Deferiprone by Taro Pharmaceuticals U.S.A., Inc. / Taro Pharmaceutical Industries, Ltd. DEFERIPRONE tablet
Deferiprone by
Drug Labeling and Warnings
Deferiprone by is a Prescription medication manufactured, distributed, or labeled by Taro Pharmaceuticals U.S.A., Inc., Taro Pharmaceutical Industries, Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DEFERIPRONE TABLETS safely and effectively. See full prescribing information for DEFERIPRONE TABLETS.
DEFERIPRONE tablets, for oral use
Initial U.S. Approval: 2011WARNING: AGRANULOCYTOSIS/NEUTROPENIA
See full prescribing information for complete boxed warning.
- Deferiprone can cause agranulocytosis that can lead to serious infections and death. Neutropenia may precede the development of agranulocytosis. (5.1)
- Measure the absolute neutrophil count (ANC) before starting deferiprone and monitor the ANC weekly on therapy. (5.1)
- Interrupt deferiprone if infection develops and monitor the ANC more frequently. (5.1)
- Advise patients taking deferiprone to report immediately any symptoms indicative of infection. (5.1)
INDICATIONS AND USAGE
Deferiprone is an iron chelator indicated for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate. (1)
Approval is based on a reduction in serum ferritin levels. There are no controlled trials demonstrating a direct treatment benefit, such as improvement in disease-related symptoms, functioning, or increased survival (1).
Limitation of Use
- Safety and effectiveness have not been established for the treatment of transfusional iron overload in patients with other chronic anemias. (1)
DOSAGE AND ADMINISTRATION
- 25 mg/kg to 33 mg/kg body weight, orally, three times per day, for a total daily dose of 75 mg/kg to 99 mg/kg body weight. (2)
DOSAGE FORMS AND STRENGTHS
- 500 mg tablets with a functional score. (3)
CONTRAINDICATIONS
- Hypersensitivity to deferiprone or to any of the excipients in the formulation. (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- The most common adverse reactions are (incidence ≥ 5%) chromaturia, nausea, vomiting and abdominal pain, alanine aminotransferase increased, arthralgia and neutropenia. (5.1, 6)
To report SUSPECTED ADVERSE REACTIONS, contact Taro Pharmaceuticals U.S.A., Inc. at 1-866-923-4914 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Avoid concomitant use with other drugs known to be associated with neutropenia or agranulocytosis; however, if this is not possible, closely monitor the absolute neutrophil count. (7.1)
- Allow at least a 4-hour interval between deferiprone and mineral supplements, and antacids that contain polyvalent cations (e.g., iron, aluminum, and zinc). (7.3)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: AGRANULOCYTOSIS/NEUTROPENIA
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Interactions with Foods, Vitamins and Antacids
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Agranulocytosis/Neutropenia
5.2 Embryofetal Toxicity
5.3 Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs Associated with Neutropenia or Agranulocytosis
7.2 UDP-Glucuronosyltransferases (UGTs)
7.3 Polyvalent Cations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: AGRANULOCYTOSIS/NEUTROPENIA
- Deferiprone can cause agranulocytosis that can lead to serious infections and death. Neutropenia may precede the development of agranulocytosis. [see Warnings and Precautions (5.1)]
- Measure the absolute neutrophil count (ANC) before starting deferiprone therapy and monitor the ANC weekly on therapy. Interrupt deferiprone therapy if neutropenia develops. [see Warnings and Precautions (5.1)]
- Interrupt deferiprone if infection develops, and monitor the ANC more frequently. [see Warnings and Precautions (5.1)]
- Advise patients taking deferiprone to report immediately any symptoms indicative of infection. [see Warnings and Precautions (5.1)]
-
1 INDICATIONS AND USAGE
Deferiprone tablets are indicated for the treatment of patients with transfusional iron overload due to thalassemia syndromes when current chelation therapy is inadequate.
Approval is based on a reduction in serum ferritin levels. There are no controlled trials demonstrating a direct treatment benefit, such as improvement in disease-related symptoms, functioning, or increased survival [see Clinical Studies (14)].
Limitation of Use:
- Safety and effectiveness have not been established for the treatment of transfusional iron overload in patients with other chronic anemias.
-
2 DOSAGE AND ADMINISTRATION
The recommended initial dose of deferiprone tablets is 25 mg/kg, orally, three times per day for a total of 75 mg/kg/day. The maximum dose is 33 mg/kg, three times per day for a total of 99 mg/kg/day.
Dose adjustments up to 33 mg/kg, orally, three times per day should be tailored to the individual patient's response and therapeutic goals (maintenance or reduction of body iron burden). The maximum recommended total daily dose is 99 mg/kg per day. The dose should be rounded by the prescriber to the nearest 250 mg (half-tablet).
Table 1a: Tablet requirement to achieve a 25 mg/kg (rounded to the nearest half-tablet) dose level for administration three times a day. Body Weight (kg) Dose (mg) Number of tablets 20 500 1 30 750 1.5 40 1000 2 50 1250 2.5 60 1500 3 70 1750 3.5 80 2000 4 90 2250 4.5 Table 1b: Tablet requirement to achieve 33 mg/kg (rounded to the nearest half-tablet) dose level for administration three times a day. Body Weight (kg) Dose (mg) Number of tablets 20 660 1.5 30 990 2 40 1320 2.5 50 1650 3.5 60 1980 4 70 2310 4.5 80 2640 5.5 90 2970 6 Monitor serum ferritin concentration every two to three months to assess the effects of deferiprone on body iron stores. Dose adjustments should be tailored to the individual patient's response and therapeutic goals (maintenance or reduction of body iron burden). If the serum ferritin falls consistently below 500 mcg/L, consider temporarily interrupting deferiprone therapy.
2.1 Interactions with Foods, Vitamins and Antacids
Allow at least a 4-hour interval between deferiprone and other medications or supplements containing polyvalent cations such as iron, aluminum, and zinc [see Drug Interactions (7.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- Deferiprone tablets are contraindicated in patients with known hypersensitivity to deferiprone or to any of the excipients in the formulation. The following reactions have been reported in association with the administration of deferiprone: Henoch-Schönlein purpura; urticaria; and periorbital edema with skin rash [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Agranulocytosis/Neutropenia
Fatal agranulocytosis can occur with deferiprone use. Deferiprone can also cause neutropenia, which may foreshadow agranulocytosis. Measure the absolute neutrophil count (ANC) before starting deferiprone therapy and monitor the ANC weekly on therapy.
Interrupt deferiprone therapy if neutropenia develops (ANC < 1.5 × 109/L).
Interrupt deferiprone if infection develops, and monitor the ANC more frequently.
Advise patients taking deferiprone to immediately interrupt therapy and report to their physician if they experience any symptoms indicative of infection.
In pooled clinical trials, the incidence of agranulocytosis was 1.7% of patients. The mechanism of deferiprone-associated agranulocytosis is unknown. Agranulocytosis and neutropenia usually resolve upon discontinuation of deferiprone, but there have been reports of agranulocytosis leading to death.
Implement a plan to monitor for and to manage agranulocytosis/neutropenia prior to initiating deferiprone treatment.
For neutropenia (ANC < 1.5 × 109/L and > 0.5 × 109/L):
Instruct the patient to immediately discontinue deferiprone and all other medications with a potential to cause neutropenia.
Obtain a complete blood cell (CBC) count, including a white blood cell (WBC) count corrected for the presence of nucleated red blood cells, an absolute neutrophil count (ANC), and a platelet count daily until recovery (ANC ≥ 1.5 × 109/L).
For agranulocytosis (ANC < 0.5 × 109/L):
Consider hospitalization and other management as clinically appropriate.
Do not resume deferiprone in patients who have developed agranulocytosis unless potential benefits outweigh potential risks. Do not rechallenge patients who develop neutropenia with deferiprone unless potential benefits outweigh potential risks.
5.2 Embryofetal Toxicity
Based on evidence of genotoxicity and developmental toxicity in animal studies, deferiprone can cause fetal harm when administered to a pregnant woman. In animal studies, administration of deferiprone during the period of organogenesis resulted in embryofetal death and malformations at doses lower than equivalent human clinical doses. If deferiprone is used during pregnancy or if the patient becomes pregnant while taking deferiprone, the patient should be apprised of the potential hazard to the fetus. Women of reproductive potential should be advised to avoid pregnancy when taking deferiprone [see Use in Specific Populations (8.1) and Nonclinical Toxicology (13.1)].
5.3 Laboratory Tests
Serum Liver Enzyme Activities
In clinical studies, 7.5% of 642 subjects treated with deferiprone developed increased ALT values. Four (0.62%) deferiprone-treated subjects discontinued the drug due to increased serum ALT levels and 1 (0.16%) due to an increase in both ALT and AST.
Monitor serum ALT values monthly during therapy with deferiprone, and consider interruption of therapy if there is a persistent increase in the serum transaminase levels.
-
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
The following adverse reactions are also discussed in other sections of the labeling: Agranulocytosis/Neutropenia [see Warnings and Precautions (5.1)]. Elevated ALT (5.3), Decreased plasma zinc concentrations (5.3).
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reaction information for deferiprone represents the pooled data collected from 642 patients who participated in single arm or active-controlled clinical studies.
The most serious adverse reaction reported in clinical trials with deferiprone was agranulocytosis [see Warnings and Precautions (5.1)].
The most common adverse reactions reported during clinical trials were chromaturia, nausea, vomiting, abdominal pain, alanine aminotransferase increased, arthralgia and neutropenia.
The table below lists the adverse drug reactions that occurred in at least 1% of patients treated with deferiprone in clinical trials.
Table 2: Adverse drug reactions occurring in ≥ 1% of 642 deferiprone-treated patients Body System % Subjects Preferred Term BLOOD AND LYMPHATIC SYSTEM DISORDERS Neutropenia 6.2 Agranulocytosis 1.7 GASTROINTESTINAL DISORDERS Nausea 12.6 Abdominal pain/discomfort 10.4 Vomiting 9.8 Diarrhea 3.0 Dyspepsia 2.0 INVESTIGATIONS Alanine Aminotransferase increased 7.5 Neutrophil count decreased 7.3 Weight increased 1.9 Aspartate Aminotransferase increased 1.2 METABOLISM AND NUTRITION DISORDERS Increased appetite 4.0 Decreased appetite 1.1 MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS Arthralgia 9.8 Back pain 2.0 Pain in extremity 1.9 Arthropathy 1.4 NERVOUS SYSTEM DISORDERS Headache 2.5 URINARY DISORDERS Chromaturia 14.6 Gastrointestinal symptoms such as nausea, vomiting, and abdominal pain were the most frequent adverse reactions reported by patients participating in clinical trials and led to the discontinuation of deferiprone therapy in 1.6% of patients.
Chromaturia (reddish/brown discoloration of the urine) is a result of the excretion of the iron in the urine.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported in patients receiving deferiprone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or to establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: thrombocytosis, pancytopenia.
Cardiac disorders: atrial fibrillation, cardiac failure.
Congenital, familial and genetic disorders: hypospadias.
Eye disorders: diplopia, papilledema, retinal toxicity.
Gastrointestinal disorders: enterocolitis, rectal hemorrhage, gastric ulcer, pancreatitis, parotid gland enlargement.
General disorders and administration site conditions: chills, pyrexia, edema peripheral, multi-organ failure.
Hepatobiliary disorders: jaundice, hepatomegaly.
Immune system disorders: anaphylactic shock, hypersensitivity.
Infections and infestations: cryptococcal cutaneous infection, enteroviral encephalitis, pharyngitis, pneumonia, sepsis, furuncle, infectious hepatitis, rash pustular, subcutaneous abscess.
Investigations: blood bilirubin increased, blood creatinine phosphokinase increased.
Metabolism and nutrition disorders: metabolic acidosis, dehydration.
Musculoskeletal and connective tissue disorders: myositis, chondropathy, trismus.
Nervous system disorders: cerebellar syndrome, cerebral hemorrhage, convulsion, gait disturbance, intracranial pressure increased, psychomotor skills impaired, pyramidal tract syndrome, somnolence.
Psychiatric disorders: bruxism, depression, obsessive-compulsive disorder.
Renal disorders: glycosuria, hemoglobinuria.
Respiratory, thoracic and mediastinal disorders: acute respiratory distress syndrome, epistaxis, hemoptysis, pulmonary embolism.
Skin, subcutaneous tissue disorders: hyperhidrosis, periorbital edema, photosensitivity reaction, pruritis, urticaria, rash, Henoch-Schönlein purpura.
Vascular disorders: hypotension, hypertension.
-
7 DRUG INTERACTIONS
7.1 Drugs Associated with Neutropenia or Agranulocytosis
Avoid concomitant use of deferiprone with other drugs known to be associated with neutropenia or agranulocytosis; however, if this is not possible, closely monitor the absolute neutrophil count [see Warnings and Precautions (5.1)].
7.2 UDP-Glucuronosyltransferases (UGTs)
Deferiprone is primarily eliminated via metabolism to the 3-O-glucuronide. In vitro studies suggest that UDP glucuronosyltransferase (UGT) 1A6 is primarily responsible for the glucuronidation of deferiprone which can be reduced up to 78% in the presence of the UGT1A6 inhibitor phenylbutazone. However, the clinical significance of coadministration of deferiprone with a UGT1A6 inhibitor (e.g. diclofenac, probenecid, or silymarin (milk thistle)) on the systemic exposure of deferiprone has not been determined. Closely monitor patients for adverse reactions that may require downward dose titration or interruption when deferiprone is concomitantly administered with a UGT1A6 inhibitor.
7.3 Polyvalent Cations
Concurrent use of deferiprone with foods, mineral supplements, and antacids that contain polyvalent cations has not been studied. However, since deferiprone has the potential to bind polyvalent cations (e.g., iron, aluminum, and zinc), allow at least a 4-hour interval between deferiprone and other medications (e.g., antacids), or supplements containing these polyvalent cations [see Dosage and Administration (2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category D [see Warnings and Precautions (5.2), Nonclinical Toxicology (13.1)]
Risk Summary
Based on evidence of genotoxicity and developmental toxicity in animal studies, deferiprone can cause fetal harm when administered to a pregnant woman. In animal studies, administration of deferiprone during the period of organogenesis resulted in embryofetal death and malformations at doses lower than equivalent human clinical doses. There are no studies in pregnant women, and available human data are limited. If deferiprone is used during pregnancy or if the patient becomes pregnant while taking deferiprone, the patient should be apprised of the potential hazard to the fetus.
Animal Data
Skeletal and soft tissue malformations occurred in offspring of rats and rabbits that received deferiprone orally during organogenesis at the lowest doses tested (25 mg/kg per day in rats; 10 mg/kg per day in rabbits). These doses were equivalent to 3% to 4% of the maximum recommended human dose (MRHD) based on body surface area. No maternal toxicity was evident at these doses. Embryofetal lethality and maternal toxicity occurred in pregnant rabbits given 100 mg/kg/day deferiprone orally during the period of organogenesis. This dose is equivalent to 32% of the MRHD based on body surface area.
8.3 Nursing Mothers
It is not known whether deferiprone is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for adverse reactions in nursing infants from deferiprone, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of deferiprone tablets for oral use in pediatric patients have not been established.
8.5 Geriatric Use
Safety and effectiveness in elderly individuals have not been established. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
An open-label, non-randomized, parallel group clinical study was conducted to evaluate the effect of impaired renal function on the safety, tolerability, and pharmacokinetics of a single 33 mg/kg oral dose of deferiprone. Subjects were categorized into 4 groups based on estimated glomerular filtration rate (eGFR): healthy volunteers (eGFR ≥ 90 mL/min/1.73m2), mild renal impairment (eGFR 60–89 mL/min/1.73m2), moderate renal impairment (eGFR 30–59 mL/min/1.73m2), and severe renal impairment (eGFR 15–29 mL/min/1.73m2). Systemic exposure to deferiprone and to its metabolite deferiprone 3-O-glucuronide was assessed by the PK parameters Cmax and AUC.
Regardless of the degree of renal impairment, the majority of the dose of deferiprone was excreted in the urine over the first 24 hours as deferiprone 3-O-glucuronide. No significant effect of renal impairment was seen on systemic exposure to deferiprone. Systemic exposure to the inactive 3-O-glucuronide increased with decreasing eGFR. Based on the results of this study, no adjustment of the deferiprone dosage regimen is required in patients with impaired renal function.
8.7 Hepatic Impairment
The influence of severe hepatic impairment on the pharmacokinetics of deferiprone and deferiprone 3-O-glucuronide has not been evaluated. Safety and efficacy of deferiprone have not been evaluated in patients with severe hepatic impairment.
An open-label, non-randomized, parallel group clinical study was conducted to evaluate the effect of impaired hepatic function on the safety, tolerability, and pharmacokinetics of a single 33 mg/kg oral dose of deferiprone. Subjects were categorized into 3 groups based on the Child-Pugh classification score: healthy volunteers, mild hepatic impairment (Class A: 5– 6 points), and moderate hepatic impairment (Class B: 7– 9 points). Systemic exposure to deferiprone and to its metabolite deferiprone 3-O-glucuronide was assessed by the PK parameters Cmax and AUC. The PK of both deferiprone and deferiprone 3-O-glucuronide was generally similar in all subjects, regardless of degree of liver impairment. A serious adverse event of acute liver and renal injury was seen in one subject with moderate hepatic impairment. Based on the results of this study, no adjustment of the deferiprone dosage regimen is required in patients with mildly or moderately impaired hepatic function.
-
10 OVERDOSAGE
No cases of acute overdose have been reported. There is no specific antidote to deferiprone overdose.
Neurological disorders such as cerebellar symptoms, diplopia, lateral nystagmus, psychomotor slowdown, hand movements and axial hypotonia have been observed in children treated with 2.5 to 3 times the recommended dose for more than one year. The neurological disorders progressively regressed after deferiprone discontinuation.
-
11 DESCRIPTION
Deferiprone tablets contain 500 mg deferiprone (3-hydroxy-1,2-dimethylpyridin-4-one), a synthetic, orally active, iron-chelating agent. Deferiprone has the following structural formula:
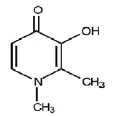
Deferiprone is a white to pinkish-white crystalline powder. It is sparingly soluble in deionized water and has a melting point range of 272°C -278°C.
Deferiprone tablets are white to pinkish-white, capsule-shaped tablets; scored on one side, engraved "T" on the left of the score line and "5" on the right and plain on the other side. The tablets can be broken in half along the score. Each tablet contains 500 mg deferiprone and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Deferiprone is a chelating agent with an affinity for ferric ion (iron III). Deferiprone binds with ferric ions to form neutral 3:1 (deferiprone:iron) complexes that are stable over a wide range of pH values. Deferiprone has a lower binding affinity for other metals such as copper, aluminum and zinc than for iron.
12.2 Pharmacodynamics
No clinical studies were performed to assess the relationship between the dose of deferiprone and the amount of iron eliminated from the body.
12.3 Pharmacokinetics
Deferiprone is rapidly absorbed from the upper part of the gastrointestinal tract, appearing in the blood within 5 to 10 minutes of oral administration. Peak serum concentrations occur approximately 1 hour after a single dose in fasted healthy subjects and patients, and up to 2 hours after a single dose in the fed state. Administration with food decreased the Cmax of deferiprone by 38% and the AUC by 10%. While a food effect cannot be ruled out, the magnitude of the exposure change does not warrant dose adjustment.
In healthy subjects, the mean maximum concentration (Cmax) of deferiprone in serum was 20 mcg/mL, and the mean total area under the concentration-time curve (AUC) was 53 mcg∙h/mL following oral administration of a 1,500 mg dose of deferiprone tablets in the fasting state. Dose proportionality over the labeled dosage range of 25 to 33 mg/kg three times per day (75 to 99 mg/kg per day) has not been studied. The elimination half life (t½) of deferiprone was 1.9 hours. The accumulation of deferiprone and its glucuronide metabolite at the highest approved dosage level of 33 mg/kg three times per day has not been studied. The volume of distribution of deferiprone is 1.6 L/kg in thalassemia patients, and approximately 1 L/kg in healthy subjects. The plasma protein binding of deferiprone in humans is less than 10%.
In humans, the majority of the deferiprone is metabolized, primarily by UGT1A6. The contribution of extrahepatic (e.g., renal) UGT1A6 is unknown. The major metabolite of deferiprone is the 3-O-glucuronide, which lacks iron binding capability. Peak serum concentration of the glucuronide occurs 2 to 4 hours after administration of deferiprone in fasting subjects.
More than 90% of deferiprone is eliminated from plasma within 5 to 6 hours of ingestion. Following oral administration, 75% to 90% is recovered in the urine in the first 24 hours, primarily as metabolite.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with deferiprone. However, in view of the genotoxicity results, and the findings of mammary gland hyperplasia and mammary gland tumors in rats treated with deferiprone in the 52-week toxicology study, tumor formation in carcinogenicity studies must be regarded as likely.
Deferiprone was positive in a mouse lymphoma cell assay in vitro. Deferiprone was clastogenic in an in vitro chromosomal aberration test in mice and in a chromosomal aberration test in Chinese Hamster Ovary cells. Deferiprone given orally or intraperitoneally was clastogenic in a bone marrow micronucleus assay in non-iron-loaded mice. A micronucleus test was also positive when mice predosed with iron dextran were treated with deferiprone. Deferiprone was not mutagenic in the Ames bacterial reverse mutation test.
A fertility and early embryonic development study of deferiprone was conducted in rats. Sperm counts, motility and morphology were unaffected by treatment with deferiprone. There were no effects observed on male or female fertility or reproductive function at the highest dose which was 25% of the MRHD based on body surface area.
-
14 CLINICAL STUDIES
In a prospective, planned, pooled analysis of patients from several studies, the efficacy of deferiprone was assessed in transfusion-dependent iron overload patients in whom previous iron chelation therapy had failed or was considered inadequate due to poor tolerance. The main criterion for chelation failure was serum ferritin > 2,500 mcg/L before treatment with deferiprone. Deferiprone therapy (35-99 mg/kg/day) was considered successful in individual patients who experienced a ≥ 20% decline in serum ferritin within one year of starting therapy.
Data from a total of 236 patients were analyzed. Of the 224 patients with thalassemia who received deferiprone monotherapy and were eligible for serum ferritin analysis, 105 (47%) were male and 119 (53%) were female. The mean age of these patients was 18.2 years.
For the patients in the analysis, the endpoint of at least a 20% reduction in serum ferritin was met in 50% (of 236 subjects), with a 95% confidence interval of 43% to 57%.
A small number of patients with thalassemia and iron overload were assessed by measuring the change in the number of milliseconds (ms) in the cardiac MRI T2* value before and after treatment with deferiprone for one year. There was an increase in cardiac MRI T2* from a mean at baseline of 11.8 ± 4.9 ms to a mean of 15.1 ± 7.0 ms after approximately one year of treatment. The clinical significance of this observation is not known.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Deferiprone Tablets, 500 mg are white to pinkish-white, capsule-shaped tablets; scored on one side, engraved "T" on the left of the score line and "5" on the right and plain on the other side. They are provided in a 100 count HDPE bottle with a child-resistant cap.
500 mg tablets, 100 tablets NDC: 51672-4196-1
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (Medication Guide)
- Inform patients of the risks of developing agranulocytosis and instruct them to immediately interrupt therapy and report to their physician if they experience any symptoms of infection such as fever, sore throat or flu-like symptoms.
- Advise patients that the amount of deferiprone prescribed is based on body weight and on the therapeutic goal (reduction or stabilization of the body iron load).
- Advise patients to take the first dose of deferiprone in the morning, the second dose at midday, and the third dose in the evening. Clinical experience suggests that taking deferiprone with meals may reduce nausea. If a dose of this medicine has been missed, take as soon as possible. However, if it is almost time for the next dose, skip the missed dose and go back to the regular dosing schedule. Do not catch-up or double doses.
- Advise patients to contact their physician in the event of overdose.
- Inform patients that their urine might show a reddish/brown discoloration due to the excretion of the iron-deferiprone complex. This is a very common sign of the desired effect of deferiprone, and it is not harmful.
- Counsel women of reproductive potential to avoid pregnancy while taking deferiprone. Advise patients to immediately notify their physician if they become pregnant, or if they plan to become pregnant during therapy.
- Inform patients that they should not breast feed while taking deferiprone.
- SPL UNCLASSIFIED SECTION
-
Medication Guide
Deferiprone (de-fer-ip-rone) Tablets
Read this Medication Guide before you start taking deferiprone tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about deferiprone tablets?
Deferiprone can cause serious side effects, including a very low white blood cell count in your blood. One type of white blood cell that is important for fighting infections is called a neutrophil. If your neutrophil count is low (neutropenia), you may be at risk of developing a serious infection that can lead to death. Neutropenia is common with deferiprone and can become severe in some people. Severe neutropenia is known as agranulocytosis. If you develop agranulocytosis, you will be at risk of developing serious infections that can lead to death.
Your healthcare provider should do a blood test before you start deferiprone and weekly during treatment to check your neutrophil count. If you develop neutropenia, your healthcare provider should check your blood counts every day until your white blood cell count improves.
Stop taking deferiprone tablets and get medical help right away if you develop any of these symptoms of infection:
- fever
- sore throat or mouth sores
- flu-like symptoms
- chills and severe shaking
See "What are the possible side effects of deferiprone tablets?" for more information about side effects.
What are deferiprone tablets?
Deferiprone tablets are a prescription medicine used to treat people with thalassemia syndromes who have iron overload from blood transfusions, when current iron removal (chelation) therapy does not work well enough.
It is not known if deferiprone tablets are safe and effective:
- to treat iron overload due to blood transfusions in people with any other type of anemia that is long lasting (chronic)
- in children
- in people with severe liver problems
Who should not take deferiprone tablets?
Do not take deferiprone tablets if you are allergic to deferiprone or any of the ingredients in deferiprone tablets. See the end of this Medication Guide for a complete list of ingredients in deferiprone tablets.
What should I tell my healthcare provider before taking deferiprone tablets?
Before you take deferiprone tablets, tell your healthcare provider if you:
- have liver problems
- have any other medical conditions.
- are pregnant or plan to become pregnant. Deferiprone can harm your unborn baby. You should avoid becoming pregnant while taking deferiprone. Tell your healthcare provider right away if you become pregnant or plan to become pregnant while taking deferiprone.
- are breastfeeding or plan to breastfeed. It is not known if deferiprone passes into your breast milk. You and your healthcare provider should decide if you will take deferiprone or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements.
Especially tell your healthcare provider if you take:
- other medicines that can cause a lowering of your neutrophil count
- antacids or mineral supplements that contain: iron, aluminum, and zinc. Allow at least 4 hours between taking deferiprone tablets and any of these products.
Ask your healthcare provider or pharmacist if your medicine is one that is listed above.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take deferiprone tablets?
- Take deferiprone tablets exactly as prescribed by your healthcare provider. Do not change your dose of deferiprone unless your healthcare provider tells you to.
- Your healthcare provider will tell you how many deferiprone tablets to take.
- Deferiprone tablets are taken 3 times each day. Take your first dose in the morning, the second dose at mid-day, and third dose in the evening.
- You can take deferiprone tablets with or without food.
- Taking deferiprone with meals may help reduce nausea.
- If you take too much deferiprone, call your healthcare provider.
- If you do miss a dose take it as soon as you remember. If it is almost time for your next dose, skip the missed dose and then continue with your regular schedule. Do not try to catch-up or take 2 doses at the same time to make up for a missed dose.
What are the possible side effects of deferiprone tablets?
Deferiprone can cause serious side effects, including:
- See "What is the most important information I should know about deferiprone tablets?"
- Increased liver enzyme levels in your blood. Your healthcare provider should do monthly blood test to check your liver function during treatment with deferiprone.
The most common side effects of deferiprone include:
- reddish-brown colored urine. This is not harmful and is expected when you are taking deferiprone.
- nausea
- vomiting
- stomach-area (abdominal) pain
- joint pain
- low neutrophil count. See "What is the most important information I should know about deferiprone tablets?"
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of deferiprone. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store deferiprone tablets?
Store deferiprone tablets at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature].
Keep deferiprone tablets and all medicines out of the reach of children.
General information about the safe and effective use of deferiprone tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use deferiprone tablets for a condition for which it was not prescribed. Do not give deferiprone tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about deferiprone tablets. If you would like more information, talk to your healthcare provider. You can ask your pharmacist or healthcare provider for information about deferiprone tablets that is written for health professionals.
For more information call 1-866-923-4914.
What are the ingredients in deferiprone tablets?
Active ingredients: deferiprone
Inactive ingredients: colloidal silicon dioxide, magnesium stearate, and microcrystalline cellulose.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Mfd. by: Taro Pharmaceutical Industries Ltd., Haifa Bay, Israel, 2624761
Dist. by: Taro Pharmaceuticals U.S.A., Inc., Hawthorne, NY, 10532
Issued: October 2019
20872-1019-0 -
PRINCIPAL DISPLAY PANEL - 500 mg Tablet Bottle Label
NDC: 51672-4196-1
100 Tablets
Deferiprone
Tablets 500 mgDISPENSE WITH THE ACCOMPANYING
MEDICATION GUIDETARO
Rx only
-
INGREDIENTS AND APPEARANCE
DEFERIPRONE
deferiprone tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 51672-4196 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Deferiprone (UNII: 2BTY8KH53L) (Deferiprone - UNII:2BTY8KH53L) Deferiprone 500 mg Inactive Ingredients Ingredient Name Strength Microcrystalline Cellulose (UNII: OP1R32D61U) Silicon Dioxide (UNII: ETJ7Z6XBU4) Magnesium Stearate (UNII: 70097M6I30) Product Characteristics Color WHITE (White to Pinkish-White) Score 2 pieces Shape OVAL Size 18mm Flavor Imprint Code T;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 51672-4196-1 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/08/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA208800 02/08/2019 Labeler - Taro Pharmaceuticals U.S.A., Inc. (145186370) Establishment Name Address ID/FEI Business Operations Taro Pharmaceutical Industries, Ltd. 600072078 MANUFACTURE(51672-4196)
Trademark Results [Deferiprone]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 DEFERIPRONE 97303966 not registered Live/Pending |
Beer, Kenneth 2022-03-09 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.