VF, 71% had coronary artery disease, and 68% had a prior myocardial infarction. The incidence of proarrhythmia in subjects with less serious or benign arrhythmias, which include subjects with an increase in frequency of PVCs, was 1.6%. Although most proarrhythmic events occurred during the first week of therapy, late events also were seen and the CAST trial [see Boxed Warning: Mortality] suggests that an increased risk of proarrythmia is present throughout treatment.
In a trial of sustained-release propafenone, there were too few deaths to assess the long-term risk to patients. There were 5 deaths; 3 in the pooled group for sustained-release propafenone (0.8%) and 2 in the placebo group (1.6%). In the overall database of 8 trials of sustained-release propafenone and immediate-release propafenone hydrochloride, the mortality rate was 2.5% per year on propafenone and 4.0% per year on placebo. Concurrent use of propafenone with other antiarrhythmic agents has not been well studied.
5.2 Unmasking Brugada Syndrome
Brugada Syndrome may be unmasked after exposure to propafenone hydrochloride. Perform an ECG after initiation of propafenone hydrochloride, and discontinue the drug if changes are suggestive of Brugada Syndrome [see Contraindications (4)].
5.3 Use with Drugs that Prolong the QT Interval and Antiarrhythmic Agents
The use of propafenone hydrochloride in conjunction with other drugs that prolong the QT interval has not been extensively studied. Such drugs may include many antiarrhythmics, some phenothiazines, tricyclic antidepressants, and oral macrolides. Withhold Class IA and III antiarrhythmic agents for at least 5 half-lives prior to dosing with propafenone hydrochloride. Avoid the use of propafenone with Class IA and III antiarrhythmic agents (including quinidine and amiodarone). There is only limited experience with the concomitant use of Class IB or IC antiarrhythmics.
5.4 Drug Interactions: Simultaneous Use with Inhibitors of Cytochrome P450 Isoenzymes 2D6 and 3A4
Propafenone is metabolized by CYP2D6, CYP3A4, and CYP1A2 isoenzymes. Approximately 6% of Caucasians in the U.S. population are naturally deficient in CYP2D6 activity and other demographic groups are deficient to a somewhat lesser extent. Drugs that inhibit these CYP pathways (such as desipramine, paroxetine, ritonavir, sertraline for CYP2D6; ketoconazole, erythromycin, saquinavir, and grapefruit juice for CYP3A4; and amiodarone and tobacco smoke for CYP1A2) can be expected to cause increased plasma levels of propafenone.
Increased exposure to propafenone may lead to cardiac arrhythmias and exaggerated beta-adrenergic blocking activity. Because of its metabolism, the combination of CYP3A4 inhibition and either CYP2D6 deficiency or CYP2D6 inhibition in users of propafenone is potentially hazardous. Therefore, avoid simultaneous use of propafenone hydrochloride with both a CYP2D6 inhibitor and a CYP3A4 inhibitor.
5.5 Use in Patients with a History of Heart Failure
Propafenone exerts a negative inotropic activity on the myocardium as well as beta-blockade effects and may provoke overt heart failure.
In clinical trial experience with propafenone hydrochloride, new or worsened congestive heart failure (CHF) has been reported in 3.7% of subjects with ventricular arrhythmia; of those 0.9% were considered probably or definitely related to propafenone HCl. Of the subjects with CHF probably related to propafenone, 80% had preexisting heart failure and 85% had coronary artery disease. CHF attributable to propafenone HCl developed rarely (less than 0.2%) in subjects with ventricular arrhythmia who had no previous history of CHF. CHF occurred in 1.9% of subjects studied with PAF or PSVT.
In a U.S. trial of sustained-release propafenone in subjects with symptomatic AF, heart failure was reported in 4 (1.0%) subjects receiving sustained-release propafenone (all doses), compared with 1 (0.8%) subject receiving placebo.
5.6 Conduction Disturbances
Propafenone slows atrioventricular conduction and may also cause dose-related first degree AV block. Average PR interval prolongation and increases in QRS duration are also dose-related. Do not give propafenone to patients with atrioventricular and intraventricular conduction defects in the absence of a pacemaker[see Contraindications (4) and Clinical Pharmacology (12.2)].
The incidence of first-degree, second-degree, and third-degree AV block observed in 2,127 subjects with ventricular arrhythmia was 2.5%, 0.6%, and 0.2%, respectively. Development of second- or third-degree AV block requires a reduction in dosage or discontinuation of propafenone HCl. Bundle branch block (1.2%) and intraventricular conduction delay (1.1%) have been reported in subjects receiving propafenone. Bradycardia has also been reported (1.5%). Experience in patients with sick sinus node syndrome is limited and these patients should not be treated with propafenone.
In a U.S. trial in 523 subjects with a history of symptomatic AF treated with sustained-release propafenone, sinus bradycardia (rate less than 50 beats/min) was reported with the same frequency with sustained-release propafenone and placebo.
5.7 Effects on Pacemaker Threshold
Propafenone may alter both pacing and sensing thresholds of implanted pacemakers and defibrillators. During and after therapy, monitor and re-program these devices accordingly.
5.8 Agranulocytosis
Agranulocytosis has been reported in patients receiving propafenone. Generally, the agranulocytosis occurred within the first 2 months of propafenone therapy and upon discontinuation of therapy the white count usually normalized by 14 days. Unexplained fever or decrease in white cell count, particularly during the initial 3 months of therapy, warrant consideration of possible agranulocytosis or granulocytopenia. Instruct patients to report promptly any signs of infection such as fever, sore throat, or chills.
5.9 Use in Patients with Hepatic Dysfunction
Propafenone is highly metabolized by the liver. Severe liver dysfunction increases the bioavailability of propafenone to approximately 70% compared with 3% to 40% in patients with normal liver function. In 8 subjects with moderate to severe liver disease, the mean half-life was approximately 9 hours. Increased bioavailability of propafenone in these patients may result in excessive accumulation. Carefully monitor patients with impaired hepatic function for excessive pharmacological effects [see Overdosage (10)].
5.10 Use in Patients with Renal Dysfunction
Approximately 50% of propafenone metabolites are excreted in the urine following administration of propafenone hydrochloride.
In patients with impaired renal function, monitor for signs of overdosage [see Overdosage (10)].
5.11 Use in Patients with Myasthenia Gravis
Exacerbation of myasthenia gravis has been reported during propafenone therapy.
5.12 Elevated ANA Titers
Positive ANA titers have been reported in patients receiving propafenone. They have been reversible upon cessation of treatment and may disappear even in the face of continued propafenone therapy. These laboratory findings were usually not associated with clinical symptoms, but there is one published case of drug-induced lupus erythematosus (positive rechallenge); it resolved completely upon discontinuation of therapy. Carefully evaluate patients who develop an abnormal ANA test and, if persistent or worsening elevation of ANA titers is detected, consider discontinuing therapy.
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions associated with propafenone hydrochloride occur most frequently in the gastrointestinal, cardiovascular, and central nervous systems. About 20% of subjects treated with propafenone hydrochloride have discontinued treatment because of adverse reactions.
Adverse reactions reported for greater than 1.5% of 474 subjects with SVT who received propafenone hydrochloride in U.S. clinical trials are presented in Table 1 by incidence and percent discontinuation, reported to the nearest percent.
| Adverse Reaction | Incidence (N = 480) | % of Subjects Who Discontinued |
| Unusual Taste | 14% | 1.3% |
| Nausea and/or Vomiting | 11% | 2.9% |
| Dizziness | 9% | 1.7% |
| Constipation | 8% | 0.2% |
| Headache | 6% | 0.8% |
| Fatigue | 6% | 1.5% |
| Blurred Vision | 3% | 0.6% |
| Weakness | 3% | 1.3% |
| Dyspnea | 2% | 1.0% |
| Wide Complex Tachycardia | 2% | 1.9% |
| CHF | 2% | 0.6% |
| Bradycardia | 2% | 0.2% |
| Palpitations | 2% | 0.2% |
| Tremor | 2% | 0.4% |
| Anorexia | 2% | 0.2% |
| Diarrhea | 2% | 0.4% |
| Ataxia | 2% | 0.0% |
In controlled trials in subjects with ventricular arrhythmia, the most common reactions reported for propafenone hydrochloride and more frequent than on placebo were unusual taste, dizziness, first-degree AV block, intraventricular conduction delay, nausea and/or vomiting, and constipation. Headache was relatively common also, but was not increased compared with placebo. Other reactions reported more frequently than on placebo or comparator and not already reported elsewhere included anxiety, angina, second-degree AV block, bundle branch block, loss of balance, congestive heart failure, and dyspepsia.
Adverse reactions reported for greater than or equal to 1% of 2,127 subjects with ventricular arrhythmia who received propafenone in U.S. clinical trials were evaluated by daily dose. The most common adverse reactions appeared dose-related (but note that most subjects spent more time at the larger doses), especially dizziness, nausea and/or vomiting, unusual taste, constipation, and blurred vision. Some less common reactions may also have been dose-related such as first degree AV block, congestive heart failure, dyspepsia, and weakness. Other adverse reactions included rash, syncope, chest pain, abdominal pain, ataxia, and hypotension.
In addition, the following adverse reactions were reported less frequently than 1% either in clinical trials or in marketing experience. Causality and relationship to propafenone therapy cannot necessarily be judged from these events.
Cardiovascular System: Atrial flutter, AV dissociation, cardiac arrest, flushing, hot flashes, sick sinus syndrome, sinus pause or arrest, supraventricular tachycardia.
Nervous System: Abnormal dreams, abnormal speech, abnormal vision, confusion, depression, memory loss, numbness, paresthesias, psychosis/mania, seizures (0.3%), tinnitus, unusual smell sensation, vertigo.
Gastrointestinal: Cholestasis, elevated liver enzymes (alkaline phosphatase, serum transaminases), gastroenteritis, hepatitis.
Hematologic: Agranulocytosis, anemia, bruising, granulocytopenia, leukopenia, purpura, thrombocytopenia.
Other: Alopecia, eye irritation, impotence, increased glucose, positive ANA (0.7%), muscle cramps, muscle weakness, nephrotic syndrome, pain, pruritus.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of propafenone hydrochloride. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal: A number of patients with liver abnormalities associated with propafenone therapy have been reported in post-marketing experience. Some appeared due to hepatocellular injury, some were cholestatic and some showed a mixed picture. Some of these reports were simply discovered through clinical chemistries, others because of clinical symptoms including fulminant hepatitis and death. One case was rechallenged with a positive outcome.
Blood and Lymphatic System: Increased bleeding time
Immune System: lupus erythematosus
Nervous System: Apnea, coma
Renal and Urinary: Hyponatremia/inappropriate ADH secretion, kidney failure
7 DRUG
INTERACTIONS
7.1 CYP2D6 and CYP3A4 Inhibitors
Drugs that inhibit CYP2D6 (such as desipramine, paroxetine, ritonavir, or sertraline) and CYP3A4 (such as ketoconazole, ritonavir, saquinavir, erythromycin, grapefruit juice) can be expected to cause increased plasma levels of propafenone. The combination of CYP3A4 inhibition and either CYP2D6 deficiency or CYP2D6 inhibition with administration of propafenone may increase the risk of adverse reactions, including proarrhythmia. Therefore, simultaneous use of propafenone hydrochloride with both a CYP2D6 inhibitor and a CYP3A4 inhibitor should be avoided [see Warnings and Precautions (5.4) and Dosage and Administration (2)].
Amiodarone: Concomitant administration of propafenone and amiodarone can affect conduction and repolarization and is not recommended.
Cimetidine: Concomitant administration of propafenone immediate-release tablets and cimetidine in 12 healthy subjects resulted in a 20% increase in steady-state plasma concentrations of propafenone.
Fluoxetine: Concomitant administration of propafenone and fluoxetine in extensive metabolizers increased the S-propafenone Cmax and AUC by 39% and 50%, respectively, and the R propafenone Cmax and AUC by 71% and 50%, respectively.
Quinidine: Small doses of quinidine completely inhibit the CYP2D6 hydroxylation metabolic pathway, making all patients, in effect, slow metabolizers [see Clinical Pharmacology (12.3)]. Concomitant administration of quinidine (50 mg three times daily) with 150 mg immediate release propafenone three times daily decreased the clearance of propafenone by 60% in extensive metabolizers, making them slow metabolizers. Steady-state plasma concentrations more than doubled for propafenone and decreased 50% for 5-OH-propafenone. A 100 mg dose of quinidine tripled steady-state concentrations of propafenone. Avoid concomitant use of propafenone and quinidine.
Rifampin: Concomitant administration of rifampin and propafenone in extensive metabolizers decreased the plasma concentrations of propafenone by 67% with a corresponding decrease of 5-OH-propafenone by 65%. The concentrations of norpropafenone increased by 30%. In slow metabolizers, there was a 50% decrease in propafenone plasma concentrations and an increase in the AUC and Cmax of norpropafenone by 74% and 20%, respectively. Urinary excretion of propafenone and its metabolites decreased significantly. Similar results were noted in elderly patients: Both the AUC and Cmax propafenone decreased by 84%, with a corresponding decrease in AUC and Cmax of 5-OH-propafenone by 69% and 57%, respectively.
7.2 Digoxin
Concomitant use of propafenone and digoxin increased steady-state serum digoxin exposure (AUC) in patients by 60% to 270%, and decreased the clearance of digoxin by 31% to 67%. Monitor plasma digoxin levels of patients receiving propafenone and adjust digoxin dosage as needed.
7.3 Warfarin
The concomitant administration of propafenone and warfarin increased warfarin plasma concentrations at steady state by 39% in healthy volunteers and prolonged the prothrombin time (PT) in patients taking warfarin. Adjust the warfarin dose as needed by monitoring INR (international normalized ratio).
7.4 Orlistat
Orlistat may limit the fraction of propafenone available for absorption. In post marketing reports, abrupt cessation of orlistat in patients stabilized on propafenone has resulted in severe adverse events including convulsions, atrioventricular block, and acute circulatory failure.
7.5 Beta-Antagonists
Concomitant use of propafenone and propranolol in healthy subjects increased propranolol plasma concentrations at steady state by 113%. In 4 patients, administration of metoprolol with propafenone increased the metoprolol plasma concentrations at steady state by 100% to 400%. The pharmacokinetics of propafenone was not affected by the coadministration of either propranolol or metoprolol. In clinical trials using propafenone immediate release tablets, subjects who were receiving beta-blockers concurrently did not experience an increased incidence of side effects.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no studies of propafenone hydrochloride in pregnant women. Available data from published case reports and several decades of postmarketing experience with use of propafenone hydrochloride in pregnancy have not identified any drug-associated risks of miscarriage, birth defects, or adverse maternal or fetal outcomes. Untreated arrhythmias during pregnancy may pose a risk to the pregnant woman and fetus (see Clinical Considerations). Propafenone and its metabolite, 5-OH-propafenone, cross the placenta in humans. In animal studies, propafenone was not teratogenic. At maternally toxic doses (ranging from 2 to 6 times the maximum recommended human dose [MRHD]), there was evidence of adverse developmental outcomes when administered to pregnant rabbits and rats during organogenesis or when administered to pregnant rats during mid-gestation through weaning of their offspring (see Data).
The estimated background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk: The incidence of VT is increased and may be more symptomatic during pregnancy. Ventricular arrhythmias most often occur in pregnant women with underlying cardiomyopathy, congenital heart disease, valvular heart disease, or mitral valve prolapse. Breakthrough arrhythmias may also occur during pregnancy, as therapeutic treatment levels may be difficult to maintain due to the increased volume of distribution and increased drug metabolism inherent in the pregnant state.
Fetal/Neonatal Adverse Reactions: Propafenone and its metabolite have been shown to cross the placenta. Adverse reactions such as fetal/neonatal arrhythmias have been associated with the use of other antiarrhythmic agents by pregnant women. Fetal/neonatal monitoring for signs and symptoms of arrhythmia is recommended during and after treatment of pregnant women with propafenone.
Labor or Delivery: Risk of arrhythmias may increase during labor and delivery. Patients treated with propafenone hydrochloride should be monitored continuously for arrhythmias during labor and delivery [seeWarnings and Precautions (5.1)].
Data
Propafenone has been shown to cause embryo-fetal mortality in rabbits and rats when given orally during organogenesis at maternally toxic doses of
150 mg/kg/day (rabbit: maternal mortality, decreased body weight gain and food consumption at approximately 3 times the MRHD on a mg/m2 basis) and
600 mg/kg/day (rat: maternal decreased body weight gain and food consumption at approximately 6 times the MRHD on a mg/m2 basis). In addition, a maternally toxic dose of 600 mg/kg/day (approximately 6 times the MRHD on a mg/m2 basis) also caused decreased fetal weights in rats. Increased placental weights and delayed ossification occurred in rabbits at a dose of 30 mg/kg/day (less than the MRHD on a mg/m2 basis) in the absence of maternal toxicity. No adverse developmental outcomes in the absence of maternal toxicity were seen following oral doses of 15 mg/kg/day to rabbits or up to 270 mg/kg/day to rats administered during organogenesis (equivalent to 0.3 times or approximately 3 times the MRHD on a mg/m2 basis, respectively). In an oral study, female rats received propafenone up to 500 mg/kg/day from mid-gestation through weaning. At 90 mg/kg/day (equivalent to the MRHD on a mg/m2 basis), there were no adverse developmental outcomes in the absence of maternal toxicity. However, doses ≥180 mg/kg/day (2 or more times the MRHD on a mg/m2 basis) produced increases in maternal deaths and resulted in reductions in neonatal survival, body weight gain, and delayed development in the presence of maternal toxicity.
8.2 Lactation
Risk Summary
Propafenone and its active metabolite, 5-OH-propafenone, are present in human milk, but the levels are likely to be low. There are no data on the effects of propafenone on the breastfed infant or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for propafenone and any potential adverse effects on the breastfed infant from propafenone or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
Males: Based on human and animal studies, propafenone hydrochloride may transiently impair spermatogenesis
in males. Evaluation of the effects on spermatogenesis was performed in 11 healthy males given oral propafenone 300 mg b.i.d. for 4 days, which was then increased to 300 mg t.i.d. for an additional 4 days. Study findings included a 28% reduction in semen sample volume on Treatment Day 8 and a 27% reduction in sperm count 64 days after treatment (both values remained within the laboratories normal reference range). These effects were not seen in followup visits up to 120 days after treatment. Reversible decreases in spermatogenesis have been demonstrated in monkeys, dogs, and rabbits after lethal or near-lethal intravenous doses of propafenone [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of propafenone in pediatric patients have not been established.
8.5 Geriatric Use
Clinical trials of propafenone hydrochloride did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger subjects. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10 OVERDOSAGE
The symptoms of overdosage may include hypotension, somnolence, bradycardia, intra-atrial and intraventricular conduction disturbances, and rarely, convulsions and high grade ventricular arrhythmias. Defibrillation, as well as infusion of dopamine and isoproterenol, has been effective in controlling abnormal rhythm and blood pressure. Convulsions have been alleviated with intravenous diazepam. General supportive measures such as mechanical respiratory assistance and external cardiac massage may be necessary.
The hemodialysis of propafenone in patients with an overdose is expected to be of limited value in the removal of propafenone as a result of both its high protein binding (greater than 95%) and large volume of distribution.
11 DESCRIPTION
Propafenone hydrochloride tablets, USP are an antiarrhythmic drug supplied in scored, film-coated tablets of 150, 225 and 300 mg for oral administration. Propafenone has some structural similarities to beta-blocking agents.
Chemically, propafenone hydrochloride (HCl) is 2’-[2-Hydroxy-3-(propylamino)- propoxy]-3-phenylpropiophenone hydrochloride, with a molecular weight of 377.92. The molecular formula is C21H27NO3HCl. The structural formula of propafenone HCl is given below:
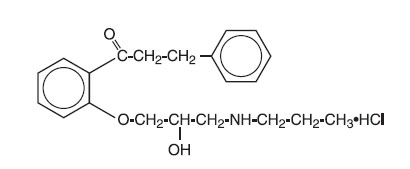
Propafenone HCl occurs as colorless crystals or white crystalline powder with a very bitter taste. It is slightly soluble in water (20°C), chloroform and ethanol. The following inactive ingredients are contained in the tablet: hypromellose, magnesium stearate, microcrystalline cellulose, povidone, pregelatinized corn starch, sodium starch glycolate, macrogol, talc, titanium dioxide, glycerol monocaprylocaprate and polyvinyl alcohol.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Propafenone is a Class 1C antiarrhythmic drug with local anesthetic effects, and a direct stabilizing action on myocardial membranes. The electrophysiological effect of propafenone manifests itself in a reduction of upstroke velocity (Phase 0) of the monophasic action potential. In Purkinje fibers, and to a lesser extent myocardial fibers, propafenone reduces the fast inward current carried by sodium ions. Diastolic excitability threshold is increased and effective refractory period prolonged. Propafenone reduces spontaneous automaticity and depresses triggered activity.
Studies in anesthetized dogs and isolated organ preparations show that propafenone has beta-sympatholytic activity at about 1/50 the potency of propranolol. Clinical studies employing isoproterenol challenge and exercise testing after single doses of propafenone indicate a beta-adrenergic blocking potency (per mg) about 1/40 that of propranolol in man. In clinical trials, resting heart rate decreases of about 8% were noted at the higher end of the therapeutic plasma concentration range. At very high concentrations in vitro, propafenone can inhibit the slow inward current carried by calcium, but this calcium antagonist effect probably does not contribute to antiarrhythmic efficacy. Moreover, propafenone inhibits a variety of cardiac potassium currents in in vitro studies (i.e., the transient outward, the delayed rectifier, and the inward rectifier current). Propafenone has local anesthetic activity approximately equal to procaine. Compared to propafenone, the main metabolite, 5-hydroxypropafenone, has similar sodium and calcium channel activity, but about 10 times less beta-blocking activity. (N-depropylpropafenone has weaker sodium channel activity but equivalent affinity for beta-receptors.)
12.2 Pharmacodynamics
Cardiac Electrophysiology: Electrophysiology trials in subjects with ventricular tachycardia have shown that propafenone prolongs atrioventricular conduction while having little or no effect on sinus node function. Both atrioventricular nodal conduction time (AH interval) and His-Purkinje conduction time (HV interval) are prolonged. Propafenone has little or no effect on the atrial functional refractory period, but AV nodal functional and effective refractory periods are prolonged. In patients with Wolff-Parkinson-White syndrome, propafenone hydrochloride reduces conduction and increases the effective refractory period of the accessory pathway in both directions.
Electrocardiograms: Propafenone prolongs the PR and QRS intervals. Prolongation of the QRS interval makes it difficult to interpret the effect of propafenone on the QT interval.
Table 2: Mean Changes in Electrocardiogram Intervalsa
| Total Daily Dose (mg) | ||||||||
| 337.5 mg | 450 mg | 675 mg | 900 mg | |||||
| Interval | msec | % | msec | % | msec | % | msec | % |
| RR | -14.5 | -1.8 | 30.6 | 3.8 | 31.5 | 3.9 | 41.7 | 5.1 |
| PR | 3.6 | 2.1 | 19.1 | 11.6 | 28.9 | 17.8 | 35.6 | 21.9 |
| QRS | 5.6 | 6.4 | 5.5 | 6.1 | 7.7 | 8.4 | 15.6 | 17.3 |
| QTc | 2.7 | 0.7 | -7.5 | -1.8 | 5.0 | 1.2 | 14.7 | 3.7 |
aChange and percent change based on mean baseline values for each treatment group.
In any individual patient, the above ECG changes cannot be readily used to predict either efficacy or plasma concentration.
Propafenone hydrochloride causes a dose-related and concentration-related decrease in the rate of single and multiple PVCs and can suppress recurrence of ventricular tachycardia. Based on the percent of patients attaining substantial (80% to 90%) suppression of ventricular ectopic activity, it appears that trough plasma levels of 0.2 to 1.5 µg/mL can provide good suppression, with higher concentrations giving a greater rate of good response.
When 600 mg/day propafenone was administered to subjects with paroxysmal atrial tachyarrhythmias, mean heart rate during arrhythmia decreased 14 beats/min and 37 beats/min for subjects with PAF and subjects with PSVT, respectively.
Hemodynamics: Trials in humans have shown that propafenone HCl exerts a negative inotropic effect on the myocardium. Cardiac catheterization trials in subjects with moderately impaired ventricular function (mean CI = 2.61 L/min/m2) utilizing intravenous propafenone infusions (loading dose of 2 mg/kg over 10 min followed by 2 mg/min for 30 min) that gave mean plasma concentrations of 3.0 mcg/mL (a dose that produces plasma levels of propafenone greater than recommended oral dosing) showed significant increases in pulmonary capillary wedge pressure, systemic and pulmonary vascular resistances and depression of cardiac output and cardiac index.
12.3 Pharmacokinetics
Absorption/Bioavailability: Propafenone HCl is nearly completely absorbed after oral administration with peak plasma levels occurring approximately 3.5 hours after administration in most individuals. Propafenone exhibits extensive saturable presystemic biotransformation (first-pass effect) resulting in a dose-dependent and dosage- form-dependent absolute bioavailability; e.g., a 150 mg tablet had absolute bioavailability of 3.4%, while a 300 mg tablet had absolute bioavailability of 10.6%. A 300 mg solution which was rapidly absorbed had absolute bioavailability of 21.4%. At still larger doses, above those recommended, bioavailability increases still further.
Propafenone HCl follows a nonlinear pharmacokinetic disposition presumably because of saturation of first-pass hepatic metabolism as the liver is exposed to higher concentrations of propafenone and shows a very high degree of inter-individual variability. For example, for an increase in daily dose from 300 to 900 mg/day there is a 10-fold increase in steady-state plasma concentration. The top 25% of subjects given 337.5 mg/day, however, had a mean concentration of propafenone larger than the bottom 25%, and about equal to the second 25%, of subjects given a dose of 900 mg. Although food increased peak blood level and bioavailability in a single-dose trial, during multiple-dose administration of propafenone to healthy volunteers, food did not change bioavailability significantly.
Distribution: Following intravenous administration of propafenone, plasma levels decline in a bi-phasic manner consistent with a 2 compartment pharmacokinetic model. The average distribution half-life corresponding to the first phase was about 5 minutes. The volume of the central compartment was about 88 liters (1.1 L/kg) and the total volume of distribution about 252 liters.
In serum, propafenone is greater than 95% bound to proteins within the concentration range of 0.5 to 2 mcg/mL.
Metabolism: There are two genetically determined patterns of propafenone metabolism. In over 90% of patients, the drug is rapidly and extensively metabolized with an elimination half-life from 2 to 10 hours. These patients metabolize propafenone into two active metabolites: 5-hydroxypropafenone which is formed by CYP2D6 and N-depropylpropafenone (norpropafenone) which is formed by both CYP3A4 and CYP1A2.
In less than 10% of patients, metabolism of propafenone is slower because the 5-hydroxy metabolite is not formed or is minimally formed. In these patients, the estimated propafenone elimination half-life ranges from 10 to 32 hours. Decreased ability to form the 5-hydroxy metabolite of propafenone is associated with a diminished ability to metabolize debrisoquine and a variety of other drugs, such as encainide, metoprolol, and dextromethorphan whose metabolism is mediated by the CYP2D6 isozyme. In these patients, the N-depropylpropafenone metabolite occurs in quantities comparable to the levels occurring in extensive metabolizers.
There are significant differences in plasma concentrations of propafenone in slow and extensive metabolizers, the former achieving concentrations 1.5 to 2.0 times those of the extensive metabolizers at daily doses of 675 to 900 mg/day. At low doses the differences are greater, with slow metabolizers attaining concentrations more than five times that of extensive metabolizers. Because the difference decreases at high doses and is mitigated by the lack of the active 5-hydroxy metabolite in the slow metabolizers, and because steady-state conditions are achieved after 4 to 5 days of dosing in all patients, the recommended dosing regimen is the same for all patients. The greater variability in blood levels require that the drug be titrated carefully in patients with close attention paid to clinical and ECG evidence of toxicity [see Dosage and Administration (2)].
Stereochemistry: Propafenone hydrochloride is a racemic mixture. The R- and S-enantiomers of propafenone display stereoselective disposition characteristics. In vitro and in vivo studies have shown that the R-isomer of propafenone is cleared faster than the S-isomer via the 5-hydroxylation pathway (CYP2D6). This results in a higher ratio of S-propafenone to R-propafenone at steady state. Both enantiomers have equivalent potency to block sodium channels; however, the S-enantiomer is a more potent beta-antagonist than the R-enantiomer. Following administration of propafenone hydrochloride immediate-release tablets, the S/R ratio for the area under the plasma concentration-time curve was about 1.7. In addition, no difference in the average values of the S/R ratios is evident between genotypes or over time.
Specific Populations: Patients with Hepatic Impairment: Decreased liver function increases the bioavailability of propafenone. Absolute bioavailability of propafenone hydrochloride immediate-release tablets is inversely related to indocyanine green clearance, reaching 60 to 70% at clearances of 7 mL/min and below. Protein binding decreases to about 88% in patients with severe hepatic dysfunction. The clearance of propafenone is reduced and the elimination half-life increased in patients with significant hepatic dysfunction[see Warnings and Precautions (5.9)].
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Lifetime maximally tolerated oral dose studies in mice (up to 360 mg/kg/day, approximately twice the MRHD on a mg/m2 basis) and rats (up to 270 mg/kg/day, about 3 times the MRHD on a mg/m2 basis) provided no evidence of a carcinogenic potential for propafenone.
Propafenone was not mutagenic in the Ames (salmonella) test and the in vivo mouse dominant lethal test. Propafenone was not clastogenic in the human lymphocyte chromosome aberration assay in vitro,the rat and Chinese hamster micronucleus tests, and other in vivo tests for chromosomal aberrations in rat bone marrow and Chinese hamster bone marrow and spermatogonia.
Propafenone, administered intravenously, has been shown to decrease spermatogenesis at lethal doses in rabbits (≥3.5 mg/kg/day) or at near-lethal dose levels in monkeys and dogs (≤5 mg/kg/day); doses were less than the MRHD on a mg/m2 basis. These effects were reversible and did not impair fertility in rabbits at an intravenous dose of 3.5 mg/kg/day (a spermatogenesis-impairing dose). Effects on spermatogenesis were not found when propafenone was administered to rats either orally or intravenously up to 360 mg/kg/day or 6 mg/kg/day, respectively, or in dogs at oral doses up to 240 mg/kg/day (up to approximately 4 or 9 times the MRHD on a mg/m2 basis in rats and dogs, respectively). Treatment of male rabbits for 10 weeks prior to mating at an oral dose of 120 mg/kg/day (approximately 2 times the MRHD on a mg/m2 basis) did not result in evidence of impaired fertility. Nor was there evidence of impaired fertility when propafenone was administered orally to male and female rats at dose levels up to 270 mg/kg/day (approximately 3 times the MRHD on a mg/m2 basis) for 10 weeks (males) or 2 weeks (females) prior to mating through mating.
13.2 Animal Toxicology and/or Pharmacology
Renal changes have been observed in the rat following 6 months of oral administration of propafenone HCl at doses of 180 and 360 mg/kg/day (about 2 and 4 times, respectively, the MRHD on a mg/m2 basis). Both inflammatory and non-inflammatory changes in the renal tubules, with accompanying interstitial nephritis, were observed. These changes were reversible, as they were not found in rats allowed to recover for 6 weeks. Fatty degenerative changes of the liver were found in rats following longer durations of administration of propafenone HCl at a dose of 270 mg/kg/day (about 3 times the MRHD on a mg/m2 basis). There were no renal or hepatic changes at 90 mg/kg/day (equivalent to the MRHD on a mg/m2 basis).
14 CLINICAL STUDIES
In two randomized, crossover, placebo-controlled, double-blind trials of 60 to 90 days duration in subjects with PAF or PSVT, propafenone reduced the rate of both arrhythmias, as shown in Table 3.
| Trial 1 | Trial 2 | |||
| Propafenone | Placebo | Propafenone | Placebo | |
|
PAF |
n = 30 |
n = 30 |
n = 9 |
n = 9 |
|
Percent attack free |
53% |
13% |
67% |
22% |
| Median time to first recurrence | > 98 days | 8 days | 62 days | 5 days |
|
PSVT |
n = 45 |
n = 45 |
n = 15 |
N = 15 |
|
Percent attack free |
47% |
16% |
38% |
7% |
| Median time to first recurrence | > 98 days | 12 days | 31 days | 8 days |
The patient population in the above trials was 50% male with a mean age of 57.3 years. Fifty percent of the subjects had a diagnosis of PAF and 50% had PSVT. Eighty percent of the subjects received 600 mg/day propafenone. No subjects died in the above 2 trials.
In U.S. long-term safety trials, 474 patients (mean age: 57.4 ± 14.5 years) with supraventricular arrhythmias [195 with PAF, 274 with PSVT and 5 with both PAF and PSVT] were treated up to 5 years (mean: 14.4 months) with propafenone. Fourteen of the subjects died. When this mortality rate was compared with the rate in a similar patient population (n = 194 subjects; mean age: 43.0 ± 16.8 years) studied in an arrhythmia clinic, there was no age-adjusted difference in mortality. This comparison was not, however, a randomized trial and the 95% confidence interval around the comparison was large, such that neither a significant adverse or favorable effect could be ruled out.
16 HOW SUPPLIED/STORAGE AND HANDLING
Propafenone hydrochloride tablets, USP are supplied as white, scored, round, film-coated tablets in three dosage strengths:
225 mg tablets debossed “5125” and “V” available as follows:
- Bottles of 300: NDC 63629-2267-1
STORE at 20º to 25ºC (68º to 77ºF) [see USP Controlled Room Temperature].
DISPENSE in a tight, light-resistant container as defined in the USP.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Instruct patients to notify their healthcare providers of any change in over-the-counter, prescription, and
supplement use.
Instruct patients to report symptoms that may be associated with altered electrolyte balance, such as excessive
or prolonged diarrhea, sweating, vomiting, or loss of appetite or thirst.
Instruct patients not to double the next dose if a dose is missed. The next dose should be taken at the usual
time.
For Patient Information Leaflet, please visit www.parpharm.com
17.1 Information for Patients
- Patients should be instructed to notify their health care providers of any change in over-the-counter, prescription and supplement use. The health care provider should assess the patients’ medication history including all over-the-counter, prescription and herbal/natural preparations for those that may affect the pharmacodynamics or kinetics of propafenone hydrochloride [see Warnings and Precautions (5.4)].
- Patients should also check with their health care providers prior to taking a new over-the-counter medicine.
- If patients experience symptoms that may be associated with altered electrolyte balance, such as excessive or prolonged diarrhea, sweating, vomiting, or loss of appetite or thirst, these conditions should be immediately reported to their health care provider.
- Patients should be instructed NOT to double the next dose if a dose is missed. The next dose should be taken at the usual time.
PATIENT INFORMATION
Propafenone Hydrochloride Tablets, USP
What are propafenone hydrochloride tablets?
Propafenone hydrochloride tablets are a prescription medicine that is used:
- in certain people who have ventricular heart rhythm disorders
- to increase the amount of time between having symptoms of heart rhythm disorders called atrial fibrillation (AF) or paroxysmal supraventricular tachycardia (PSVT)
It is not known if propafenone hydrochloride tablets are safe and effective in children.
Who should not take propafenone hydrochloride tablets?
Do not take propafenone hydrochloride tablets if you have:
- heart failure (weak heart)
- had a recent heart attack
- a heart rate that is too slow, and you do not have a pacemaker
- a heart condition called Brugada Syndrome
- very low blood pressure
- certain breathing problems that make you short of breath or wheeze
- certain abnormal body salt (electrolyte) levels in your blood
Talk to your doctor before taking propafenone hydrochloride tablets if you think you have any of the conditions listed above.
What should I tell my doctor before taking propafenone hydrochloride tablets?
Before you take propafenone hydrochloride tablets, tell your doctor if you:
- have liver or kidney problems
- have breathing problems
- have symptoms including diarrhea, sweating, vomiting, or loss of appetite or thirst that are severe. These symptoms may be a sign of abnormal electrolyte levels in your blood.
- have myasthenia gravis
- have lupus erythematosus
- have been told you have or had an abnormal blood test called Antinuclear Antibody Test or ANA Test
- have any other medical conditions
- are pregnant or plan to become pregnant.
- are breastfeeding or plan to breastfeed. Propafenone hydrochloride tablets can pass into your milk. You and your doctor should discuss the best way to feed your baby during this time.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Propafenone hydrochloride tablets and certain other medicines can affect (interact with) each other and cause serious side effects. You can ask your pharmacist for a list of medicines that interact with propafenone hydrochloride tablets.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.
How should I take propafenone hydrochloride tablets?
- Take propafenone hydrochloride tablets exactly as prescribed. Your doctor will tell you how many tablets to take and how often to take them.
- To help reduce the chance of certain side effects, your doctor may start you with a low dose of propafenone hydrochloride tablets, and then slowly increase the dose.
- You should not drink grapefruit juice during treatment with propafenone hydrochloride tablets.
- If you miss a dose of propafenone hydrochloride tablets, take your next dose at the usual time. Do not take 2 doses at the same time.
- If you take too much propafenone hydrochloride tablets, call your doctor or go to the nearest hospital emergency room right away.
- Call your doctor if your heart problems get worse.
What are possible side effects of propafenone hydrochloride tablets?
Propafenone hydrochloride tablets can cause serious side effects including:
- New or worsened abnormal heart beats that can cause sudden death or be life-threatening. Your doctor may do an electrocardiogram (ECG or EKG) before and during treatment to check your heart for these problems.
-
New or worsened heart failure. Tell your doctor about any changes in your heart symptoms, including:
- any new or increased swelling in your arms or legs
- trouble breathing
- sudden weight gain
- Effects on pacemaker function. Propafenone hydrochloride tablets may affect how an implanted pacemaker or defibrillator works. Your doctor should check how your pacemaker or defibrillator is working during and after treatment with propafenone hydrochloride tablets. They may need to be re-programmed.
-
Very low white blood cell levels in your blood (agranulocytosis). Your bone marrow may not produce enough of a certain type of white blood cells called neutrophils. If this happens, you are more likely to get infections. Tell your doctor right away if you have any of these symptoms, especially during the first 3 months of treatment:
- fever
- sore throat
- chills
- Worsening of myasthenia gravis in people who already have this condition. Tell your doctor about any change in your symptoms.
- Propafenone hydrochloride tablets may cause lower sperm counts in men. This could affect the ability to father a child. Talk to your doctor if this is a concern for you.
Common side effects of propafenone hydrochloride tablets include:
- unusual taste
- nausea
- vomiting
- dizziness
- constipation
- headache
- tiredness
- irregular heart beats
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of propafenone hydrochloride tablets. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store propafenone hydrochloride tablets?
- Store propafenone hydrochloride tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep the bottle tightly closed.
Keep propafenone hydrochloride tablets and all medicines out of the reach of children.
General information about propafenone hydrochloride tablets
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. Do not use propafenone hydrochloride tablets for a condition for which it was not prescribed. Do not give propafenone hydrochloride tablets to other people, even if they have the same symptoms you have. It may harm them.
If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about propafenone hydrochloride tablets that is written for health professionals. For more information about propafenone hydrochloride tablets, call 1-800-828-9393.
What are the ingredients in propafenone hydrochloride tablets?
Active ingredient: propafenone hydrochloride
Inactive ingredients: hypromellose, magnesium stearate, microcrystalline cellulose, povidone, pregelatinized corn starch, sodium starch glycolate, macrogol, talc, titanium dioxide, glycerol monocaprylocaprate and polyvinyl alcohol.
This Patient Information has been approved by the U.S. Food and Drug Administration.
For Patient Information Leaflet, please visit www.parpharm.com
Distributed by:
Par Pharmaceutical
Chestnut Ridge, NY 10977
Revised: 02/2020 PD5448H-01-1-02

| PROPAFENONE HYDROCHLORIDE
propafenone hydrochloride tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Bryant Ranch Prepack (171714327) |
| Registrant - Bryant Ranch Prepack (171714327) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bryant Ranch Prepack | 171714327 | REPACK(63629-2267) , RELABEL(63629-2267) | |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.