BIKTARVY- bictegravir sodium, emtricitabine, and tenofovir alafenamide fumarate tablet
Biktarvy by
Drug Labeling and Warnings
Biktarvy by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BIKTARVY safely and effectively. See full prescribing information for BIKTARVY.
BIKTARVY ® (bictegravir, emtricitabine, and tenofovir alafenamide) tablets, for oral use
Initial U.S. Approval: 2018WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Closely monitor hepatic function in these patients. If appropriate, anti-hepatitis B therapy may be warranted. (5.1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BIKTARVY is a three-drug combination of bictegravir (BIC), a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI), and emtricitabine (FTC) and tenofovir alafenamide (TAF), both HIV-1 nucleoside analog reverse transcriptase inhibitors (NRTIs), and is indicated as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 25 kg who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of BIKTARVY. (1)
DOSAGE AND ADMINISTRATION
- Testing: Prior to or when initiating BIKTARVY test for hepatitis B virus infection. Prior to or when initiating BIKTARVY, and during treatment, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. (2.1)
- Recommended dosage: One tablet taken once daily with or without food in patients weighing at least 25 kg. (2.2)
- Renal impairment: BIKTARVY is not recommended in patients with estimated creatinine clearance below 30 mL per minute. (2.3)
- Hepatic impairment: BIKTARVY is not recommended in patients with severe hepatic impairment. (2.4)
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg of bictegravir (equivalent to 52.5 mg of bictegravir sodium), 200 mg of emtricitabine, and 25 mg of tenofovir alafenamide (equivalent to 28 mg of tenofovir alafenamide fumarate). (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.3)
- New onset or worsening renal impairment: Assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein when initiating BIKTARVY and during therapy as clinically appropriate in all patients. Also assess serum phosphorus in patients with chronic kidney disease. (5.4)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than or equal to 5%, all grades) are diarrhea, nausea, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-GILEAD-5 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Testing When Initiating and During Treatment with BIKTARVY
2.2 Recommended Dosage
2.3 Not Recommended in Patients with Severe Renal Impairment
2.4 Not Recommended in Patients with Severe Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
5.2 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
5.3 Immune Reconstitution Syndrome
5.4 New Onset or Worsening Renal Impairment
5.5 Lactic Acidosis/Severe Hepatomegaly with Steatosis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Other Antiretroviral Medications
7.2 Potential for BIKTARVY to Affect Other Drugs
7.3 Potential Effect of Other Drugs on One or More Components of BIKTARVY
7.4 Drugs Affecting Renal Function
7.5 Established and Potentially Significant Drug Interactions
7.6 Drugs without Clinically Significant Interactions with BIKTARVY
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
14.2 Clinical Trial Results in HIV-1 Subjects with No Antiretroviral Treatment History
14.3 Clinical Trial Results in HIV-1 Virologically-Suppressed Subjects Who Switched to BIKTARVY
14.4 Clinical Trial Results in HIV-1 Infected Pediatric Subjects Between the Ages of 6 to Less than 18 Years
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: POST TREATMENT ACUTE EXACERBATION OF HEPATITIS B
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing emtricitabine (FTC) and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY.
Closely monitor hepatic function with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue BIKTARVY. If appropriate, anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
BIKTARVY is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 25 kg who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of BIKTARVY.
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing When Initiating and During Treatment with BIKTARVY
Prior to or when initiating BIKTARVY, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.4)].
2.2 Recommended Dosage
BIKTARVY is a three-drug fixed dose combination product containing 50 mg of bictegravir (BIC), 200 mg of emtricitabine (FTC), and 25 mg of tenofovir alafenamide (TAF). The recommended dosage of BIKTARVY is one tablet taken orally once daily with or without food in adults and pediatric patients weighing at least 25 kg [see Clinical Pharmacology (12.3)].
2.3 Not Recommended in Patients with Severe Renal Impairment
BIKTARVY is not recommended in patients with estimated creatinine clearance below 30 mL per minute [see Use in Specific Populations (8.6)].
2.4 Not Recommended in Patients with Severe Hepatic Impairment
BIKTARVY is not recommended in patients with severe hepatic impairment (Child-Pugh Class C) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
Each BIKTARVY tablet contains 50 mg of bictegravir (BIC) (equivalent to 52.5 mg of bictegravir sodium), 200 mg of emtricitabine (FTC), and 25 mg of tenofovir alafenamide (TAF) (equivalent to 28 mg of tenofovir alafenamide fumarate). The tablets are purplish brown, capsule-shaped, film-coated, and debossed with "GSI" on one side and "9883" on the other side.
-
4 CONTRAINDICATIONS
BIKTARVY is contraindicated to be co-administered with:
- dofetilide due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events [see Drug Interactions (7.5)].
- rifampin due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to BIKTARVY [see Drug Interactions (7.5)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Severe Acute Exacerbation of Hepatitis B in Patients Coinfected with HIV-1 and HBV
Patients with HIV-1 should be tested for the presence of chronic hepatitis B virus (HBV) infection before or when initiating antiretroviral therapy [see Dosage and Administration (2.1)].
Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in patients who are coinfected with HIV-1 and HBV and have discontinued products containing FTC and/or tenofovir disoproxil fumarate (TDF), and may occur with discontinuation of BIKTARVY. Patients coinfected with HIV-1 and HBV who discontinue BIKTARVY should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.2 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of BIKTARVY with certain other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Contraindications (4) and Drug Interactions (7.5)]:
- Loss of therapeutic effect of BIKTARVY and possible development of resistance.
- Possible clinically significant adverse reactions from greater exposures of concomitant drugs.
See Table 3 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during BIKTARVY therapy; review concomitant medications during BIKTARVY therapy; and monitor for the adverse reactions associated with the concomitant drugs.
5.3 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections [such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis], which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.4 New Onset or Worsening Renal Impairment
Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of tenofovir prodrugs in both animal toxicology studies and human trials. In clinical trials of BIKTARVY, there have been no cases of Fanconi syndrome or Proximal Renal Tubulopathy (PRT). In clinical trials of BIKTARVY in subjects with no antiretroviral treatment history with eGFRs greater than 30 mL per minute, and in virologically suppressed subjects switched to BIKTARVY with eGFRs greater than 50 mL per minute, renal serious adverse events were encountered in less than 1% of subjects treated with BIKTARVY through Week 48 [see Adverse Reactions (6.1)]. BIKTARVY is not recommended in patients with estimated creatinine clearance below 30 mL per minute.
Patients taking tenofovir prodrugs who have impaired renal function and those taking nephrotoxic agents including non-steroidal anti-inflammatory drugs are at increased risk of developing renal-related adverse reactions.
Prior to or when initiating BIKTARVY, and during treatment with BIKTARVY, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients as clinically appropriate. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue BIKTARVY in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
5.5 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including emtricitabine, a component of BIKTARVY, and tenofovir DF, another prodrug of tenofovir, alone or in combination with other antiretrovirals. Treatment with BIKTARVY should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B [see Warnings and Precautions (5.1)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.3)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.4)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.5)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials in Adults with No Antiretroviral Treatment History
The primary safety assessment of BIKTARVY was based on Week 48 data from two randomized, double-blind, active-controlled trials, Trial 1489 and Trial 1490, that enrolled 1274 HIV-1 infected adult subjects with no antiretroviral treatment history. A total of 634 subjects received one tablet of BIKTARVY once daily [see Clinical Studies (14.2)].
The most common adverse reactions (all Grades) reported in at least 5% of subjects in the BIKTARVY group in either Trial 1489 or Trial 1490 were diarrhea, nausea, and headache. The proportion of subjects who discontinued treatment with BIKTARVY, abacavir [ABC]/dolutegravir [DTG]/ lamivudine [3TC]), or DTG + FTC/TAF, due to adverse events, regardless of severity, was 1%, 1%, and <1%, respectively. Table 1 displays the frequency of adverse reactions (all Grades) greater than or equal to 2% in the BIKTARVY group.
Table 1 Adverse Reactions* (All Grades) Reported in ≥ 2% of HIV-1 Infected Adults with No Antiretroviral Treatment History Receiving BIKTARVY in Trials 1489 or 1490 (Week 48 analysis) Trial 1489 Trial 1490 Adverse Reactions BIKTARVY
N=314ABC/DTG/3TC
N=315BIKTARVY
N=320DTG + FTC/TAF
N=325- * Frequencies of adverse reactions are based on all adverse events attributed to trial drugs by the investigator. No adverse reactions of Grade 2 or higher occurred in ≥ 1% of subjects treated with BIKTARVY.
Diarrhea 6% 4% 3% 3% Nausea 5% 17% 3% 5% Headache 5% 5% 4% 3% Fatigue 3% 3% 2% 2% Abnormal dreams 3% 3% <1% 1% Dizziness 2% 3% 2% 1% Insomnia 2% 3% 2% <1% Additional adverse reactions (all Grades) occurring in less than 2% of subjects administered BIKTARVY in Trials 1489 and 1490 included vomiting, flatulence, dyspepsia, abdominal pain, rash, and depression.
Suicidal ideation, suicide attempt, and depression suicidal occurred in <1% of subjects administered BIKTARVY; all events were serious and primarily occurred in subjects with a preexisting history of depression, prior suicide attempt or psychiatric illness.
The majority (87%) of adverse events associated with BIKTARVY were Grade 1.
Clinical Trials in Virologically Suppressed Adults
The safety of BIKTARVY in virologically-suppressed adults was based on Week 48 data from 282 subjects in a randomized, double-blind, active-controlled trial (Trial 1844) in which virologically-suppressed subjects were switched from either DTG + ABC/3TC or ABC/DTG/3TC to BIKTARVY; and Week 48 data from 290 subjects in an open-label, active-controlled trial in which virologically-suppressed subjects were switched from a regimen containing atazanavir (ATV) (given with cobicistat or ritonavir) or darunavir (DRV) (given with cobicistat or ritonavir) plus either FTC/TDF or ABC/3TC, to BIKTARVY (Trial 1878). Overall, the safety profile in virologically suppressed adult subjects in Trials 1844 and 1878 was similar to that in subjects with no antiretroviral treatment history [see Clinical Studies (14.3)].
Laboratory Abnormalities
The frequency of laboratory abnormalities (Grades 3–4) occurring in at least 2% of subjects receiving BIKTARVY in Trials 1489 and 1490 are presented in Table 2.
Table 2 Laboratory Abnormalities (Grades 3–4) Reported in ≥ 2% of Subjects Receiving BIKTARVY in Trials 1489 or 1490 (Week 48 analysis) Trial 1489 Trial 1490 Laboratory Parameter Abnormality* BIKTARVY
N=314ABC/DTG/3TC
N=315BIKTARVY
N=320DTG + FTC/TAF
N=325ULN = Upper limit of normal - * Frequencies are based on treatment-emergent laboratory abnormalities.
Amylase (>2.0 × ULN) 2% 2% 2% 2% ALT (>5.0 × ULN) 1% 1% 2% 1% AST (>5.0 × ULN) 2% 1% 1% 3% Creatine Kinase (≥10.0 × ULN) 4% 3% 4% 2% Neutrophils (<750 mm3) 2% 3% 2% 1% LDL-cholesterol (fasted) (>190 mg/dL) 2% 3% 3% 3% Changes in Serum Creatinine: BIC has been shown to increase serum creatinine due to inhibition of tubular secretion of creatinine without affecting renal glomerular function [see Clinical Pharmacology (12.2)]. Increases in serum creatinine occurred by Week 4 of treatment and remained stable through Week 48. In Trials 1489 and 1490, median (Q1, Q3) serum creatinine increased by 0.10 (0.03, 0.17) mg per dL from baseline to Week 48 in the BIKTARVY group and was similar to the comparator groups who received ABC/DTG/3TC, or DTG + FTC/TAF. There were no discontinuations due to renal adverse events through Week 48 in BIKTARVY clinical trials.
Changes in Bilirubin: In Trials 1489 and 1490, total bilirubin increases were observed in 12% of subjects administered BIKTARVY through Week 48. Increases were primarily Grade 1 (1.0 to 1.5 × ULN) (9%) and Grade 2 (1.5 to 2.5 × ULN) (3%). Graded bilirubin increases in the ABC/DTG/3TC, and DTG + FTC/TAF groups, were 4% and 6%, respectively. Increases were primarily Grade 1 (3% ABC/DTG/3TC and 5% DTG + FTC/TAF) or Grade 2 (1% ABC/DTG/3TC and 1% DTG + FTC/TAF). There were no discontinuations due to hepatic adverse events through Week 48 in BIKTARVY clinical studies.
Clinical Trials in Pediatric Subjects
The safety of BIKTARVY was evaluated in HIV-1 infected virologically-suppressed subjects between the ages of 12 to less than 18 years and weighing at least 35 kg (N=50) through Week 48 (cohort 1), and in virologically-suppressed subjects between the ages of 6 to less than 12 years and weighing at least 25 kg (N=50) through Week 24 (cohort 2) in an open label clinical trial (Trial 1474) [see Clinical Studies (14.4)]. No new adverse reactions or laboratory abnormalities were identified compared to those observed in adults. Adverse reactions were reported in 10% of pediatric subjects. The majority (85%) of adverse reactions were Grade 1. No Grade 3 or 4 adverse reactions were reported. The adverse reaction reported by more than one subject (regardless of severity) was abdominal pain (n=2). One subject (1%) had Grade 2 adverse reactions of insomnia and anxiety that led to discontinuation of BIKTARVY. The other adverse reactions that occurred in single subjects were similar to those seen in adults.
6.2 Postmarketing Experience
The following events have been identified during post approval use of products containing TAF. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
7 DRUG INTERACTIONS
7.1 Other Antiretroviral Medications
Because BIKTARVY is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended [see Indications and Usage (1)]. Comprehensive information regarding potential drug-drug interactions with other antiretroviral medications is not provided because the safety and efficacy of concomitant HIV-1 antiretroviral therapy is unknown.
7.2 Potential for BIKTARVY to Affect Other Drugs
BIC inhibits organic cation transporter 2 (OCT2) and multidrug and toxin extrusion transporter 1 (MATE1) in vitro. Coadministration of BIKTARVY with drugs that are substrates of OCT2 and MATE1 (e.g., dofetilide) may increase their plasma concentrations (see Table 3).
7.3 Potential Effect of Other Drugs on One or More Components of BIKTARVY
BIC is a substrate of CYP3A and UGT1A1. A drug that is a strong inducer of CYP3A and also an inducer of UGT1A1 can substantially decrease the plasma concentrations of BIC which may lead to loss of therapeutic effect of BIKTARVY and development of resistance [see Clinical Pharmacology (12.3)].
The use of BIKTARVY with a drug that is a strong inhibitor of CYP3A and also an inhibitor of UGT1A1 may significantly increase the plasma concentrations of BIC.
TAF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Co-administration of drugs that inhibit P-gp and BCRP may increase the absorption and plasma concentrations of TAF [see Clinical Pharmacology (12.3)]. Co-administration of drugs that induce P-gp activity are expected to decrease the absorption of TAF, resulting in decreased plasma concentration of TAF, which may lead to loss of therapeutic effect of BIKTARVY and development of resistance (see Table 3).
7.4 Drugs Affecting Renal Function
Because FTC and tenofovir are primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion, coadministration of BIKTARVY with drugs that reduce renal function or compete for active tubular secretion may increase concentrations of FTC, tenofovir, and other renally eliminated drugs and this may increase the risk of adverse reactions. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.4)].
7.5 Established and Potentially Significant Drug Interactions
Table 3 provides a listing of established or potentially clinically significant drug interactions with recommended prevention or management strategies. The drug interactions described are based on studies conducted with either BIKTARVY, the components of BIKTARVY (BIC, FTC, and TAF) as individual agents, or are drug interactions that may occur with BIKTARVY [see Contraindications (4), Warnings and Precautions (5.2), and Clinical Pharmacology (12.3)].
Table 3 Established and Potentially Significant* Drug Interactions: Alteration in Regimen May be Recommended Concomitant Drug Class: Drug Name Effect on Concentration† Clinical Comment - * Table is not all inclusive.
- † ↑ = Increase, ↓ = Decrease.
- ‡ Drug-drug interaction study was conducted with either BIKTARVY or its components as individual agents.
- § Strong inducer of CYP3Aand P-gp, and inducer of UGT1A1.
- ¶ The induction potency of St. John's wort may vary widely based on preparation.
Antiarrhythmics:
dofetilide↑ Dofetilide Coadministration is contraindicated due to the potential for serious and/or life-threatening events associated with dofetilide therapy [see Contraindications (4)]. Anticonvulsants:
carbamazepine‡
oxcarbazepine
phenobarbital
phenytoin↓ BIC
↓ TAFCoadministration with alternative anticonvulsants should be considered. Antimycobacterials:
rifabutin‡
rifampin‡,§
rifapentine↓ BIC
↓ TAFCoadministration with rifampin is contraindicated due to the effect of rifampin on the BIC component of BIKTARVY [see Contraindications (4)].
Coadministration with rifabutin or rifapentine is not recommended.Herbal Products:
St. John's wort¶↓ BIC
↓ TAFCoadministration with St. John's wort is not recommended. Medications or oral supplements containing polyvalent cations (e.g., Mg, Al, Ca, Fe):
Calcium or iron supplements‡
Cation-containing antacids or laxatives‡
Sucralfate
Buffered medications↓ BIC Antacids containing Al/Mg:
BIKTARVY can be taken at least 2 hours before or 6 hours after taking antacids containing Al/Mg.
Routine administration of BIKTARVY together with, or 2 hours after, antacids containing Al/Mg is not recommended.
Supplements or antacids containing Calcium or Iron:
BIKTARVY and supplements or antacids containing calcium or iron can be taken together with food.
Routine administration of BIKTARVY under fasting conditions together with, or 2 hours after, supplements or antacids containing calcium or iron is not recommended.Metformin ↑ Metformin Refer to the prescribing information of metformin for assessing the benefit and risk of concomitant use of BIKTARVY and metformin. 7.6 Drugs without Clinically Significant Interactions with BIKTARVY
Based on drug interaction studies conducted with BIKTARVY or the components of BIKTARVY, no clinically significant drug interactions have been observed when BIKTARVY is combined with the following drugs: ethinyl estradiol, ledipasvir/sofosbuvir, midazolam, norgestimate, sertraline, sofosbuvir, sofosbuvir/velpatasvir, and sofosbuvir/velpatasvir/voxilaprevir.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to BIKTARVY during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
There are insufficient human data on the use of BIKTARVY during pregnancy to inform a drug-associated risk of birth defects and miscarriage. Dolutegravir, another integrase inhibitor, has been associated with neural tube defects (NTDs) (see Data). Discuss the benefit-risk of using BIKTARVY with individuals of childbearing potential, particularly if pregnancy is being planned. Bictegravir (BIC) and tenofovir alafenamide (TAF) use in women during pregnancy has not been evaluated; however, emtricitabine (FTC) use during pregnancy has been evaluated in a limited number of women reported to the APR. Available data from the APR show no difference in the overall risk of major birth defects for FTC compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data). The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in the clinically recognized pregnancies in the U.S. general population is 15–20%. Methodological limitations of the APR include the use of MACDP as the external comparator group. The MACDP population is not disease-specific, evaluates women and infants from a limited geographic area, and does not include outcomes for births that occurred at less than 20 weeks gestation.
In animal reproduction studies, no evidence of adverse developmental outcomes was observed with the components of BIKTARVY at exposures that were either not maternally toxic (rabbits) or greater than (rats and mice) those in humans at the recommended human dose (RHD) (see Data). During organogenesis, systemic exposures (AUC) to BIC were approximately 36 (rats) and 0.6 times (rabbits), to FTC were approximately 60 (mice) and 108 times (rabbits), and to TAF were approximately 2 (rats) and 78 times (rabbits) the exposure at the RHD of BIKTARVY. In rat pre/postnatal development studies, maternal systemic exposures (AUC) were 30 times (BIC), 60 times (FTC), and 19 times (TDF) the exposures of each component in humans at the RHD.
Data
Human Data
Bictegravir: Data from an observational study in Botswana showed that dolutegravir, another integrase inhibitor, was associated with increased risk of neural tube defects when administered at the time of conception and in early pregnancy. Data available to date from other sources including the APR, clinical trials, and postmarketing data are insufficient to address this risk with BIC.
Emtricitabine: Based on prospective reports to the APR of 3,406 exposures to FTC-containing regimens during pregnancy resulting in live births (including 2,326 exposed in the first trimester and 1,080 exposed in the second/third trimester), there was no difference between FTC and overall birth defects compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. The prevalence of birth defects in live births was 2.3% (95% CI: 1.7% to 3.0%) with first trimester exposure to FTC-containing regimens and 2.0% (95% CI: 1.3% to 3.1%) with the second/third trimester exposure to FTC-containing regimens.
Animal Data
Bictegravir: BIC was administered orally to pregnant rats (5, 30, or 300 mg/kg/day) and rabbits (100, 300, or 1000 mg/kg/day) on gestation days 7 through 17, and 7 through 19, respectively. No adverse embryo-fetal effects were observed in rats and rabbits at BIC exposures (AUC) of up to approximately 36 (rats) and 0.6 (rabbits) times the exposure in humans at the RHD of BIKTARVY. Spontaneous abortion, increased clinical signs [fecal changes, thin body, and cold-to-touch], and decreased body weight were observed at a maternally toxic dose in rabbits (1000 mg/kg/day; approximately 1.4 times higher than human exposure at the RHD).
In a pre/postnatal development study, BIC was administered orally to pregnant rats (up to 300 mg/kg/day) from gestation days 6 to lactation/post-partum day 24. No significant adverse effects were observed in the offspring exposed daily from before birth (in utero) through lactation at maternal and pup exposures (AUC) of approximately 30 and 11 times higher, respectively, than human exposures at the RHD.
Emtricitabine: FTC was administered orally to pregnant mice (250, 500, or 1000 mg/kg/day) and rabbits (100, 300, or 1000 mg/kg/day) through organogenesis (on gestation days 6 through 15, and 7 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with emtricitabine in mice at exposures approximately 60 times higher and in rabbits at approximately 108 times higher than human exposures at the RHD.
In a pre/postnatal development study with FTC, mice were administered doses up to 1000 mg/kg/day; no significant adverse effects directly related to drug were observed in the offspring exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the RHD.
Tenofovir alafenamide: TAF was administered orally to pregnant rats (25, 100, or 250 mg/kg/day) and rabbits (10, 30, or 100 mg/kg/day) through organogenesis (on gestation days 6 through 17, and 7 through 20, respectively). No adverse embryo-fetal effects were observed in rats and rabbits at TAF exposures of approximately 2 (rats) and 78 (rabbits) times higher than the exposure in humans at the recommended daily dose of BIKTARVY. TAF is rapidly converted to tenofovir; the observed tenofovir exposure in rats and rabbits were 55 (rats) and 86 (rabbits) times higher than human tenofovir exposures at the RHD. Since TAF is rapidly converted to tenofovir and lower tenofovir exposures in rats and mice were observed after TAF administration compared to TDF administration, a pre/postnatal development study in rats was conducted only with TDF. Doses up to 600 mg/kg/day were administered through lactation; no adverse effects were observed in the offspring on gestation day 7 [and lactation day 20] at tenofovir exposures of approximately 12 [19] times higher than the exposures in humans at the RHD of BIKTARVY.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1-infected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection.
It is not known whether BIKTARVY or all of the components of BIKTARVY are present in human breast milk, affects human milk production, or has effects on the breastfed infant. Based on published data, FTC has been shown to be present in human breast milk. BIC was detected in the plasma of nursing rat pups likely due to the presence of BIC in milk, and tenofovir has been shown to be present in the milk of lactating rats and rhesus monkeys after administration of TDF (see Data). It is unknown if TAF is present in animal milk.
Because of the potential for 1) HIV transmission (in HIV-negative infants); 2) developing viral resistance (in HIV-positive infants); and 3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are receiving BIKTARVY.
Data
Animal Data
Bictegravir: BIC was detected in the plasma of nursing rat pups in the pre/postnatal development study (post-natal day 10), likely due to the presence of BIC in milk.
Tenofovir alafenamide: Studies in rats and monkeys have demonstrated that tenofovir is secreted in milk. Tenofovir was excreted into the milk of lactating rats following oral administration of TDF (up to 600 mg/kg/day) at up to approximately 24% of the median plasma concentration in the highest dosed animals at lactation day 11. Tenofovir was excreted into the milk of lactating monkeys following a single subcutaneous (30 mg/kg) dose of tenofovir at concentrations up to approximately 4% of plasma concentration, resulting in exposure (AUC) of approximately 20% of plasma exposure.
8.4 Pediatric Use
The safety and effectiveness of BIKTARVY for the treatment of HIV-1 infection was established in pediatric patients with body weight greater than or equal to 25 kg [see Indications and Usage (1) and Dosage and Administration (2.2)].
Use of BIKTARVY in pediatric patients between the ages of 6 to less than 18 years and weighing at least 25 kg is supported by trials in adults and by an open-label trial in virologically-suppressed pediatric subjects aged 12 to less than 18 years and weighing at least 35 kg receiving BIKTARVY through Week 48 (cohort 1 of Trial 1474, N=50) and in virologically-suppressed pediatric subjects aged 6 to less than 12 years and weighing at least 25 kg receiving BIKTARVY through Week 24 (cohort 2 of Trial 1474, N=50). The safety and efficacy of BIKTARVY in these pediatric subjects was similar to that in adults, and there was no clinically significant change in exposure for the components of BIKTARVY [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.4)].
Safety and effectiveness of BIKTARVY in pediatric patients weighing less than 25 kg have not been established.
8.5 Geriatric Use
Clinical trials of BIKTARVY did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
BIKTARVY is not recommended in patients with severe renal impairment (estimated creatinine clearance (CLcr) below 30 mL per minute, estimated by Cockcroft-Gault (C-G). No dosage adjustment of BIKTARVY is recommended in patients with CLcr greater than or equal to 30 mL per minute [see Dosage and Administration (2.3)].
8.7 Hepatic Impairment
No dosage adjustment of BIKTARVY is recommended in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. BIKTARVY has not been studied in patients with severe hepatic impairment (Child-Pugh Class C). Therefore, BIKTARVY is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
No data are available on overdose of BIKTARVY in patients. If overdose occurs, monitor the patient for evidence of toxicity. Treatment of overdose with BIKTARVY consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient.
Hemodialysis treatment removes approximately 30% of the FTC dose over a 3-hour dialysis period starting within 1.5 hours of FTC dosing (blood flow rate of 400 mL per minute and a dialysate flow rate of 600 mL per minute). It is not known whether FTC can be removed by peritoneal dialysis.
Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%.
-
11 DESCRIPTION
BIKTARVY (bictegravir, emtricitabine, and tenofovir alafenamide) is a fixed dose combination tablet containing bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) for oral administration.
- BIC is an integrase strand transfer inhibitor (INSTI).
- FTC, a synthetic nucleoside analog of cytidine, is an HIV nucleoside analog reverse transcriptase inhibitor (HIV NRTI).
- TAF, an HIV NRTI, is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate.
Each tablet contains 50 mg of BIC (equivalent to 52.5 mg of bictegravir sodium), 200 mg of FTC, and 25 mg of TAF (equivalent to 28 mg of tenofovir alafenamide fumarate) and the following inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose. The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
Bictegravir: The chemical name of bictegravir sodium is 2,5-Methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-, sodium salt (1:1), (2R,5S,13aR)-.
Bictegravir sodium has a molecular formula of C21H17F3N3NaO5 and a molecular weight of 471.4 and has the following structural formula:
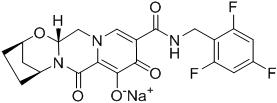
Bictegravir sodium is an off-white to yellow solid with a solubility of 0.1 mg per mL in water at 20 °C.
Emtricitabine: The chemical name of FTC is 4-amino-5-fluoro-1-(2R-hydroxymethyl-1,3-oxathiolan-5S-yl)-(1H)-pyrimidin-2-one. FTC is the (-)enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5 position.
FTC has a molecular formula of C8H10FN3O3S and a molecular weight of 247.2 and has the following structural formula:
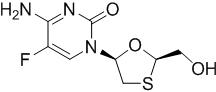
FTC is a white to off-white powder with a solubility of approximately 112 mg per mL in water at 25 °C.
Tenofovir alafenamide: The chemical name of tenofovir alafenamide fumarate drug substance is L-alanine, N-[(S)-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]phenoxyphosphinyl]-, 1-methylethyl ester, (2E)-2-butenedioate (2:1).
Tenofovir alafenamide fumarate has an empirical formula of C21H29O5N6P∙½(C4H4O4) and a formula weight of 534.5 and has the following structural formula:
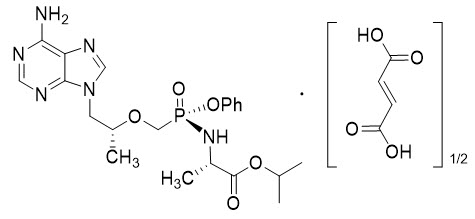
Tenofovir alafenamide fumarate is a white to off-white or tan powder with a solubility of 4.7 mg per mL in water at 20 °C.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
BIKTARVY is a fixed dose combination of antiretroviral drugs bictegravir (BIC), emtricitabine (FTC), and tenofovir alafenamide (TAF) [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a thorough QT/QTc trial in 48 healthy subjects, BIC at doses 1.5 and 6 times the recommended dose did not affect the QT/QTc interval and did not prolong the PR interval. In a thorough QT/QTc trial in 48 healthy subjects, TAF at the recommended dose or at a dose 5 times the recommended dose, did not affect the QT/QTc interval and did not prolong the PR interval. The effect of FTC on the QT interval is not known.
Effects on Serum Creatinine
Mean change from baseline in serum creatinine in healthy subjects who received BIC 75 mg (1.5 times the approved recommended dosage) once daily with food for 14 days was 0.1 mg per dL on Days 7 and 14 compared to placebo. BIC did not have a significant effect on the estimated creatinine clearance or on the actual glomerular filtration rate (determined by the clearance of probe drug, iohexol).
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of BIKTARVY components are provided in Table 4. The multiple dose PK parameters of BIKTARVY components (based on population pharmacokinetic analysis) are provided in Table 5.
Table 4 Pharmacokinetic Properties of the Components of BIKTARVY Bictegravir (BIC) Emtricitabine (FTC) Tenofovir Alafenamide (TAF) PBMCs=peripheral blood mononuclear cells; CES1=carboxylesterase 1 - * Values reflect administration of BIKTARVY with or without food.
- † Values refer to geometric mean ratio [high-fat meal/fasting] in PK parameters and (90% confidence interval). High fat meal is approximately 800 kcal, 50% fat.
- ‡ t 1/2 values refer to median (Q1, Q3) terminal plasma half-life. Note that the active metabolite of TAF, tenofovir diphosphate, has a half-life of 150–180 hours within PBMCs.
- § In vivo, TAF is hydrolyzed within cells to form tenofovir (major metabolite), which is phosphorylated to the active metabolite, tenofovir diphosphate. In vitro studies have shown that TAF is metabolized to tenofovir by cathepsin A in PBMCs and macrophages; and by CES1 in hepatocytes.
- ¶ Dosing in mass balance studies: single dose administration of [ 14C] BIC; single dose administration of [ 14C] FTC after multiple dosing of FTC for ten days; single dose administration of [ 14C] TAF.
Absorption Tmax (h)* 2.0–4.0 1.5–2.0 0.5–2.0 Effect of high-fat meal (relative to fasting)† AUC ratio 1.24 (1.16, 1.33) 0.96 (0.93, 0.99) 1.63 (1.43, 1.85) Cmax ratio 1.13 (1.06, 1.20) 0.86 (0.78, 0.93) 0.92 (0.73, 1.14) Distribution % bound to human plasma proteins >99 <4 ~80 Blood-to-plasma ratio 0.64 0.6 1.0 Elimination t1/2 (h)‡ 17.3 (14.8, 20.7) 10.4 (9.0, 12.0) 0.51 (0.45, 0.62)‡ Metabolism Metabolic pathway(s) CYP3A
UGT1A1Not significantly metabolized Cathepsin A§ (PBMCs)
CES1 (hepatocytes)Excretion Major route of elimination Metabolism Glomerular filtration and active tubular secretion Metabolism % of dose excreted in urine¶ 35 70 <1 % of dose excreted in feces¶ 60.3 13.7 31.7 Table 5 Multiple Dose PK Parameters of BIC, FTC, and TAF Following Oral Administration of BIKTARVY in HIV-Infected Adults Parameter Mean (CV%) Bictegravir Emtricitabine Tenofovir Alafenamide CV=Coefficient of Variation; NA=Not Applicable Cmax
(microgram per mL)6.15 (22.9) 2.13 (34.7) 0.121 (15.4) AUCtau
(microgram∙h per mL)102 (26.9) 12.3 (29.2) 0.142 (17.3) Ctrough
(microgram per mL)2.61 (35.2) 0.096 (37.4) NA Specific Populations
Patients with Renal Impairment
No clinically relevant differences in the pharmacokinetics of BIC, TAF, or its metabolite tenofovir were observed between subjects with severe renal impairment (CLcr 15 to 29 mL per minute estimated by Cockcroft-Gault method) and healthy subjects.
Patients with Hepatic Impairment
Bictegravir: Clinically relevant changes in the pharmacokinetics of BIC were not observed in subjects with moderate (Child-Pugh Class B) hepatic impairment.
Emtricitabine: The pharmacokinetics of FTC has not been studied in subjects with hepatic impairment; however, FTC is not significantly metabolized by liver enzymes, so the impact of hepatic impairment should be limited.
Tenofovir Alafenamide: Clinically relevant changes in the pharmacokinetics of TAF or its metabolite tenofovir were not observed in subjects with mild or moderate (Child-Pugh Class A and B) hepatic impairment [see Use in Specific Populations (8.7)].
Hepatitis B and/or Hepatitis C Virus Coinfection
The pharmacokinetics of BIC, FTC, and TAF have not been evaluated in subjects coinfected with hepatitis B and/or C virus.
Geriatric Patients
The pharmacokinetics of BIC, FTC, and TAF have not been fully evaluated in the elderly (65 years of age and older). Population pharmacokinetics analysis of HIV-infected subjects in Phase 3 trials of BIKTARVY showed that age did not have a clinically relevant effect on exposures of BIC and TAF up to 74 years of age [see Use in Specific Populations (8.5)].
Pediatric Patients
Mean BIC Ctrough was lower in 50 pediatric patients aged 12 to less than 18 years and weighing at least 35 kg who received BIKTARVY in Trial 1474 relative to adults following administration of BIKTARVY, but was not considered clinically significant based on exposure-response relationships; exposures of FTC and TAF in these pediatric patients were similar to those in adults (Table 6).
Table 6 Multiple Dose PK Parameters of BIC, FTC, and TAF Following Oral Administration of BIKTARVY in HIV-Infected Pediatric Subjects Aged 12 to less than 18 years Parameter Mean (CV%) Bictegravir* Emtricitabine† Tenofovir Alafenamide* CV=Coefficient of Variation; NA=Not Applicable - * From Population PK analysis of cohort 1 of Trial 1474 (n=50 for BIC; n=49 for TAF).
- † From Intensive PK analysis of cohort 1 of Trial 1474 (n=24).
Cmax
(microgram per mL)6.24 (27.1) 2.69 (34.0) 0.133 (70.2) AUCtau
(microgram∙h per mL)89.1 (31.0) 13.6 (21.7) 0.196 (50.3) Ctrough
(microgram per mL)1.78 (44.4) 0.064 (25.0) NA Mean BIC Cmax, and exposures of FTC and TAF (AUCtau and Cmax) achieved in 50 pediatric patients between the ages of 6 to less than 12 years and weighing at least 25 kg who received BIKTARVY in Trial 1474 were higher than exposures in adults; however, the increase was not considered clinically significant as the safety profiles were similar in adult and pediatric patients (Table 7) [see Use in Specific Populations (8.4)].
Table 7 Multiple Dose PK Parameters of BIC, FTC, and TAF Following Oral Administration of BIKTARVY in HIV-Infected Pediatric Subjects Aged 6 to less than 12 years Parameter Mean (CV%) Bictegravir* Emtricitabine† Tenofovir Alafenamide* CV=Coefficient of Variation; NA=Not Applicable - * From Population PK analysis of cohort 2 of Trial 1474 (n=50 for BIC; n=47 for TAF).
- † From Intensive PK analysis of cohort 2 of Trial 1474 (n=25 except n=24 for Ctrough).
Cmax
(microgram per mL)9.46 (24.3) 3.89 (31.0) 0.205 (44.6) AUCtau
(microgram∙h per mL)128 (27.8) 17.6 (36.9) 0.278 (40.3) Ctrough
(microgram per mL)2.36 (39.0) 0.227 (323) NA Drug Interaction Studies
As BIKTARVY is a complete regimen for the treatment of HIV-1 infection, comprehensive information regarding potential drug-drug interactions with other antiretroviral agents is not provided.
BIC is a substrate of CYP3A and UGT1A1.
BIC is an inhibitor of OCT2 and MATE1. At clinically relevant concentrations, BIC is not an inhibitor of hepatic transporters OATP1B1, OATP1B3, OCT1, BSEP, renal transporters OAT1 and OAT3, or CYP (including CYP3A) or UGT1A1 enzymes.
TAF is a substrate of P-gp and BCRP.
At clinically relevant concentrations, TAF is not an inhibitor of drug transporters P-gp, BCRP, hepatic transporters OATP1B1, OATP1B3, OCT1, BSEP, renal transporters OAT1, OAT3, OCT2, MATE1, or CYP (including CYP3A) or UGT1A1 enzymes.
Drug interaction studies were conducted with BIKTARVY or its components. Tables 8 and 9 summarize the pharmacokinetic effects of other drugs on BIC and TAF, respectively. Table 10 summarizes the pharmacokinetic effects of BIKTARVY or its components on other drugs.
Effect of Other Drugs on BIKTARVY Components
Table 8 Effect of Other Drugs on BIC* Coadministered Drug Dose of Coadministered Drug (mg) BIC (mg) Mean Ratio of BIC Pharmacokinetic Parameters (90% CI); No effect = 1.00 Cmax AUC Cmin NA= Not Applicable - * All interaction studies conducted in healthy volunteers.
- † Study conducted with additional voxilaprevir 100 mg to achieve voxilaprevir exposures expected in HCV-infected patients.
- ‡ Maximum strength antacid contained 80 mg aluminum hydroxide, 80 mg magnesium hydroxide, and 8 mg simethicone, per mL.
- § Reference treatment administered under fasted conditions.
Ledipasvir/ Sofosbuvir (fed) 90/400 once daily 75 once daily 0.98
(0.94, 1.03)1.00
(0.97, 1.03)1.04
(0.99, 1.09)Rifabutin (fasted) 300 once daily 75 once daily 0.80
(0.67, 0.97)0.62
(0.53, 0.72)0.44
(0.37, 0.52)Rifampin (fed) 600 once daily 75 single dose 0.72
(0.67, 0.78)0.25
(0.22, 0.27)NA Sofosbuvir/ velpatasvir/ voxilaprevir (fed) 400/100/100+100 voxilaprevir† once daily 50 once daily 0.98
(0.94, 1.01)1.07
(1.03, 1.10)1.10
(1.05, 1.17)Voriconazole (fasted) 300 twice daily 75 single dose 1.09
(0.96, 1.23)1.61
(1.41, 1.84)NA Maximum strength antacid (simultaneous administration, fasted) 20 mL‡ single dose (oral) 50 single dose 0.20
(0.16, 0.24)0.21
(0.18, 0.26)NA Maximum strength antacid (2 h after BIKTARVY fasted) 20 mL‡ single dose (oral) 50 single dose 0.93
(0.88, 1.00)0.87
(0.81, 0.93)NA Maximum strength antacid (2 h before BIKTARVY fasted) 20 mL‡ single dose (oral) 50 single dose 0.42
(0.33, 0.52)0.48
(0.38, 0.59)NA Maximum strength antacid (simultaneous administration, fed§) 20 mL‡ single dose (oral) 50 single dose 0.51
(0.43, 0.62)0.53
(0.44, 0.64)NA Calcium carbonate (simultaneous administration, fasted) 1200 single dose 50 single dose 0.58
(0.51, 0.67)0.67
(0.57, 0.78)NA Calcium carbonate (simultaneous administration, fed§) 1200 single dose 50 single dose 0.90
(0.78, 1.03)1.03
(0.89, 1.20)NA Ferrous fumarate (simultaneous administration, fasted) 324 single dose 50 single dose 0.29
(0.26, 0.33)0.37
(0.33, 0.42)NA Ferrous fumarate (simultaneous administration, fed§) 324 single dose 50 single dose 0.75
(0.65, 0.87)0.84
(0.74, 0.95)NA Table 9 Effect of Other Drugs on TAF* Coadministered Drug Dose of Coadministered Drug (mg) Tenofovir Alafenamide (mg) Mean Ratio of Tenofovir Alafenamide Pharmacokinetic Parameters (90% CI); No effect = 1.00 Cmax AUC Cmin NA= Not Applicable - * All interaction studies conducted in healthy volunteers.
- † Study conducted with emtricitabine/tenofovir alafenamide.
- ‡ Study conducted with additional voxilaprevir 100 mg to achieve voxilaprevir exposures expected in HCV-infected patients.
Carbamazepine 300 twice daily 25 single dose† 0.43
(0.36, 0.51)0.46
(0.40, 0.54)NA Ledipasvir/sofosbuvir 90/400 once daily 25 once daily 1.17
(1.00, 1.38)1.27
(1.19, 1.34)NA Sofosbuvir/ velpatasvir/ voxilaprevir 400/100/100 +100 voxilaprevir‡ once daily 25 once daily 1.28
(1.09, 1.51)1.57
(1.44, 1.71)NA Effect of BIKTARVY Components on Other Drugs
Table 10 Effect of Components of BIKTARVY on Other Drugs* Coadministered Drug Dose of Coadministered Drug (mg) BIC (mg) TAF (mg) Mean Ratio of Coadministered Drug Pharmacokinetic Parameters (90% CI); No effect = 1.00 Cmax AUC Cmin NA= Not Applicable - * All interaction studies conducted in healthy volunteers.
- † The predominant circulating nucleoside metabolite of sofosbuvir.
- ‡ Study conducted with emtricitabine/tenofovir alafenamide.
- § Study conducted with elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide.
- ¶ Study conducted with additional voxilaprevir 100 mg to achieve voxilaprevir exposures expected in HCV-infected patients.
Ledipasvir 90/400 once daily 75 once daily 25 once daily 0.85
(0.81, 0.90)0.87
(0.83, 0.92)0.90
(0.84, 0.96)Sofosbuvir 1.11
(1.00, 1.24)1.07
(1.01, 1.13)NA GS-331007† 1.10
(1.07, 1.13)1.11
(1.08, 1.14)1.02
(0.99, 1.06)Metformin 500 twice daily 50 once daily 25 once daily 1.28
(1.21, 1.36)1.39
(1.31, 1.48)1.36
(1.21, 1.53)Midazolam 2 single dose 50 once daily 25 once daily 1.03
(0.87, 1.23)1.15
(1.00, 1.31)NA Norelgestromin norgestimate 0.180/0.215/0.250 once daily / ethinyl estradiol 0.025 once daily 75 once daily - 1.23
(1.14, 1.32)1.08
(1.05, 1.10)1.10
(1.05, 1.15)Norgestrel 1.15
(1.10, 1.21)1.13
(1.07, 1.19)1.14
(1.06, 1.22)Ethinyl estradiol 1.15
(1.03, 1.27)1.04
(0.99, 1.10)1.05
(0.95, 1.14)Norelgestromin norgestimate 0.180/0.215/0.250 once daily / ethinyl estradiol 0.025 once daily - 25 once daily‡ 1.17
(1.07,1.26)1.12
(1.07,1.17)1.16
(1.08, 1.24)Norgestrel 1.10
(1.02, 1.18)1.09
(1.01, 1.18)1.11
(1.03, 1.20)Ethinyl estradiol 1.22
(1.15, 1.29)1.11
(1.07, 1.16)1.02
(0.92, 1.12)Sertraline 50 single dose - 10 once daily§ 1.14
(0.94, 1.38)0.93
(0.77, 1.13)NA Sofosbuvir 400/100/100+ 100¶ once daily 50 once daily 25 once daily 1.14
(1.04,1.25)1.09
(1.02, 1.15)NA GS-331007† 1.03
(0.99,1.06)1.03
(1.00,1.06)1.01
(0.98, 1.05)Velpatasvir 0.96
(0.91,1.01)0.96
(0.90, 1.02)0.94
(0.88, 1.01)Voxilaprevir 0.90
(0.76, 1.06)0.91
(0.80, 1.03)0.97
(0.88, 1.06)12.4 Microbiology
Mechanism of Action
Bictegravir: BIC inhibits the strand transfer activity of HIV-1 integrase (integrase strand transfer inhibitor; INSTI), an HIV-1 encoded enzyme that is required for viral replication. Inhibition of integrase prevents the integration of linear HIV-1 DNA into host genomic DNA, blocking the formation of the HIV-1 provirus and propagation of the virus.
Emtricitabine: FTC, a synthetic nucleoside analog of cytidine, is phosphorylated by cellular enzymes to form emtricitabine 5'-triphosphate. Emtricitabine 5'-triphosphate inhibits the activity of the HIV-1 reverse transcriptase by competing with the natural substrate deoxycytidine 5'-triphosphate and by being incorporated into nascent viral DNA which results in chain termination. Emtricitabine 5′-triphosphate is a weak inhibitor of mammalian DNA polymerases α, β, Ɛ, and mitochondrial DNA polymerase γ.
Tenofovir Alafenamide: TAF is a phosphonamidate prodrug of tenofovir (2′-deoxyadenosine monophosphate analog). Plasma exposure to TAF allows for permeation into cells and then TAF is intracellularly converted to tenofovir through hydrolysis by cathepsin A. Tenofovir is subsequently phosphorylated by cellular kinases to the active metabolite tenofovir diphosphate. Tenofovir diphosphate inhibits HIV-1 replication through incorporation into viral DNA by the HIV reverse transcriptase, which results in DNA chain-termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases that include mitochondrial DNA polymerase γ and there is no evidence of toxicity to mitochondria in cell culture.
Antiviral Activity in Cell Culture
The triple combination of BIC, FTC, and TAF was not antagonistic with respect to antiviral activity in cell culture.
Bictegravir: The antiviral activity of BIC against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+ T-lymphocytes. In MT-4 cells (human lymphoblastoid T-cell line) acutely infected with HIV-1 IIIB, the mean 50% effective concentration (EC50) was 2.4±0.4 nM, and the protein-adjusted EC95 value was 361 nM (0.162 micrograms per mL). BIC displayed antiviral activity in activated PBMCs against clinical isolates of HIV-1 representing groups M, N, and O, including subtypes A, B, C, D, E, F, and G, with a median EC50 value of 0.55 nM (range <0.05 to 1.71 nM). The EC50 value against a single HIV-2 isolate was 1.1 nM.
Emtricitabine: The antiviral activity of FTC against laboratory and clinical isolates of HIV-1 was assessed in T lymphoblastoid cell lines, the MAGI-CCR5 cell line, and PBMCs. In PBMCs acutely infected with HIV-1 subtypes A, B, C, D, E, F, and G, the median EC50 value for FTC was 9.5 nM (range 1 to 30 nM) and against HIV-2 was 7 nM.
Tenofovir Alafenamide: The antiviral activity of TAF against laboratory and clinical isolates of HIV-1 subtype B was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells and CD4-T lymphocytes. The EC50 values for TAF ranged from 2.0 to 14.7 nM. TAF displayed antiviral activity in cell culture against all HIV-1 groups (M, N, O), including subtypes A, B, C, D, E, F, and G (EC50 values ranged from 0.1 to 12 nM) and strain specific activity against HIV-2 (EC50 values ranged from 0.9 to 2.6 nM).
Resistance
In Cell Culture
Bictegravir: HIV-1 isolates with reduced susceptibility to BIC have been selected in cell culture. In one selection with BIC, a virus pool emerged expressing amino acid substitutions M50I and R263K in the HIV-1 integrase. M50I, R263K, and M50I+R263K substitutions, when introduced into a wild-type virus by site-directed mutagenesis, conferred 1.3-, 2.2-, and 2.9-fold reduced susceptibility to BIC, respectively. In a second selection, emergence of amino acid substitutions T66I and S153F was detected, and 0.4-, 1.9-, and 0.5-fold reductions in BIC susceptibility were observed with T66I, S153F, and T66I+S153F, respectively. In addition, S24G and E157K substitutions emerged during the selection process.
Tenofovir Alafenamide: HIV-1 isolates with reduced susceptibility to TAF were selected in cell culture. HIV-1 isolates selected by TAF expressed a K65R substitution in HIV-1 RT, sometimes in the presence of S68N or L429I substitutions; in addition, a K70E substitution in HIV-1 RT was observed.
In Clinical Trials
In Subjects with No Antiretroviral Treatment History: Pooled genotypic resistance analyses were performed on paired baseline and on-treatment HIV-1 isolates from subjects receiving BIKTARVY through Week 48 in Trials 1489 and 1490 [see Clinical Studies (14.2)] who had HIV-1 RNA greater than or equal to 200 copies/mL at the time of confirmed virologic failure, Week 48, or early study drug discontinuation. No specific amino acid substitutions emerged consistently in the 8 treatment failure subjects with evaluable genotypic resistance data and failed to establish an association with genotypic BIC resistance. There were no treatment-emergent NRTI resistance-associated substitutions detected in the 8 evaluated treatment failure isolates. Phenotypic resistance analyses of failure isolates found fold-changes in drug susceptibility below the biological or clinical cutoffs for BIC, FTC, and TFV, compared to wild-type reference HIV-1.
In Virologically Suppressed Adult Subjects: In 2 switch trials, Trials 1844 and 1878 [see Clinical Studies (14.3)], of virologically suppressed HIV-1 infected subjects (n=572), only one subject with virologic rebound in the resistance analysis population had IN genotypic and phenotypic data, and 2 rebounders had RT genotypic and phenotypic data. No subjects had HIV-1 with treatment-emergent genotypic or phenotypic resistance to BIC, FTC, or TAF.
In Virologically Suppressed Pediatric Subjects: In Trial 1474 [see Clinical Studies (14.4)], two of 50 subjects in cohort 1 were evaluated for the development of resistance through Week 48; no amino acid substitutions known to be associated with resistance to BIC, FTC, or TFV were detected. No subjects in cohort 2 met the criteria for resistance analyses.
Cross-Resistance
Bictegravir: Cross-resistance has been observed among INSTIs. The susceptibility of BIC was tested against 64 clinical isolates expressing known INSTI resistance-associated substitutions listed by IAS-USA (20 with single substitutions and 44 with 2 or more substitutions). Isolates with a single INSTI-resistance substitution including E92Q, T97A, Y143C/R, Q148R, and N155H showed less than 2-fold reduced susceptibility to BIC. All isolates (n=14) with more than 2.5-fold reduced susceptibility to BIC (above the biological cutoff for BIC) contained G140A/C/S and Q148H/R/K substitutions; the majority (64.3%, 9/14) had a complex INSTI resistance pattern with an additional INSTI-resistance substitution L74M, T97A, or E138A/K. Of those evaluated isolates containing G140A/C/S and Q148H/R/K substitutions in the absence of additional INSTI-resistance substitutions, 38.5% (5/13) showed more than 2.5-fold reduction. In addition, site-directed mutant viruses with G118R (dolutegravir and raltegravir treatment-emergent substitution) and G118R+T97A had 3.4- and 2.8-fold reduced susceptibility to BIC, respectively.
BIC demonstrated equivalent antiviral activity with less than 2-fold reductions in susceptibility against HIV-1 variants expressing substitutions associated with resistance to NNRTIs, NRTIs, and PIs, compared with the wild-type virus.
Emtricitabine: Cross-resistance has been observed among NRTIs. FTC-resistant viruses with an M184V/I substitution in HIV-1 RT were cross-resistant to lamivudine. HIV-1 isolates containing the K65R RT substitution, selected in vivo by abacavir, didanosine, and tenofovir, demonstrated reduced susceptibility to inhibition by FTC.
Tenofovir Alafenamide: Cross-resistance has been observed among NRTIs. Tenofovir resistance substitutions K65R and K70E result in reduced susceptibility to abacavir, didanosine, emtricitabine, lamivudine, and tenofovir. HIV-1 with multiple thymidine analog substitutions (M41L, D67N, K70R, L210W, T215F/Y, K219Q/E/N/R), or multinucleoside resistant HIV-1 with a T69S double insertion mutation or with a Q151M substitution complex including K65R, showed reduced susceptibility to TAF in cell culture.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Bictegravir
BIC was not carcinogenic in a 6-month rasH2 transgenic mouse study at doses of up to 100 mg/kg/day in males and 300 mg/kg/day in females. BIC was not carcinogenic in a 2-year rat study at doses up to 300 mg/kg/day, which resulted in exposures of approximately 31 times the exposure in humans at the recommended dose of BIKTARVY.
BIC was not genotoxic in the reverse mutation bacterial test (Ames test), mouse lymphoma or rat micronucleus assays.
BIC did not affect fertility, reproductive performance or embryonic viability in male and female rats at 29 times higher exposures (AUC) than in humans at the recommended dose of BIKTARVY.
Emtricitabine
In long-term carcinogenicity studies of FTC, no drug-related increases in tumor incidence were found in mice at doses up to 750 mg per kg per day (25 times the human systemic exposure at the recommended dose of BIKTARVY) or in rats at doses up to 600 mg per kg per day (30 times the human systemic exposure at the recommended dose of BIKTARVY).
FTC was not genotoxic in the reverse mutation bacterial test (Ames test), mouse lymphoma or mouse micronucleus assays.
FTC did not affect fertility in male rats at approximately 140 times or in male and female mice at approximately 60 times higher exposures (AUC) than in humans given the recommended dose of BIKTARVY. Fertility was normal in the offspring of mice exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the recommended dose of BIKTARVY.
Tenofovir Alafenamide
Since TAF is rapidly converted to tenofovir and a lower tenofovir exposure in rats and mice was observed after TAF administration compared to TDF administration, carcinogenicity studies were conducted only with TDF. Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 10 times (mice) and 4 times (rats) those observed in humans following a 300 mg dose of TDF. The tenofovir exposure in these studies was approximately 151 times (mice) and 51 times (rat) those observed in humans after administration of the daily recommended dose of BIKTARVY. At the high dose in female mice, liver adenomas were increased at tenofovir exposures approximately 151 times the exposure observed in humans at the recommended dose of BIKTARVY. In rats, the study was negative for carcinogenic findings.
TAF was not genotoxic in the reverse mutation bacterial test (Ames test), mouse lymphoma or rat micronucleus assays.
There were no effects on fertility, mating performance or early embryonic development when TAF was administered to male rats at a dose equivalent to 155 times the human dose of BIKTARVY based on body surface area comparisons for 28 days prior to mating and to female rats for 14 days prior to mating through Day 7 of gestation.
13.2 Animal Toxicology and/or Pharmacology
Minimal to slight infiltration of mononuclear cells in the posterior uvea was observed in dogs with similar severity after three and nine month administration of TAF; reversibility was seen after a three month recovery period. No eye toxicity was observed in the dog at systemic exposures of 7 (TAF) and 14 (tenofovir) times the exposure seen in humans with the recommended daily dose of BIKTARVY.
-
14 CLINICAL STUDIES
14.1 Description of Clinical Trials
The efficacy and safety of BIKTARVY were evaluated in the trials summarized in Table 11.
Table 11 Trials Conducted with BIKTARVY in Subjects with HIV-1 Infection Trial Population Trial Arms (N) Timepoint (Week) - * Randomized, double blind, active controlled trial.
- † HIV-1 RNA less than 50 copies per mL.
- ‡ Randomized, open label, active controlled trial.
- § Open label trial.
Trial 1489*
(NCT 02607930)Adults with no antiretroviral treatment history BIKTARVY (314)
ABC/DTG/3TC (315)48 Trial 1490*
(NCT 02607956)BIKTARVY (320)
DTG + FTC/TAF (325)48 Trial 1844*
(NCT 02603120)Virologically-suppressed† adults BIKTARVY (282)
ABC/DTG/3TC (281)48 Trial 1878‡
(NCT 02603107)BIKTARVY (290)
ATV or DRV (with cobicistat or ritonavir) plus either FTC/TDF or ABC/3TC (287)48 Trial 1474§ (cohort 1)
(NCT 02881320)Virologically-suppressed† adolescents between the ages of 12 to less than 18 years (at least 35 kg) BIKTARVY (50) 48 Trial 1474§ (cohort 2)
(NCT 02881320)Virologically-suppressed† children between the ages of 6 to less than 12 years (at least 25 kg) BIKTARVY (50) 24 14.2 Clinical Trial Results in HIV-1 Subjects with No Antiretroviral Treatment History
In Trial 1489, subjects were randomized in a 1:1 ratio to receive either BIKTARVY (N=314) or ABC/DTG/3TC (600 mg/50 mg/300 mg) (N=315) once daily. In Trial 1490, subjects were randomized in a 1:1 ratio to receive either BIKTARVY (N=320) or DTG + FTC/TAF (50 mg + 200 mg/25 mg) (N=325) once daily.
In Trial 1489, the mean age was 34 years (range 18–71), 90% were male, 57% were White, 36% were Black, and 3% were Asian. 22% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 1.3–6.5). The mean baseline CD4+ cell count was 464 cells per mm3 (range 0–1424) and 11% had CD4+ cell counts less than 200 cells per mm3. 16% of subjects had baseline viral loads greater than 100,000 copies per mL.
In Trial 1490, the mean age was 37 years (range 18–77), 88% were male, 59% were White, 31% were Black, and 3% were Asian. 25% of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 2.3–6.6). The mean baseline CD4+ cell count was 456 cells per mm3 (range 2–1636) and 12% had CD4+ cell counts less than 200 cells per mm3. 19% of subjects had baseline viral loads greater than 100,000 copies per mL.
In both trials, subjects were stratified by baseline HIV-1 RNA (less than or equal to 100,000 copies per mL, greater than 100,000 copies per mL to less than or equal to 400,000 copies per mL, or greater than 400,000 copies per mL), by CD4 count (less than 50 cells per mm3, 50-199 cells per mm3, or greater than or equal to 200 cells per mm3), and by region (US or ex-US).
Treatment outcomes of Trials 1489 and 1490 through Week 48 are presented in Table 12.
Table 12 Virologic Outcomes of Randomized Treatment in Trials 1489 and 1490 at Week 48* in Subjects with No Antiretroviral Treatment History Trial 1489 Trial 1490 BIKTARVY
(N=314)ABC/DTG/3TC
(N=315)BIKTARVY
(N=320)DTG + FTC/TAF
(N=325)- * Week 48 window was between Day 295 and 378 (inclusive).
- † Includes subjects who had ≥ 50 copies/mL in the Week 48 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than an adverse event (AE), death or lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
- ‡ Includes subjects who discontinued due to AE or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window.
- § Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
HIV-1 RNA < 50 copies/mL 92% 93% 89% 93% Treatment Difference (95% CI) BIKTARVY vs. Comparator -0.6% (-4.8% to 3.6%) -3.5% (-7.9% to 1.0%) HIV-1 RNA ≥ 50 copies/mL† 1% 3% 4% 1% No Virologic Data at Week 48 Window 7% 4% 6% 6% Discontinued Study Drug Due to AE or Death‡ 0 1% 1% 1% Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA <50 copies/mL§ 5% 3% 3% 4% Missing Data During Window but on Study Drug 2% <1% 2% 1% Treatment outcomes were similar across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
In Trials 1489 and 1490, the mean increase from baseline in CD4+ count at Week 48 was 233 and 229 cells per mm3 in the BIKTARVY and ABC/DTG/3TC groups, respectively, and 180 and 201 cells per mm3 in the BIKTARVY and DTG + FTC/TAF groups, respectively.
14.3 Clinical Trial Results in HIV-1 Virologically-Suppressed Subjects Who Switched to BIKTARVY
In Trial 1844, the efficacy and safety of switching from a regimen of DTG + ABC/3TC or ABC/DTG/3TC to BIKTARVY were evaluated in a randomized, double-blind trial of virologically-suppressed (HIV-1 RNA less than 50 copies per mL) HIV-1 infected adults (N=563, randomized and dosed). Subjects must have been stably suppressed (HIV-1 RNA less than 50 copies per mL) on their baseline regimen for at least 3 months prior to trial entry and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY at baseline (N=282), or stay on their baseline antiretroviral regimen (N=281). Subjects had a mean age of 45 years (range 20–71), 89% were male, 73% were White, and 22% were Black. 17% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 723 cells per mm3 (range 124–2444).
In Trial 1878, the efficacy and safety of switching from either ABC/3TC or FTC/TDF (200/300 mg) plus ATV or DRV (given with either cobicistat or ritonavir) to BIKTARVY were evaluated in a randomized, open-label study of virologically-suppressed HIV-1 infected adults (N=577, randomized and dosed). Subjects must have been stably suppressed on their baseline regimen for at least 6 months, must not have been previously treated with any INSTI, and had no history of treatment failure. Subjects were randomized in a 1:1 ratio to either switch to BIKTARVY (N=290) or stay on their baseline antiretroviral regimen (N=287). Subjects had a mean age of 46 years (range 20–79), 83% were male, 66% were White, and 26% were Black. 19% of subjects identified as Hispanic/Latino. The mean baseline CD4+ cell count was 663 cells per mm3 (range 62–2582). Subjects were stratified by prior treatment regimen. At screening, 15% of subjects were receiving ABC/3TC plus ATV or DRV (given with either cobicistat or ritonavir) and 85% of subjects were receiving FTC/TDF plus ATV or DRV (given with either cobicistat or ritonavir).
Treatment outcomes of Trials 1844 and 1878 through Week 48 are presented in Table 13.
Table 13 Virologic Outcomes of Trials 1844 and 1878 at Week 48* in Virologically-Suppressed Subjects who Switched to BIKTARVY Trial 1844 Trial 1878 BIKTARVY
(N=282)ABC/DTG/3TC
(N=281)BIKTARVY
(N=290)ATV- or DRV-based regimen†
(N=287)- * Week 48 window was between Day 295 and 378 (inclusive).
- † ATV given with cobicistat or ritonavir or DRV given with cobicistat or ritonavir plus either FTC/TDF or ABC/3TC.
- ‡ Includes subjects who had ≥ 50 copies/mL in the Week 48 window; subjects who discontinued early due to lack or loss of efficacy; subjects who discontinued for reasons other than lack or loss of efficacy and at the time of discontinuation had a viral value of ≥ 50 copies/mL.
- § Includes subjects who discontinued for reasons other than an AE, death, or lack or loss of efficacy, e.g., withdrew consent, loss to follow-up, etc.
HIV-1 RNA ≥ 50 copies/mL‡ 1% <1% 2% 2% Treatment Difference (95% CI) 0.7% (-1.0% to 2.8%) 0.0% (-2.5% to 2.5%) HIV-1 RNA < 50 copies/mL 94% 95% 92% 89% No Virologic Data at Week 48 Window 5% 5% 6% 9% Discontinued Study Drug Due to AE or Death and Last Available HIV-1 RNA < 50 copies/mL 2% 1% 1% 1% Discontinued Study Drug Due to Other Reasons and Last Available HIV-1 RNA < 50 copies/mL§ 2% 3% 3% 7% Missing Data During Window but on Study Drug 2% 1% 2% 2% In Trial 1844, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was -31 cells per mm3 in subjects who switched to BIKTARVY and 4 cells per mm3 in subjects who stayed on ABC/DTG/3TC.
In Trial 1878, treatment outcomes between treatment groups were similar across subgroups by age, sex, race, and region. The mean change from baseline in CD4+ count at Week 48 was 25 cells per mm3 in patients who switched to BIKTARVY and 0 cells per mm3 in patients who stayed on their baseline regimen.
14.4 Clinical Trial Results in HIV-1 Infected Pediatric Subjects Between the Ages of 6 to Less than 18 Years
In Trial 1474, an open-label, single arm trial the efficacy, safety, and pharmacokinetics of BIKTARVY in HIV-1 infected pediatric subjects were evaluated in virologically-suppressed adolescents between the ages of 12 to less than 18 years weighing at least 35 kg (N=50) and in virologically-suppressed children between the ages of 6 to less than 12 years weighing at least 25 kg (N=50).
Cohort 1: Virologically-suppressed adolescents (12 to less than 18 years; at least 35 kg)
Subjects in cohort 1 treated with BIKTARVY once daily had a mean age of 14 years (range: 12 to 17) and a mean baseline weight of 51.7 kg (range: 35 to 123), 64% were female, 27% were Asian and 65% were black. At baseline, median CD4+ cell count was 750 cells per mm3 (range: 337 to 1207), and median CD4+% was 33% (range: 19% to 45%).
After switching to BIKTARVY, 98% (49/50) of subjects in cohort 1 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. The mean change from baseline in CD4+ cell count at Week 48 was -22 cells per mm3.
Cohort 2: Virologically-suppressed children (6 to less than 12 years; at least 25 kg)
Subjects in cohort 2 treated with BIKTARVY once daily had a mean age of 10 years (range: 6 to 11) and a mean baseline weight of 31.9 kg (range: 25 to 69), 54% were female, 22% were Asian and 72% were black. At baseline, median CD4+ cell count was 898 cells per mm3 (range 390 to 1991) and median CD4+% was 37% (range: 19% to 53%).
After switching to BIKTARVY, 100% (50/50) of subjects in cohort 2 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 24. The mean change from baseline in CD4+ cell count at Week 24 was -24 cells per mm3.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
BIKTARVY tablets are purplish brown, capsule-shaped, and film-coated with "GSI" debossed on one side and "9883" on the other side. Each bottle contains 30 tablets (NDC: 61958-2501-1), a silica gel desiccant, polyester coil, and is closed with a child-resistant closure. Each BIKTARVY tablet contains 50 mg of bictegravir (BIC), 200 mg of emtricitabine (FTC), and 25 mg of tenofovir alafenamide (TAF).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Post-treatment Acute Exacerbation of Hepatitis B in Patients with HBV Coinfection
Severe acute exacerbations of hepatitis B have been reported in patients who are coinfected with HBV and HIV-1 and have discontinued products containing FTC and/or TDF, and may likewise occur with discontinuation of BIKTARVY [see Warnings and Precautions (5.1)]. Advise the patient to not discontinue BIKTARVY without first informing their healthcare provider.
Drug Interactions
BIKTARVY may interact with certain drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or non-prescription medication or herbal products including St. John's wort [see Contraindications (4) and Drug Interactions (7)].
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any symptoms of infection, as in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.3)].
Renal Impairment
Advise patients to avoid taking BIKTARVY with concurrent or recent use of nephrotoxic agents. Renal impairment including cases of acute renal failure has been reported in association with the use of tenofovir prodrugs [see Warnings and Precautions (5.4)].
Lactic Acidosis and Severe Hepatomegaly
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with use of drugs similar to BIKTARVY. Advise patients that they should stop BIKTARVY if they develop clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [see Warnings and Precautions (5.5)].
Missed Dosage
Inform patients that it is important to take BIKTARVY on a regular dosing schedule with or without food and to avoid missing doses as it can result in development of resistance [see Dosage and Administration (2.2)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant women exposed to BIKTARVY [see Use in Specific Populations (8.1)].
Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration Issued: 06/2019 Patient Information
BIKTARVY® (bik-TAR-vee)
(bictegravir, emtricitabine,
and tenofovir alafenamide)
tabletsImportant: Ask your healthcare provider or pharmacist about medicines that should not be taken with BIKTARVY. For more information, see "What should I tell my healthcare provider before taking BIKTARVY?" What is the most important information I should know about BIKTARVY?
BIKTARVY can cause serious side effects, including:-
Worsening of Hepatitis B virus infection. If you have hepatitis B virus (HBV) infection and take BIKTARVY, your HBV may get worse (flare-up) if you stop taking BIKTARVY. A "flare-up" is when your HBV infection suddenly returns in a worse way than before.
- Do not run out of BIKTARVY. Refill your prescription or talk to your healthcare provider before your BIKTARVY is all gone.
- Do not stop taking BIKTARVY without first talking to your healthcare provider.
If you stop taking BIKTARVY, your healthcare provider will need to check your health often and do blood tests regularly for several months to check your HBV infection. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking BIKTARVY.
What is BIKTARVY?
BIKTARVY is a prescription medicine that is used without other anti-HIV-1 medicines to treat Human Immunodeficiency Virus-1 (HIV-1) in adults and children who weigh at least 55 pounds (25 kg):- who have not received anti-HIV-1 medicines in the past, or
- to replace their current anti-HIV-1 medicines for people whose healthcare provider determines that they meet certain requirements.
BIKTARVY contains the medicines bictegravir, emtricitabine, and tenofovir alafenamide.
It is not known if BIKTARVY is safe and effective in children who weigh less than 55 pounds (25 kg).Do not take BIKTARVY if you also take a medicine that contains: - dofetilide
- rifampin
What should I tell my healthcare provider before taking BIKTARVY?
Before taking BIKTARVY, tell your healthcare provider about all your medical conditions, including if you:- have liver problems, including hepatitis B virus infection
- have kidney problems
- are pregnant or plan to become pregnant. It is not known if BIKTARVY can harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with BIKTARVY.
Pregnancy Registry: There is a pregnancy registry for women who take BIKTARVY during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk with your healthcare provider about how you can take part in this registry. - are breastfeeding or plan to breastfeed. Do not breastfeed if you take BIKTARVY.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- At least one of the medicines in BIKTARVY can pass to your baby in your breast milk. It is not known if the other medicines in BIKTARVY can pass into your breast milk.
Some medicines may interact with BIKTARVY. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.- You can ask your healthcare provider or pharmacist for a list of medicines that interact with BIKTARVY.
- Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take BIKTARVY with other medicines.
How should I take BIKTARVY? - Take BIKTARVY exactly as your healthcare provider tells you to take it. BIKTARVY is taken by itself (not with other HIV-1 medicines) to treat HIV-1 infection.
- Take BIKTARVY 1 time each day with or without food.
- Do not change your dose or stop taking BIKTARVY without first talking with your healthcare provider. Stay under a healthcare provider's care during treatment with BIKTARVY.
- If you take antacids that contain aluminum or magnesium, take BIKTARVY at least 2 hours before or 6 hours after you take these antacids.
- If you take supplements or antacids that contain iron or calcium, take BIKTARVY with food at the same time that you take these supplements or antacids.
- Do not miss a dose of BIKTARVY.
- If you take too much BIKTARVY, call your healthcare provider or go to the nearest hospital emergency room right away.
- When your BIKTARVY supply starts to run low, get more from your healthcare provider or pharmacy. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to BIKTARVY and become harder to treat.
What are the possible side effects of BIKTARVY?
BIKTARVY may cause serious side effects, including:- See "What is the most important information I should know about BIKTARVY?"
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having any new symptoms after starting your HIV-1 medicine.
- New or worse kidney problems, including kidney failure. Your healthcare provider should do blood and urine tests to check your kidneys when starting and during treatment with BIKTARVY. Your healthcare provider may tell you to stop taking BIKTARVY if you develop new or worse kidney problems.
- Too much lactic acid in your blood (lactic acidosis). Too much lactic acid is a serious but rare medical emergency that can lead to death. Tell your healthcare provider right away if you get these symptoms: weakness or being more tired than usual, unusual muscle pain, being short of breath or fast breathing, stomach pain with nausea and vomiting, cold or blue hands and feet, feel dizzy or lightheaded, or a fast or abnormal heartbeat.
- Severe liver problems. In rare cases, severe liver problems can happen that can lead to death. Tell your healthcare provider right away if you get these symptoms: skin or the white part of your eyes turns yellow, dark "tea-colored" urine, light-colored stools, loss of appetite for several days or longer, nausea, or stomach-area pain.
These are not all the possible side effects of BIKTARVY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store BIKTARVY? - Store BIKTARVY below 86°F (30°C).
- Keep BIKTARVY in its original container.
- Keep the container tightly closed.
General information about the safe and effective use of BIKTARVY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use BIKTARVY for a condition for which it was not prescribed. Do not give BIKTARVY to other people, even if they have the same symptoms you have. It may harm them. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about BIKTARVY that is written for health professionals.
For more information, call 1-800-445-3235 or go to www.BIKTARVY.com.What are the ingredients in BIKTARVY?
Active ingredients: bictegravir, emtricitabine, and tenofovir alafenamide.
Inactive ingredients: croscarmellose sodium, magnesium stearate, and microcrystalline cellulose.
The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
Manufactured and distributed by: Gilead Sciences, Inc. Foster City, CA 94404
BIKTARVY is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2019 Gilead Sciences, Inc. All rights reserved. 210251-GS-002 -
Worsening of Hepatitis B virus infection. If you have hepatitis B virus (HBV) infection and take BIKTARVY, your HBV may get worse (flare-up) if you stop taking BIKTARVY. A "flare-up" is when your HBV infection suddenly returns in a worse way than before.
-
PRINCIPAL DISPLAY PANEL - 50 mg/200 mg/25 mg Tablet Bottle Label
NDC: 61958-2501-1
30 tabletsBiktarvy®
(bictegravir, emtricitabine, and
tenofovir alafenamide) tablets
50 mg /200 mg /25 mgNote to pharmacist:
Do not cover ALERT box with pharmacy label.ALERT: Find out about medicines that
should NOT be taken with Biktarvy
-
INGREDIENTS AND APPEARANCE
BIKTARVY
bictegravir sodium, emtricitabine, and tenofovir alafenamide fumarate tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-2501 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BICTEGRAVIR SODIUM (UNII: 4L5MP1Y7W7) (BICTEGRAVIR - UNII:8GB79LOJ07) BICTEGRAVIR 50 mg EMTRICITABINE (UNII: G70B4ETF4S) (EMTRICITABINE - UNII:G70B4ETF4S) EMTRICITABINE 200 mg TENOFOVIR ALAFENAMIDE FUMARATE (UNII: FWF6Q91TZO) (TENOFOVIR ANHYDROUS - UNII:W4HFE001U5) TENOFOVIR ALAFENAMIDE 25 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) WATER (UNII: 059QF0KO0R) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color brown (purplish-brown) Score no score Shape OVAL (capsule-shaped) Size 15mm Flavor Imprint Code GSI;9883 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-2501-1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 02/07/2018 2 NDC: 61958-2501-2 7 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 07/26/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210251 02/07/2018 Labeler - Gilead Sciences, Inc. (185049848)
Trademark Results [Biktarvy]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BIKTARVY 87226465 5344455 Live/Registered |
Gilead Sciences Ireland UC 2016-11-04 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.