DECNUPAZ- pivekimab sunirine-pvzy injection, powder, lyophilized, for solution
Drug Labeling and Warnings
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DECNUPAZ safely and effectively. See full prescribing information for DECNUPAZ.
DECNUPAZTM (pivekimab sunirine-pvzy) for injection, for intravenous use
Initial U.S. Approval: 2026
WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME)
See full prescribing information for complete boxed warning.
-
VOD, a severe form of hepatotoxicity, has been reported in patients with BPDCN treated with DECNUPAZ, including severe or fatal hepatic VOD. (5.1)
-
Closely monitor for signs and symptoms of VOD. Monitor liver tests and total bilirubin prior to each dose. (5.1)
- Discontinue DECNUPAZ for patients who experience VOD. (2.4, 5.1)
INDICATIONS AND USAGE
DECNUPAZ is a CD123-directed antibody and alkylating agent conjugate indicated for the treatment of adult patients with blastic plasmacytoid dendritic cell neoplasm (BPDCN). (1)
DOSAGE AND ADMINISTRATION
For intravenous infusion only. (2.6)
- The recommended dose of DECNUPAZ is 0.045 mg/kg once every 3 weeks until disease progression or unacceptable toxicity. (2.2)
- Premedicate with a corticosteroid on the day prior to infusion and premedicate with a corticosteroid, antihistamine, and an antipyretic at least 30 to 60 minutes prior to DECNUPAZ infusion. (2.3)
- DECNUPAZ requires reconstitution followed by two dilutions prior to administration. See full Prescribing Information for instructions on preparation and administration. (2.5, 2.6)
DOSAGE FORMS AND STRENGTHS
- For injection: 2 mg of pivekimab sunirine-pvzy as a lyophilized cake in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
-
Infusion-related reactions (IRR): DECNUPAZ can cause serious, life-threatening IRR. Premedicate with a corticosteroid the day before infusion, and premedicate with a corticosteroid, antihistamine, and an antipyretic prior to DECNUPAZ infusion. Monitor patients for IRR. Interrupt, reduce the rate of infusion, or permanently discontinue DECNUPAZ based on the severity. (5.2)
-
Edema: Monitor for the development of edema and fluid retention. Depending on severity, delay, consider resuming at a lower dose, or permanently discontinue DECNUPAZ. (5.3)
-
Sulfite Allergic Reactions: DECNUPAZ contains sodium metabisulfite, which may cause allergic type reactions, including anaphylactic symptoms and asthmatic episodes in certain susceptible people. (5.4)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.5)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) were edema, fatigue, musculoskeletal pain, hemorrhage, infusion-related reactions, nausea, and diarrhea. (6.1)
The most common Grade 3 or 4 laboratory abnormalities (≥10%) were neutrophils decreased, platelets decreased, lymphocyte count decreased, white blood cells decreased, hemoglobin decreased, and glucose increased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchDRUG INTERACTIONS
Strong and Moderate CYP3A Inhibitors: Closely monitor for DECNUPAZ adverse reactions (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2026
-
VOD, a severe form of hepatotoxicity, has been reported in patients with BPDCN treated with DECNUPAZ, including severe or fatal hepatic VOD. (5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME)
1. INDICATIONS AND USAGE
2. DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Recommended Dosage
2.3 Premedications
2.4 Dosage Modifications for Adverse Reactions
2.5 Instructions for Preparation
2.6 Administration
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome)
5.2 Infusion-Related Reactions
5.3 Edema
5.4 Sulfite Allergic Reactions
5.5 Embryo-Fetal Toxicity
6. ADVERSE REACTIONS
6.1 Clinical Trials Experience
7. DRUG INTERACTIONS
7.1 Effect of Other Drugs on DECNUPAZ
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
14.1 Treatment-naïve Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
14.2 Relapsed or Refractory Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
15. REFERENCES
16. HOW SUPPLIED/STORAGE AND HANDLING
17. PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATOTOXICITY, INCLUDING HEPATIC VENO-OCCLUSIVE DISEASE (VOD) (ALSO KNOWN AS SINUSOIDAL OBSTRUCTION SYNDROME)
DECNUPAZ can cause hepatotoxicity, including severe or fatal hepatic VOD (also known as sinusoidal obstruction syndrome) [see Warnings and Precautions (5.1)].
Closely monitor patients for signs and symptoms of VOD including elevations in liver tests, hepatomegaly (which may be painful), rapid weight gain, and ascites [see Warnings and Precautions (5.1)].
Monitor liver tests, including ALT, AST, and total bilirubin, prior to each dose of DECNUPAZ [see Warnings and Precautions (5.1)].
Delay DECNUPAZ dosage for liver test elevation. Permanently discontinue DECNUPAZ for patients who experience VOD [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
- 1. INDICATIONS AND USAGE
-
2.
DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
DECNUPAZ requires reconstitution followed by two dilutions prior to administration.
Read the entire preparation instructions carefully before preparing and administering DECNUPAZ.
2.2 Recommended Dosage
The recommended dose of DECNUPAZ in adult patients with BPDCN is 0.045 mg/kg intravenously over approximately 15-30 minutes once every 3 weeks (21-day cycle) until disease progression or unacceptable toxicity. Calculate the dose based on the patient’s actual body weight [see Dosage and Administration (2.5)].
2.3 Premedications
Administer the premedications in Table 1 the day prior to and the day of the infusion of DECNUPAZ to reduce the risk of infusion-related reactions (IRRs) [see Warnings and Precautions (5.2)].
Table 1. Recommended Premedications Prior to Each DECNUPAZ Infusion Administration Time Prior to DECNUPAZ Infusion Premedication Route of Administration Dose (or equivalent) Day before DECNUPAZ infusion Corticosteroid Oral or intravenous Dexamethasone 8 mg
twice daily30 to 60 minutes prior to infusion Corticosteroid Intravenous Dexamethasone 8 mg Antihistamine Intravenous Diphenhydramine 25 mg to 50 mg Antipyretic Oral Acetaminophen 325 mg to 650 mg 2.4 Dosage Modifications for Adverse Reactions
Table 2 provides recommended dosage modifications for DECNUPAZ due to adverse reactions.
Table 2. Recommended Dosage Modifications for Adverse Reactions Adverse Reaction Severity of Adverse Reactiona Dose Modification Guidelines Veno-occlusive disease (VOD) [see Warnings and Precautions (5.1)] Any Grade - Permanently discontinue DECNUPAZ
Increased aspartate aminotransferase (AST) or alanine aminotransferase (ALT) [see Warnings and Precautions (5.1)] Either AST or ALT is >2.5 x ULN - Delay further DECNUPAZ dosing until AST or ALT have returned to ≤2.5 × ULN
Increased bilirubin [see Warnings and Precautions (5.1)] Total bilirubin > 1.5 × ULN - Delay further DECNUPAZ dosing until total bilirubin has returned to ≤1.5 × ULN
Infusion-related reactions [see Warnings and Precautions (5.2)] Grade 2 - Interrupt DECNUPAZ infusion and institute appropriate medical management
- After full resolution of symptoms, resume DECNUPAZ infusion at 50% of the previous rate and if no further symptoms appear, increase rate as appropriate until infusion is completed
Grade 3 - Stop DECNUPAZ infusion and institute appropriate medical management
- After full resolution of symptoms, resume the infusion at 50% of the previous rate
- If symptoms recur, permanently discontinue
Grade 4 - Permanently discontinue DECNUPAZ
Edema [see Warnings and Precautions (5.3)] Grade 1 (5-10% inter-limb discrepancy in volume or circumference,
4 kg weight gain, or 1+ pitting edema (2 mm))- Follow weekly weights
- Consider administering diuretic therapy
Grade 2 (10-30% inter-limb discrepancy in volume or circumference, >4 kg weight gain, or 2+ pitting edema (4 mm)) - Administer diuretic therapy
- Manage hypoalbuminemia as needed
- Delay further DECNUPAZ dosing until edema has returned to Grade 0-1 or baseline
- If delayed more than 2 weeks, consider dose reduction before resuming
Grade 3 (> 30% inter-limb discrepancy in volume, or 3+/4+ pitting edema (>6 mm))
- Consider combination diuretic therapy
- Manage hypoalbuminemia as needed
- Delay further DECNUPAZ dosing until edema has returned to Grade 0-1 or baseline
- Consider resuming DECNUPAZ infusion at 0.015 mg/kg intravenously once every 3 weeks
Grade 4 (life threatening) - Permanently discontinue DECNUPAZ
Other Non-hematologic Adverse Reactions [see Adverse Reactions (6.1)] Grade 3 - Delay further DECNUPAZ dosing until resolved to ≤ Grade 2 or baseline
Grade 4 - Permanently discontinue DECNUPAZ
ULN=upper limit of normal
aNational Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03; Grade 1 is mild, Grade 2 is moderate, Grade 3 is severe, and Grade 4 is life-threatening.
2.5 Instructions for Preparation
Preparation - Use aseptic technique to prepare DECNUPAZ.
- DECNUPAZ is a hazardous drug. Follow applicable special handling and disposal procedures in accordance with local requirements.1
- Determine the dose and the number of DECNUPAZ vials needed. More than one vial may be needed to achieve a full dose.
- Remove the DECNUPAZ vial(s) from the refrigerator and allow the vial(s) to reach room temperature 15°C to 30°C (59℉ to 86°F) before use.
Reconstitution - Using a sterile syringe, reconstitute DECNUPAZ by slowly injecting 1.1 mL of Sterile Water for Injection into each vial to obtain a concentration of 2 mg/mL. Each single-dose vial contains 1 mL (2 mg) of withdrawable DECNUPAZ.
- Gently swirl the vial in a circular motion. Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution in each vial should appear clear to slightly opalescent, colorless to slightly yellow and free of visible contaminants, particles and/or particulates. Do not use if discoloration or particulate matter is present.
- DECNUPAZ contains no preservative. Use reconstituted solution immediately. If not used immediately, store the reconstituted DECNUPAZ vials in a refrigerator at 2°C to 8°C (36°F to 46°F) for up to 4 hours from the time of reconstitution. Do not freeze.
First Dilution - DECNUPAZ must be diluted with 5% Dextrose Injection.
- Add 4.8 mL of 5% Dextrose Injection to the reconstituted solution in the vial. Mix the diluted solution by gentle inversion. Do not shake.
- The resulting diluted solution concentration is 0.4 mg/mL.
- Calculate the required administration volume needed from the first dilution of vial(s).
Volume needed from vial(s) after first dilution (mL) = Dose (mg) / 0.4 mg/mL
- DECNUPAZ requires a second dilution prior to administration.
Second Dilution - Withdraw the calculated volume from the first dilution of vial(s) using an appropriately sized syringe.
- Withdraw an equal volume of 5% Dextrose Injection in an appropriately sized second syringe.
- Attach a connector to the two syringes and transfer DECNUPAZ into the syringe containing 5% Dextrose Injection.
- Disconnect the syringes. Draw air into the syringe containing the DECNUPAZ diluted solution and close.
- Gently invert the syringe to mix the solution. Do not shake.
- Remove air bubbles from the syringe before administration.
- The final concentration is 0.2 mg/mL.
- Discard any unused drug remaining in the vial(s).
- Do not use an in-line filter due to increased drug loss. If an in-line filter is required, a 1.2 micron polyethersulfone (PES) filter can be used.
Storage of Diluted Solution
- Immediately use diluted DECNUPAZ solution.
- The total storage time from the start of dose preparation to completion of administration should not exceed 24 hours. Store the diluted solution of DECNUPAZ under refrigeration at 2°C to 8°C (36°F to 46°F) for no more than 24 hours, including up to 8 hours at room temperature at 9°C to 25°C (48°F to 77°F), from the time of reconstitution to completion of the intravenous infusion.
- Discard diluted infusion solution if storage time exceeds these limits.
- If refrigerated, allow approximately 30 minutes for the diluted solution to come to room temperature prior to administration. Do not shake.
- Do not freeze the diluted infusion solution.
- Protect from light during storage.
- Do not shake.
2.6 Administration
- After preparing the dose for infusion, visually inspect the syringe content for particulates and discard if present.
- Do not mix DECNUPAZ with any other drugs or any intravenous fluids other than 5% Dextrose Injection.
- Protect the intravenous syringe from light using a light-blocking cover during infusion. The infusion line does not need to be protected from light.
- Administer DECNUPAZ as an intravenous infusion only. Do not administer as an intravenous push or bolus.
- The first infusion of DECNUPAZ should be administered at a rate of 0.8 mL/min (0.165 mg/min) for the first 30 minutes.
- If well tolerated, the infusion rate can be increased after 30 minutes to 1.7 mL/min (0.33 mg/min), if necessary.
- Subsequent infusions may be delivered at the highest tolerated rate.
- The first infusion of DECNUPAZ should be administered at a rate of 0.8 mL/min (0.165 mg/min) for the first 30 minutes.
- Following the infusion, flush the intravenous line with sufficient volume of 5% Dextrose Injection to ensure delivery of the full dose. Do not use any other intravenous fluids for flushing.
- 3. DOSAGE FORMS AND STRENGTHS
- 4. CONTRAINDICATIONS
-
5.
WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity, Including Hepatic Veno-occlusive Disease (VOD) (also known as Sinusoidal Obstruction Syndrome)
DECNUPAZ can cause hepatotoxicity, including VOD, a severe form of hepatotoxicity. In CADENZA, VOD occurred in 6% (7/116) of adult patients during treatment or following a subsequent hematopoietic stem cell transplantation (HSCT). Of the 7 total patients who developed VOD, 3 patients had treatment-naïve BPDCN and 4 patients had relapsed/refractory BPDCN. Among all 116 patients treated with DECNUPAZ at 0.045 mg/kg, VOD occurred in 2/116 (2%) during treatment, with onset up to 30 days after the last dose. Among 19 patients with BPDCN who proceeded to HSCT, VOD occurred in 5/19 patients (26%), including two fatal cases. The median time from subsequent HSCT to onset of VOD was 11 days (range: 7 – 25 days).
After receiving DECNUPAZ, patients should be closely monitored for signs and symptoms of VOD including elevations in ALT, AST, total bilirubin, hepatomegaly (which may be painful), rapid weight gain, and ascites. Monitor liver tests, including ALT, AST, and total bilirubin, prior to each dose of DECNUPAZ. Based on elevations of liver tests, delay DECNUPAZ. In patients who experience VOD, discontinue DECNUPAZ and treat according to standard medical practice [see Dosage and Administration (2.4)].
5.2 Infusion-Related Reactions
DECNUPAZ can cause serious, life-threatening infusion-related reactions (IRR); signs and symptoms of IRR include dyspnea, flushing, fever, chills, nausea, chest discomfort, hypotension, and vomiting. In CADENZA, IRRs occurred in 26% (30/116) of patients during treatment with DECNUPAZ at 0.045 mg/kg once every three weeks, including Grade 1 in 4.3% (5/116), Grade 2 in 16% (19/116), and Grade 3 in 5% (6/116) of patients. IRR occurred in Cycle 1 in 25% (29/116) of patients with decreasing frequency in subsequent cycles. IRR led to discontinuation in one patient.
Premedicate with a corticosteroid the day before infusion, and premedicate with a corticosteroid, antihistamine, and antipyretic prior to dosing [see Dosage and Administration (2.3)]. Premedication the day before infusion and prior to dosing led to reduced frequency and severity of IRRs.
Monitor patients closely for potential IRR during the infusion and for at least four hours, or longer as clinically indicated, after the first infusion and for at least 1 hour after subsequent infusions.
Interrupt infusion of DECNUPAZ and institute appropriate medical management if an infusion-related reaction occurs. Depending on the severity of the infusion-related reaction, reduce infusion rate or permanently discontinue [see Dosage and Administration (2.4)].
5.3 Edema
DECNUPAZ can cause edema and fluid retention, including serious events. In CADENZA, Grade 3-4 edema occurred in 16% (18/116) of patients treated with DECNUPAZ, including Grade 3-4 generalized edema in 2.6% (3/116) of patients [see Adverse Reactions (6.1)].
Monitor patients for new or worsening edema. For Grade 2 or Grade 3 edema, delay further dosing of DECNUPAZ until edema has returned to Grade 0-1 or baseline. For Grade 3 edema or Grade 2 edema with dose delay for more than 2 weeks, consider resuming at a lower dose. For Grade 4 edema, permanently discontinue. Institute appropriate medical management for edema [see Dosage and Administration (2.4)].
5.4 Sulfite Allergic Reactions
DECNUPAZ contains sodium metabisulfite, a sulfite that may cause allergic type reactions, including anaphylactic symptoms and life-threatening or less severe asthmatic episodes in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in non-asthmatic people.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action, DECNUPAZ can cause embryo-fetal harm when administered to a pregnant woman because it contains a genotoxic compound (FGN849) and affects actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)].
Advise patients of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with DECNUPAZ and for 7 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with DECNUPAZ, and for 4 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6.
ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Hepatotoxicity, Including Hepatic VOD (also known as Sinusoidal Obstruction Syndrome) [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- Edema [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of DECNUPAZ was evaluated in CADENZA, a single-arm, open-label study that included 116 adults with newly diagnosed or relapsed/refractory myeloid malignancies, including 84 with BPDCN, treated with DECNUPAZ 0.045 mg/kg once every three weeks.
The median number of cycles administered was 3 (range: 1 to 34) in the overall population, and 3.5 (range: 1 to 34) in patients with BPDCN.
Serious adverse reactions occurred in 55% of patients treated with DECNUPAZ. The most common (≥2%) serious adverse reactions were febrile neutropenia, pneumonia, edema, sepsis, hemorrhage, thrombosis, infusion-related reactions, viral infection, pneumonitis, infections without specified pathogens, pyrexia, and musculoskeletal pain. Fatal adverse reactions occurred in 4.3% of patients who received DECNUPAZ, including cardiac arrest (0.9%), clostridium difficile infection (0.9%), failure to thrive (0.9%), depressed level of consciousness (0.9%), and respiratory failure (0.9%).
Permanent discontinuation due to adverse reactions occurred in 10% of patients who received DECNUPAZ. Adverse reactions which resulted in permanent discontinuation of DECNUPAZ in ≥1% of patients included veno-occlusive disease and pneumonitis.
Dosage interruptions of DECNUPAZ due to adverse reactions occurred in 37% of patients. Adverse reactions which resulted in dosage interruptions in ≥2% of patients included edema, pneumonia, infusion-related reaction, bacterial infections, fatigue, hemorrhage, neutropenia, pneumonitis, and pyrexia.
Dose reductions of DECNUPAZ due to an adverse reaction occurred in 6% of patients. Adverse reactions which required dose reductions in ≥2% of patients included edema.
The most common adverse reactions (≥20%) were edema, fatigue, musculoskeletal pain, hemorrhage, infusion-related reactions, nausea, and diarrhea. The most common Grade 3 to 4 laboratory abnormalities (≥10%) were neutrophils decreased, platelets decreased, lymphocyte count decreased, white blood cells decreased, hemoglobin decreased, and glucose increased.
Table 3 summarizes the common adverse reactions (≥10%) in patients treated with DECNUPAZ in CADENZA.
Table 3. Adverse Reactions (≥10%) in Patients Who Received DECNUPAZ in CADENZA DECNUPAZ
(N=116)Adverse Reaction§ All Grades
(%)Grade 3 or 4
(%)General disorders and administration site conditions Edemaa 52 16 Fatigueb 34 5 Pyrexiab 16 0.9 Chills 11 0 Musculoskeletal and connective tissue disorders Musculoskeletal painb 34 8 Vascular disorders Hemorrhageb 28 6 Thrombosisb 13 5 Injury, poisoning and procedural complications Infusion-related reactions 26 5 Fall 13 1.7 Gastrointestinal disorders Nauseab 24 0.9 Diarrheab 21 0.9 Constipation 19 0 Abdominal painb 14 0.9 Respiratory, thoracic and mediastinal disorders Dyspneab 19 1.7 Coughb 15 0 Skin and subcutaneous tissue disorders Rashc 19 0 Nervous system disorders Neuropathy peripherald 18 1.7 Headacheb 16 2.6 Dizzinessb 10 0.9 Metabolism and nutrition disorders Decreased appetiteb 16 0.9 Infections and infestations Infections without specified pathogensb 16 6 Viral infectionse 13 6 Bacterial infectionsf 12 5 Pneumoniag 11 9 Psychiatric disorders Insomnia 15 0 Blood and lymphatic system disorders Febrile neutropenia 11 11 § Adverse reactions were graded based on CTCAE Version 4.03
a. Edema includes acute pulmonary edema, face edema, generalized edema, hypervolemia, edema, edema genital, edema peripheral, pericardial effusion, peripheral swelling, pleural effusion, pulmonary edema, swelling face, weight increased, ascites.
b. Consists of multiple related terms.
c. Rash includes erythema, erythema nodosum, guttate psoriasis, photosensitivity reaction, psoriasis, rash, rash erythematous, rash macular, rash maculo-papular, rash pruritic, skin lesion, skin lesion inflammation, stasis dermatitis.
d. Neuropathy peripheral includes burning sensation, dysesthesia, facial nerve disorder, hypoesthesia, IIIrd nerve disorder, neuralgia, neuropathy peripheral, paresthesia, sciatica.
e. Viral infections includes COVID-19, cytomegalovirus infection, HCoV-229E infection, herpes simplex, herpes zoster, herpes zoster disseminated, influenza, ophthalmic herpes simplex, oral herpes.
f. Bacterial infections includes cellulitis, clostridium difficile infection, erysipelas, folliculitis, vulval abscess.
g. Pneumonia includes pneumocystis jirovecii pneumonia, pneumonia, pneumonia viral.
Clinically relevant adverse reactions occurring in <10% of patients who received DECNUPAZ in CADENZA included:
Vascular disorders: capillary leak syndrome (9%)a, hypotensionb (7%)
Gastrointestinal disorders: stomatitisb (6%)
Infections and infestations: sepsisc (7%), fungal infectionsd (5%)
Respiratory, thoracic and mediastinal disorders: pneumonitis (5%)
Cardiac disorders: arrhythmiae (6%)
Renal and urinary disorders: acute kidney injuryb (6%)
Hepatobiliary disorders: veno-occlusive disease (1.7%)
a. At least 2 of the following new onset signs and symptoms within 7 days of each other: hypoalbuminemia (including albumin <3.0 g/dL), edema (including weight increase >5 kg), hypotension (including systolic blood pressure <90 mmHg).
b. Consists of multiple related terms.
c. Includes bacteremia, klebsiella bacteremia, pulmonary sepsis, sepsis, and streptococcal bacteremia.
d. Includes candida infection, fungal balanitis, fungal foot infection, fungal skin infection.
e. Includes arrhythmia, atrial fibrillation, atrioventricular block, bradycardia, cardiac arrest, tachyarrhythmia.
Table 4 summarizes laboratory abnormalities in CADENZA.
Table 4. Select Laboratory Abnormalities (≥10%) That Worsened from Baseline in Patients Who Received DECNUPAZ in CADENZA Laboratory Abnormality* DECNUPAZa All Grades
(%)Grade 3 or 4
(%)Chemistry Creatinine increased 76 0 Glucose increased 53 10 Albumin decreased 50 1.8 Phosphate decreased 39 8 Calcium decreased 34 1.8 Alanine aminotransferase increased 32 4.4 Aspartate aminotransferase increased 29 0.9 Sodium decreased 28 1.8 Potassium decreased 26 3.5 Alkaline phosphatase increased 20 0.9 Magnesium decreased 18 0 Bilirubin increased 16 0.9 Hematology Platelets decreased 64 40 Neutrophils decreased 63 45 Lymphocyte count decreased 62 39 White blood cells decreased 55 34 Hemoglobin decreased 40 20 * Laboratory abnormalities were graded based on CTCAE Version 4.03.
a The denominator used to calculate the rate varied from 78 to 114 based on the number of patients with at least one post-baseline value.
- Hepatotoxicity, Including Hepatic VOD (also known as Sinusoidal Obstruction Syndrome) [see Warnings and Precautions (5.1)]
-
7.
DRUG INTERACTIONS
7.1 Effect of Other Drugs on DECNUPAZ
Strong and moderate CYP3A inhibitors
Closely monitor patients for adverse reactions with DECNUPAZ when used concomitantly with strong and moderate CYP3A inhibitors.
FGN849 is a substrate of CYP3A [see Clinical Pharmacology (12.3)]. Concomitant use of DECNUPAZ with strong and moderate CYP3A inhibitors may increase unconjugated FGN849 exposure, which may increase the risk of DECNUPAZ adverse reactions.
-
8.
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, DECNUPAZ can cause embryo-fetal harm when administered to a pregnant woman because it contains a genotoxic compound (FGN849) and affects actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. There are no available data on the use of DECNUPAZ in pregnant women to inform a drug-associated risk. Advise patients of the potential risks to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Animal reproductive or developmental toxicity studies were not conducted with pivekimab sunirine-pvzy. The cytotoxic component of DECNUPAZ, FGN849, is a DNA-alkylating agent that is toxic to rapidly dividing cells, indicating it has the potential to cause embryofetal lethality and teratogenicity.
8.2 Lactation
Risk Summary
There are no data on the presence of pivekimab sunirine-pvzy or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with DECNUPAZ and for 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
DECNUPAZ can cause fetal harm when administered to a pregnant patient [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiation of DECNUPAZ.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with DECNUPAZ and for 7 months after the last dose.
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment with DECNUPAZ and for 4 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on its mechanism of action, DECNUPAZ may impair male and female reproductive function and fertility.
8.4 Pediatric Use
The safety and effectiveness of DECNUPAZ have not been established in pediatric patients.
8.5 Geriatric Use
Of the 116 patients who were treated in CADENZA, 70% of patients were ≥65 years of age and 28% were ≥75 years of age. No overall differences in safety or effectiveness of DECNUPAZ have been observed between patients 65 years of age and older and younger adult patients.
Age does not have a clinically meaningful effect on the pharmacokinetics of DECNUPAZ [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Avoid use of DECNUPAZ in patients with moderate to severe renal impairment (CLcr <60 mL/min, estimated by Cockcroft-Gault) or patients with end stage renal disease. A higher incidence of Grade ≥3 adverse events, serious adverse events, and dose delays was observed in patients with moderate renal impairment (CLcr 30 to <60 mL/min). DECNUPAZ has not been studied in patients with severe renal impairment (CLcr <30 mL/min) or end stage renal disease.
No dosage adjustment of DECNUPAZ is recommended for patients with mild renal impairment (CLcr 60 to <90 mL/min, estimated by Cockcroft-Gault) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Avoid use of DECNUPAZ in patients with moderate to severe hepatic impairment (total bilirubin >1.5 x ULN with any AST). Limited data are available in patients with moderate hepatic impairment (total bilirubin >1.5 to 3 times ULN and any AST). DECNUPAZ has not been studied in patients with severe hepatic impairment.
No dosage adjustment of DECNUPAZ is recommended for patients with mild hepatic impairment (total bilirubin ≤ULN and AST >ULN or total bilirubin ≤1.5 times ULN and any AST) [see Clinical Pharmacology (12.3)].
-
11.
DESCRIPTION
Pivekimab sunirine-pvzy is a CD123-directed antibody and alkylating agent conjugate created by conjugating the IgG1 monoclonal antibody G4723A to the DGN549C linker-payload. The antibody-drug conjugate (ADC) contains approximately two DGN549C molecules sulfonated prior to conjugation and bound to the heavy chains (HC) of the G4723A antibody. Pivekimab sunirine-pvzy has an approximate molecular weight of 148 kDa. Pivekimab sunirine-pvzy is produced by site directed chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
Pivekimab sunirine-pvzy has the following structure:
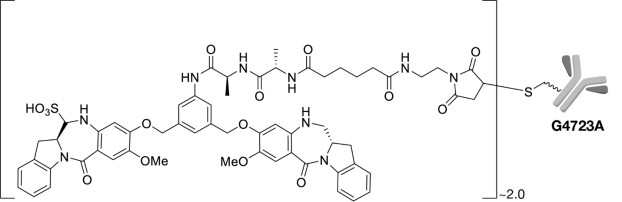
DECNUPAZ (pivekimab sunirine-pvzy) for injection is a sterile, lyophilized cake in a single-dose vial for reconstitution and dilution. DECNUPAZ is supplied as 2 mg per vial and requires reconstitution with Sterile Water for Injection, USP (1.1 mL) to obtain a concentration of 2 mg/mL. Following reconstitution, each mL delivers 2 mg of pivekimab sunirine-pvzy, methionine (0.45 mg), polysorbate 20 (0.1 mg), sodium hydroxide (0.2 mg), sodium metabisulfite (0.0048 mg), succinic acid (1.2 mg), trehalose (71.7 mg), and Sterile Water for Injection. The pH is 4.2. -
12.
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pivekimab sunirine-pvzy is a CD123 (alpha-subunit of the interleukin-3 receptor)-directed antibody-drug conjugate (ADC). The antibody is a humanized anti-CD123 IgG1. Pivekimab sunirine-pvzy binds to CD123 expressing cells and upon intracellular processing releases a cell membrane-permeable payload, FGN849, leading to DNA alkylation, single-strand DNA breaks, apoptosis, and cell death. The payload, FGN849, is a member of the indolinobenzodiazepine pseudodimer (IGN) class of cytotoxic molecules. Pivekimab sunirine-pvzy exhibited antitumor activity in in vitro and in vivo models of BPDCN.
12.2 Pharmacodynamics
Exposure-Response Relationships
Higher FGN849 (cytotoxic component of DECNUPAZ) exposure was associated with increased rates of Grade ≥2 infusion-related reactions.
Cardiac Electrophysiology
There is insufficient information to characterize the effect of pivekimab sunirine-pvzy on the QTc interval.
In 116 patients who received DECNUPAZ 0.045 mg/kg once every 3 weeks in CADENZA, 0.9% of patients had QTcF greater than 500 ms.
12.3 Pharmacokinetics
Pivekimab sunirine-pvzy and FGN849 pharmacokinetics were observed at Cycle 1 in patients in CADENZA at the approved recommended dosage and are presented as mean (CV%), unless otherwise specified. The exposure parameters of pivekimab sunirine-pvzy (ADC) and unconjugated FGN849 are summarized in Table 5. The Cmax and AUC of the ADC increased more than proportionally over a dose range of 0.045 to 0.18 mg/kg (the approved recommended dose to 4 times the recommended dose). ADC time to maximum concentrations (Tmax) occurred approximately at the end of the infusion, while FGN849 Tmax occurred approximately 2 hours after the end of infusion. There was no accumulation of the ADC or FGN849 in Cycle 3.
Table 5. Cycle 1 Exposure parameters of pivekimab sunirine-pvzy and unconjugated FGN849 at DECNUPAZ 0.045 mg/kg Every 3 Weeks Pivekimab sunirine-pvzy
Geometric mean (%CV)Unconjugated FGN849
Geometric mean (%CV)Cmax 442 (169) ng/mL 58.8 (110) pg/mL AUClast 892 (101) ngh/mL 171 (128) pgh/mL Cmax=maximum concentration, AUClast=area under the concentration from time zero to the last quantifiable concentration
Distribution
Pivekimab sunirine-pvzy volume of distribution is 5.4 L (39).
FGN849 plasma protein binding is 99.6% in vitro.
Elimination
Pivekimab sunirine-pvzy elimination half-life is approximately 1.5 hours at the 0.045 mg/kg every 3 weeks dosage.
Pivekimab sunirine-pvzy clearance is 1.8 L/hour (76).
Metabolism
Pivekimab sunirine-pvzy is expected to be catabolized into small peptides and amino acids. FGN849 is primarily metabolized by CYP3A.
Specific Populations
No clinically significant differences in the pharmacokinetics of pivekimab sunirine-pvzy or FGN849 were observed for age (19 to 91 years), body weight (45 to 160 kg), mild hepatic impairment (total bilirubin >ULN to 1.5 times ULN and any AST), or CLcr 60 to <90 mL/min (estimated by Cockcroft-Gault). Higher pivekimab sunirine-pvzy exposure and lower FGN849 exposure were observed in female patients compared to those in male patients.
The pharmacokinetics of pivekimab sunirine-pvzy in patients with moderate to severe hepatic impairment (total bilirubin >1.5 times ULN with any AST) or CLcr <60 mL/min is unknown.
Drug Interaction Studies
No dedicated clinical studies evaluating the drug-drug interaction potential of pivekimab sunirine-pvzy have been conducted.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: FGN849 is primarily a CYP3A substrate but is not a significant substrate of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP2D6. FGN849 is an inhibitor of CYP3A and CYP2C8 but is not an inhibitor of CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6.
Transporter systems: FGN849 is a substrate of P-gp and BCRP but is not a substrate of MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, or OCT2. FGN849 is not an inhibitor of P-gp, BCRP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, or OCT2.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the study described below with the incidence of ADA in other studies, including those of pivekimab sunirine-pvzy.
Following administration of pivekimab sunirine-pvzy in CADENZA in BPDCN patients, 19/80 (24%) of patients tested positive for treatment emergent antibodies against pivekimab sunirine-pvzy. Of those who tested positive for anti-drug antibody, neutralizing antibodies were detected in 14/18 (78%) of evaluable patients. There was no identified clinically significant effect of anti-drug antibodies on pharmacokinetics, safety, or effectiveness of pivekimab sunirine-pvzy.
-
13.
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with pivekimab sunirine-pvzy or the small molecule FGN849.
Mutagenesis
FGN849 was mutagenic in the bacterial reverse mutation (Ames) assay and clastogenic in the in vitro and in vivo (rat) micronucleus assays. These results are consistent with the pharmacological mechanism of action of DNA alkylation that induces G2-M phase arrest of dividing cells resulting in cell death.
Impairment of Fertility
Fertility studies have not been conducted with pivekimab sunirine-pvzy or FGN849.
-
14.
CLINICAL STUDIES
14.1 Treatment-naïve Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
CADENZA (NCT03386513) was a multicenter, open-label, single-arm, clinical trial that included 33 adult patients with treatment-naїve BPDCN with no central nervous system (CNS) involvement. Treatment consisted of DECNUPAZ 0.045 mg/kg intravenously once every three weeks. Patient baseline characteristics are presented in Table 6.
Table 6. Baseline Demographics of Patients with Treatment-naїve BPDCN Parameter N=33 Gender, N (%) Male 27 (82) Female 6 (18) Race, N (%) White 27 (82) Black or African American 1 (3) Not Reported 5 (15) Ethnicity, N (%) Hispanic or Latino 4 (12) Non-Hispanic or Latino 28 (85) Unknown 1 (3) Age (years) Median (Range) 73 (48, 84) ECOG, N (%) 0 12 (36) 1 21 (64) BPDCN at Baseline, N (%) Skin 31 (94) Bone Marrow 16 (48) Peripheral Blood 3 (9) Lymph Nodes 12 (36) Viscera 0 Disease subgroup, N (%) BPDCN de novo 22 (67) BPDCN with PCHMa 11 (33) aPCHM is defined as any previously diagnosed or concurrently present hematologic malignancy at the time of BPDCN diagnosis
The efficacy of DECNUPAZ in patients with treatment-naїve BPDCN was based on the rate of complete remission or clinical complete remission (CR/CRc). Key efficacy measures are presented in Table 7. The median follow-up was 21.5 months (range: 4.2, 27). The median time to CR/CRc was 1.8 months (range: <0.5 to 4). Among the 33 patients with treatment-naїve BPDCN, 13 (39.4%) patients were able to receive post-study treatment HSCT.
Table 7. Efficacy Results in Patients with Treatment-naїve BPDCN Endpoint N=33 CR/CRca Rate, N (%)b,c 23 (69.7) (95% CI) (51.3, 84.4) CR, N (%) 16 (48.5) (95% CI) (30.8, 66.5) CRc, N (%) 7 (21.2) (95% CI) (9.0, 38.9) Duration of CR/CRc (months)d,e,f Median 9.7 95% CI (2.9, NE) CI = confidence interval; NE = not estimable
aCRc is defined as complete remission with residual skin abnormality not indicative of active disease.
bCR/CRc rate was 77.3% (95% CI: 54.6, 92.2) in patients with de novo BPDCN (N=22).
cCR/CRc rate was 54.5% (95% CI: 23.4, 83.3) in patients with PCHM (N=11).
dKaplan-Meier estimate.
eMedian duration of CR/CRc was 9.8 months (range: 0.9, 23.9+) in 17 patients with de novo BPDCN who achieved CR/CRc.
fMedian duration of CR/CRc was 6.3 months (range: 2.4, 23.2+) in 6 patients with BPDCN with PCHM who achieved CR/CRc.
14.2 Relapsed or Refractory Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN)
CADENZA (NCT03386513) was a multicenter, open-label, single-arm, clinical trial that included 51 adult patients with relapsed or refractory BPDCN without evidence of active CNS disease treated with DECNUPAZ 0.045 mg/kg intravenously once every three weeks. Patient baseline characteristics are presented in Table 8.
Table 8. Baseline Demographics of Patients with Relapsed or Refractory BPDCN Parameter N=51 Gender, N (%) Male 42 (82) Female 9 (18) Race, N (%) White 42 (82) Black or African American 2 (4) Asian 1 (2) Not Reported 6 (12) Ethnicity, N (%) Hispanic or Latino 8 (16) Non-Hispanic or Latino 38 (75) Unknown 5 (10) Age (years) Median (Range) 69 (19, 85) ECOG, N (%) 0 18 (35) 1 30 (59) 2 2 (4) 3 1 (2) BPDCN at Baseline, N (%) Skin 34 (67) Bone Marrow 24 (47) Peripheral Blood 10 (20) Lymph Nodes 18 (35) Viscera 3 (6) Number of prior lines of therapy, median (Range) 1 (1, 4) Prior stem cell transplantation, N (%) 16 (31) The efficacy of DECNUPAZ in patients with relapsed/refractory BPDCN was based on the rate of CR/CRc. Key efficacy measures are presented in Table 9. The median follow-up was 24.1 months (range: 0.2 to 30.4). The median time to CR/CRc was 1.7 months (range: 1 to 6). Among the 51 patients with relapsed/refractory BPDCN, 6 (11.8%) patients were able to receive post-study treatment HSCT.
Table 9. Efficacy Results in Patients with Relapsed or Refractory BPDCN Endpoint N=51 CR/CRca Rate, N (%) 8 (15.7) (95% CI) (7.0, 28.6) CR, N (%) 7 (13.7) (95% CI) (5.7, 26.3) CRc, N (%) 1 (2.0) (95% CI) (0.1, 10.5) Duration of CR/CRc (months)b Median 9.2 Range (2.7, 27.6+) CI = confidence interval; + = Censor indicator
aCRc is defined as complete remission with residual skin abnormality not indicative of active disease.
bKaplan-Meier estimate.
-
15.
REFERENCES
1“OSHA Hazardous Drugs.” OSHA http://www.osha.gov/SLTC/hazardousdrugs/index.html
-
16.
HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
DECNUPAZ (pivekimab sunirine-pvzy) for injection is a sterile, preservative-free, white to off-white lyophilized cake, supplied in a single-dose glass vial. The DECNUPAZ vial stoppers are not made with natural rubber latex.
- 2 mg single-dose vial with dark grey flip-top (NDC: 0074-0282-02)
Storage and Handling
Store DECNUPAZ vials upright in a refrigerator at 2°C to 8°C (36°F to 46°F) until time of preparation in the original carton to protect from light.
Do not freeze or shake.
DECNUPAZ is a hazardous product. Follow applicable special handling and disposal procedures1.
-
17.
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Veno-occlusive Disease (VOD)
Advise patients that DECNUPAZ can cause VOD. Advise patients to immediately contact their healthcare provider if they experience symptoms of VOD, which may include jaundice, rapid weight gain, dark urine, and abdominal pain or distention. Inform patients that liver problems may require dosing interruption or permanent discontinuation of DECNUPAZ [see Warnings and Precautions (5.1)].
Infusion-Related Reactions
Advise patients that DECNUPAZ can cause infusion-related reactions. Advise patients to immediately contact their healthcare provider for any signs or symptoms of infusion-related reactions, which may include chills, tachycardia, hypotension, fever, tachypnea, and dyspnea [see Warnings and Precautions (5.2)].
Edema
Advise patients that DECNUPAZ can cause edema. Advise patients to contact their healthcare provider if they experience swelling, weight gain, shortness of breath or difficulty breathing, or fluid retention [see Warnings and Precautions (5.3)].
Sulfite Allergic Reactions
Advise patients about potential for sulfite sensitivity. Inform patients that DECNUPAZ contains sodium metabisulfite, which may cause allergic type reactions including anaphylactic symptoms and life-threatening or less severe asthmatic episodes, and to seek immediate medical care if they experience these signs or symptoms [see Warnings and Precautions (5.4)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise female patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with DECNUPAZ [see Warnings and Precautions (5.5) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with DECNUPAZ and for 7 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with DECNUPAZ and for 4 months after the last dose [see Use in Specific Populations (8.3)].
Infertility
Advise females and males of reproductive potential that DECNUPAZ may impair reproductive function and fertility [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment and for 1 month after the last dose of DECNUPAZ [see Use in Specific Populations (8.2)].
Manufactured by:
AbbVie Inc.
North Chicago, IL 60064, U.S.A.
U.S. License No. 1889
© 2026 AbbVie. All rights reserved.
DECNUPAZ and its design are trademarks of ImmunoGen, Inc., an AbbVie company.20098137
-
MEDICATION GUIDE
MEDICATION GUIDE
DECNUPAZ (DEK-nuh-paz)
(pivekimab sunirine-pvzy)
for injection, for intravenous useWhat is the most important information I should know about DECNUPAZ?
DECNUPAZ can cause serious side effects, including:- Liver problems (hepatotoxicity), including veno-occlusive disease (blockage of the small veins in the liver) that can be severe, life-threatening, or may lead to death. Your healthcare provider will do blood tests before each dose of DECNUPAZ and during treatment with DECNUPAZ to check for liver problems. Tell your healthcare provider right away if you develop signs or symptoms of liver problems, including:
○ yellowing of the skin or eyes ○ pain in your stomach (abdomen) ○ fast weight gain ○ swelling of your stomach ○ dark urine Your healthcare provider will check you for liver problems during your treatment with DECNUPAZ and may provide treatment for your side effects. Your healthcare provider may also delay or stop treatment with DECNUPAZ if you have severe liver problems.
See “What are the possible side effects of DECNUPAZ?” for more information about side effects.What is DECNUPAZ?
DECNUPAZ is a prescription medicine used to treat adults with blastic plasmacytoid dendritic cell neoplasm (BPDCN).
It is not known if DECNUPAZ is safe and effective in children.Before receiving DECNUPAZ, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems
- are allergic to sulfites
- have asthma
- have kidney problems
- are pregnant or plan to become pregnant. DECNUPAZ can harm your unborn baby.
○ Your healthcare provider will check for pregnancy before you start treatment with DECNUPAZ.
○ Use effective birth control (contraception) during treatment with DECNUPAZ and for 7 months after your last dose.
○ Tell your healthcare provider if you become pregnant or think that you may be pregnant during treatment with DECNUPAZ.
Males who have female partners who are able to become pregnant:
○ Use an effective birth control during treatment with DECNUPAZ and for 4 months after your last dose DECNUPAZ.
- are breastfeeding or plan to breastfeed. It is not known if DECNUPAZ passes into your breast milk. Do not breastfeed during treatment with DECNUPAZ and for 1 month after the last dose.
Certain medicines may affect DECNUPAZ and increase your risk of side effects.How will I receive DECNUPAZ? - Your healthcare provider will give you DECNUPAZ into your vein through an intravenous (IV) line over about 15 to 30 minutes.
- DECNUPAZ is given 1 time every three weeks (21-day treatment cycle).
- You will receive your first infusion over 30 minutes. If you do not have problems with your first infusion, you may receive your next infusions over 15 minutes.
- Your healthcare provider will decide how many treatments of DECNUPAZ you will receive.
- Your healthcare provider will give you medicines the day before and on the day of your infusion to help reduce infusion-related reactions. See “What are the possible side effects of DECNUPAZ?”
- Your healthcare provider may slow down your infusion of DECNUPAZ or permanently stop treatment with DECNUPAZ if you have an infusion-related reaction.
What are the possible side effects of DECNUPAZ?
DECNUPAZ can cause serious side effects, including:- See “What is the most important information I should know about DECNUPAZ?”
- Infusion-related reactions (IRR). DECNUPAZ can cause serious, life-threatening infusion-related reactions. Your healthcare provider will give you medicines the day before and on the day of your infusion of DECNUPAZ to help reduce infusion-related reactions. Your healthcare provider will check you for symptoms of infusion-related reactions during your infusion and for at least four hours or longer if needed, after your first infusion, and for at least one hour after each of your next infusions. Tell your healthcare provider right away if you develop signs or symptoms of infusion-related reactions, including:
○ shortness of breath ○ nausea ○ flushing ○ chest pain ○ fever ○ feeling faint or lightheaded ○ chills ○ vomiting -
Fluid retention (edema). DECNUPAZ can cause your body to hold too much fluid during treatment. Your healthcare provider may prescribe water pills (diuretic) if you develop edema. Tell your healthcare provider if you develop new or worsening edema, including:
○ swelling of your ankles or legs
○ shortness of breath or difficulty breathing
○ unusual weight gain
- Sulfite allergic reactions. DECNUPAZ contains sodium metabisulfite, a sulfite that may cause severe, life-threatening allergic reactions in some people. Sulfite allergic reactions are more common in people with asthma than in people without asthma. Get medical help right away if you develop hives, itching, rash, swelling of the eyes, tongue, lips, chest pain, trouble breathing, or swallowing.
The most common side effects of DECNUPAZ include:fluid retention (edema) bleeding feeling tired infusion-related reactions muscle, bone and joint pain nausea diarrhea The most common severe abnormal laboratory test results with DECNUPAZ include: decreased white blood cell counts decreased red blood cell counts decreased platelet counts increased blood sugar level Your healthcare provider may decrease your dose, delay your infusion or permanently stop treatment with DECNUPAZ if you have side effects.
DECNUPAZ may cause fertility problems in males and females, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility.
These are not all the possible side effects of DECNUPAZ.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of DECNUPAZ.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about DECNUPAZ that is written for healthcare professionals.What are the ingredients in DECNUPAZ?
Active ingredient: pivekimab sunirine-pvzy
Inactive ingredients: methionine, polysorbate 20, sodium hydroxide, sodium metabisulfite, succinic acid, trehalose and sterile water for injection
Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
U.S. License Number: 1889
Marketed by: AbbVie Inc., North Chicago, IL 60064 U.S.A.
©2026 AbbVie. All rights reserved.
DECNUPAZ and its design are trademarks of ImmunoGen, Inc., an AbbVie company.
For more information, go to www.DECNUPAZ.com or call 1-800-633-9110This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: 5/2026
20098137
-
PRINCIPAL DISPLAY PANEL
NDC: 0074-0282-02
DecnupazTM
pivekimab sunirine-pvzy
for injection
2 mg per vial
WARNING: Hazardous Drug
For intravenous infusion after
reconstitution and two dilutions
1 Single-dose Vial
Discard Unused Portion
Dispense the enclosed Medication
Guide to each patient
Rx Only
abbvie

-
INGREDIENTS AND APPEARANCE
DECNUPAZ
pivekimab sunirine-pvzy injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0074-0282 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PIVEKIMAB SUNIRINE (UNII: L15LO3W1XX) (PIVEKIMAB SUNIRINE - UNII:L15LO3W1XX) PIVEKIMAB SUNIRINE 2 mg in 1 mL Inactive Ingredients Ingredient Name Strength SUCCINIC ACID (UNII: AB6MNQ6J6L) 1.2 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 0.2 mg in 1 mL SODIUM METABISULFITE (UNII: 4VON5FNS3C) 0.0048 mg in 1 mL TREHALOSE (UNII: B8WCK70T7I) 71.7 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.1 mg in 1 mL WATER (UNII: 059QF0KO0R) METHIONINE (UNII: AE28F7PNPL) 0.45 mg in 1 mL Product Characteristics Color white (white to off-white) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0074-0282-02 1 in 1 CARTON 05/27/2026 1 1.15 mL in 1 VIAL, SINGLE-DOSE; Type 6: Drug/Biologic Combination Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761460 05/27/2026 Labeler - AbbVie Inc. (078458370)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.