LUMASON- sulfur hexafluoride kit
Lumason by
Drug Labeling and Warnings
Lumason by is a Prescription medication manufactured, distributed, or labeled by BRACCO DIAGNOSTICS INC, Bracco Suisse SA, AIRGAS THERAPEUTICS LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LUMASON® safely and effectively. See full prescribing information for LUMASON.
LUMASON (sulfur hexafluoride lipid-type A microspheres) for injectable suspension, for intravenous use or intravesical use
Initial U.S. Approval: 2014WARNING: SERIOUS CARDIOPULMONARY REACTIONS
See full prescribing information for complete boxed warning
Serious cardiopulmonary reactions, including fatalities, have occurred uncommonly during or following the injection of ultrasound contrast agents, including sulfur hexafluoride lipid microspheres (5.1). Most serious reactions occur within 30 minutes of administration (5.1).
INDICATIONS AND USAGE
Lumason is an ultrasound contrast agent indicated for use
- in echocardiography to opacify the left ventricular chamber and to improve the delineation of the left ventricular endocardial border in adult patients with suboptimal echocardiograms (1)
- in ultrasonography of the liver for characterization of focal liver lesions in adult and pediatric patients (1)
- in ultrasonography of the urinary tract for the evaluation of suspected or known vesicoureteral reflux in pediatric patients (1)
DOSAGE AND ADMINISTRATION
Avoid intra-arterial injection (2.1, 5.3)
See Full Prescribing Information for reconstitution instructions (2.3)
For intravenous injection:
- Echocardiography: After reconstitution, administer 2 mL as an intravenous injection in adult patients (2.2, 2.4)
- Echocardiography in pediatric patients: After reconstitution, administer 0.03 mL per kg as an intravenous injection up to a maximum of 2 mL per injection (2.2, 2.4)
- Ultrasonography of the liver in adults: After reconstitution, administer 2.4 mL as an intravenous injection (2.2, 2.4)
- Ultrasonography of the liver in pediatric patients: After reconstitution, administer 0.03 mL per kg as an intravenous injection, up to a maximum of 2.4 mL per injection (2.2, 2.4)
- May repeat dose one time during a single examination (2.2, 2.4)
- Follow each injection with an intravenous flush of 0.9% Sodium Chloride Injection (2.2, 2.4)
For intravesical administration in pediatric patients:
- Ultrasonography of the urinary tract: After reconstitution, administer 1 mL via sterile 6-8F urinary catheter. Bladder should be first emptied and then partially filled with saline before injection of Lumason (2.2, 2.4)
- After Lumason administration, continue filling the bladder with saline until the patient has the urge to micturate or at the first sign of back pressure to the infusion (2.4)
DOSAGE FORMS AND STRENGTHS
- For injectable suspension: 25 mg of lipid-type A lyophilized powder with headspace fill of 60.7 mg sulfur hexafluoride in a single-patient use vial for reconstitution (3)
CONTRAINDICATIONS
- History of hypersensitivity reactions to sulfur hexafluoride lipid microsphere components or to any of the inactive ingredients in Lumason (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 0.5%) are headache and nausea (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Bracco Diagnostics Inc at 1-800-257-5181 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS CARDIOPULMONARY REACTIONS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Recommended Dosage
2.3 Reconstitution Instructions
2.4 Administration Instructions
2.5 Imaging Guidelines
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Cardiopulmonary Reactions
5.2 Hypersensitivity Reactions
5.3 Systemic Embolization
5.4 Ventricular Arrhythmia Related to High Mechanical Index
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Echocardiography
14.2 Ultrasonography of the Liver
14.3 Ultrasonography of the Urinary Tract
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS CARDIOPULMONARY REACTIONS
Serious cardiopulmonary reactions, including fatalities, have occurred uncommonly during or following the injection of ultrasound contrast agents, including sulfur hexafluoride lipid microspheres [see Warnings and Precautions (5.1)]. Most serious reactions occur within 30 minutes of administration [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
Lumason is indicated for use in adult and pediatric patients with suboptimal echocardiograms to opacify the left ventricular chamber and to improve the delineation of the left ventricular endocardial border.
Lumason is indicated for use with ultrasound of the liver in adult and pediatric patients to characterize focal liver lesions.
Ultrasonography of the Urinary Tract
Lumason is indicated for use in ultrasonography of the urinary tract in pediatric patients for the evaluation of suspected or known vesicoureteral reflux.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Do not administer Lumason by intra-arterial injection [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage
The recommended dose of Lumason after reconstitution is 2 mL administered as an intravenous bolus injection during echocardiography. During a single examination, a second injection of 2 mL may be administered to prolong contrast enhancement. Follow each Lumason injection with an intravenous flush using 5 mL of 0.9% Sodium Chloride Injection.
The recommended dose of Lumason after reconstitution in pediatric patients is 0.03 mL per kg administered as an intravenous injection during echocardiography. During a single examination, a second injection of 0.03 mL per kg may be administered, if needed. Do not exceed 2 mL per injection. Follow Lumason injection with an intravenous flush using 5 mL of 0.9% Sodium Chloride Injection.
The recommended dose of Lumason after reconstitution in adult patients is 2.4 mL administered as an intravenous injection during ultrasonography of the liver. During a single examination, a second injection of 2.4 mL may be administered, if needed. Follow Lumason injection with an intravenous flush using 5 mL of 0.9% Sodium Chloride Injection.
The recommended dose of Lumason after reconstitution in pediatric patients is 0.03 mL per kg administered an intravenous injection during ultrasonography of the liver. During a single examination, a second injection of 0.03 mL per kg may be administered, if needed. Do not exceed 2.4 mL per injection. Follow Lumason injection with an intravenous flush of 0.9% Sodium Chloride Injection.
Ultrasonography of the Urinary Tract
The recommended dose of Lumason after reconstitution is 1 mL. The bladder may be refilled with normal saline for a second cycle of voiding and imaging, without the need of a second Lumason administration.
2.3 Reconstitution Instructions
- Inspect the Lumason kit and its components for signs of damage. Do not use the kit if the protective caps on the Lumason vial and prefilled syringe with 5 mL Sodium Chloride 0.9% Injection are not intact or if the kit shows other signs of damage.
- Under aseptic conditions, reconstitute Lumason by injecting
the prefilled syringe with 5 mL Sodium Chloride 0.9% Injection into
the Lumason vial using the following illustrated steps:
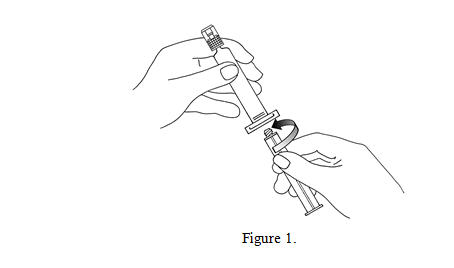
1. Connect the plunger rod to the prefilled syringe barrel by screwing it clockwise into the syringe (see Figure 1).
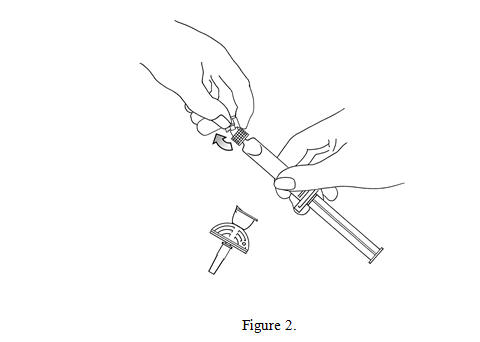
2. Open the Mini-Spike blister and remove the syringe tip cap (see Figure 2).
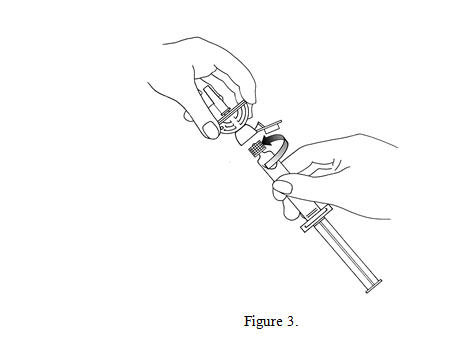
3. Open the Mini-Spike green cap and connect the syringe to the Mini-Spike by screwing it in clockwise (see Figure 3).
4. Remove the flip cap plastic protective cap from the vial, remove the Mini-Spike spike protection and position the spike in the center of the rubber stopper of the vial. Press firmly inward until the spike is fully inserted in the stopper (see Figure 4).
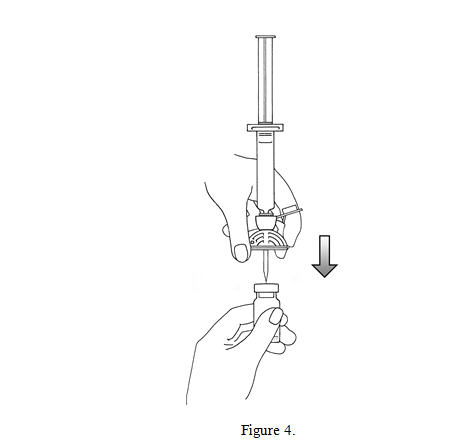
5. Empty the content of the syringe into the vial by pushing on the plunger rod (see Figure 5).
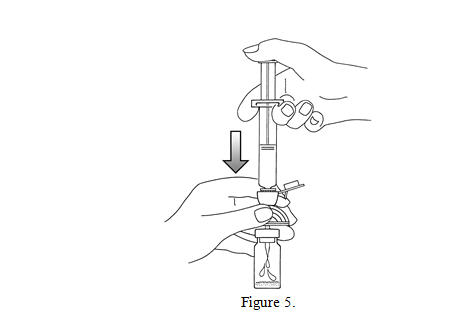
6. Shake vigorously for 20 seconds, mixing all the contents in the vial (see Figure 6). A homogeneous white milky liquid indicates formation of sulfur hexafluoride lipid microspheres.
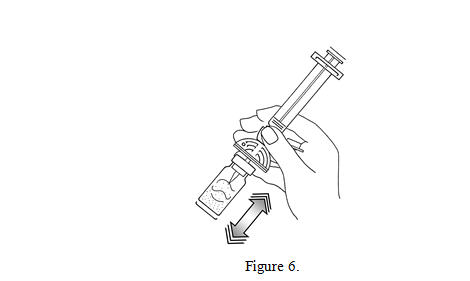
7. For preparation of doses greater than or equal to 1 mL, invert the system and slowly withdraw the intended volume of suspension into the syringe (see Figure 7). For preparation of doses less than 1 mL, withdraw 2 mL of the reconstituted suspension into the 5 mL syringe and measure the volume of Lumason to inject by using the 0.2 mL graduations between the 1 mL and 2 mL marks.
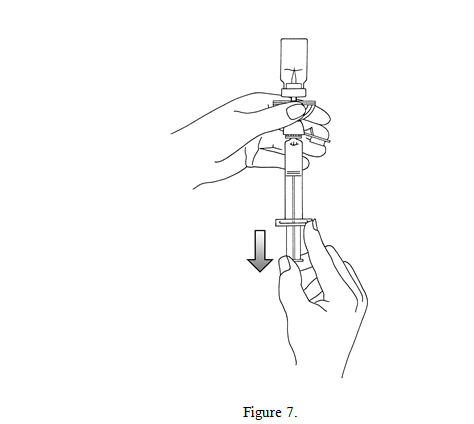
8. Unscrew the syringe from the Mini-Spike (see Figure 8). Peel and remove the diluent label to display the reconstituted product label. For intravenous administration, immediately connect the syringe to a dose administration line (20 G) and administer as directed under the Administration Instructions below. For intravesical administration, immediately connect the syringe to a sterile urinary catheter (6 french to 8 french) and administer as directed under the Administration Instructions below.

- Following reconstitution, Lumason suspension contains 1.5 to 5.6 x108 microspheres/mL with 45 mcg/mL of sulfur hexafluoride.
- Use immediately after reconstitution. If the suspension is not used immediately after reconstitution, resuspend the microspheres for a few seconds by hand agitation before the suspension is drawn into the syringe. Reconstituted suspension within a vial may be used for up to 3 hours from the time of its reconstitution. Maintain the vial containing the reconstituted suspension at room temperature.
2.4 Administration Instructions
Inspect visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted suspension is milky-white, and does not contain visible particulate matter. Do not use the single-patient use vial for more than one patient.
Administer Lumason as an intravenous bolus injection.
Intravesical Administration in Pediatric Patients
- Insert a sterile 6 french to 8 french urinary catheter into the bladder under sterile conditions;
- Empty the bladder of urine, and then fill the bladder with saline (sterile 0.9% sodium chloride solution) to approximately one third or half of its predicted total volume. The total bladder volume in children is calculated as [(age in years + 2) x 30] mL;
- Administer Lumason as an intravesical bolus injection through the urinary catheter;
- Continue filling the bladder with saline until the patient has the urge to micturate or at the first sign of back pressure to the infusion.
- Immediately following the first voiding, the bladder may be refilled with normal saline for a second cycle of voiding and imaging, without the need of a second Lumason administration
2.5 Imaging Guidelines
After baseline non-contrast echocardiography is complete, adjust the mechanical index for the ultrasound device to 0.8 or lower. Continue ultrasound imaging following Lumason injection.
After identification of the target focal lesion on non-contrast ultrasound examination, hold transducer still while switching scanner to low mechanical index (≤ 0.4) contrast-specific imaging. Continue ultrasound imaging following Lumason injection.
Ultrasonography of the Urinary Tract
After baseline non-contrast ultrasound examination of the kidney and bladder, switch the scanner to low mechanical index (≤0.4) contrast specific imaging. Perform continuous alternate ultrasound imaging of the bladder, ureters, and kidneys during filling and voiding of the bladder.
-
3 DOSAGE FORMS AND STRENGTHS
For injectable suspension: Lumason is supplied as a 3-part single-patient use kit comprised of:
- one Lumason clear vial containing 25 mg of lipid-type A sterile white lyophilized powder with headspace filled with 60.7 mg of sulfur hexafluoride gas
- one prefilled syringe containing 5 mL Sodium Chloride 0.9% Injection, USP (Diluent)
- one Mini-Spike
Following reconstitution, Lumason is a homogeneous, milky white suspension containing1.5 to 5.6 x108 microspheres/ mL with 45 mcg/mL of sulfur hexafluoride.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Cardiopulmonary Reactions
Serious cardiopulmonary reactions, including fatalities have occurred uncommonly during or shortly following administration of ultrasound contrast agents, including Lumason. These reactions typically occurred within 30 minutes of administration. The risk for these reactions may be increased among patients with unstable cardiopulmonary conditions (acute myocardial infarction, acute coronary artery syndromes, worsening or unstable congestive heart failure, or serious ventricular arrhythmias). Always have cardiopulmonary resuscitation personnel and equipment readily available prior to Lumason administration and monitor all patients for acute reactions.
The reported reactions that may follow the administration of ultrasound contrast agents include: fatal cardiac or respiratory arrest, shock, syncope, symptomatic arrhythmias (atrial fibrillation, tachycardia, bradycardia, supraventricular tachycardia, ventricular fibrillation, and ventricular tachycardia), hypertension, hypotension, dyspnea, hypoxia, chest pain, respiratory distress, stridor, wheezing, loss of consciousness, and convulsions.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions such as skin erythema, rash, urticaria, flushing, throat tightness, dyspnea, or anaphylactic shock have uncommonly been observed following the injection of Lumason. These reactions may occur in patients with no history of prior exposure to sulfur hexafluoride lipid containing microspheres. Always have cardiopulmonary resuscitation personnel and equipment readily available prior to Lumason administration and monitor all patients for hypersensitivity reactions.
5.3 Systemic Embolization
When administering Lumason to patients with cardiac shunt, microspheres can bypass filtering by the lung and enter the arterial circulation. Assess patients with shunts for embolic phenomena following Lumason administration. Lumason is only for intravenous and/or intravesical administration; do not administer Lumason by intra- arterial injection [see Dosage and Administration (2.1)].
5.4 Ventricular Arrhythmia Related to High Mechanical Index
High ultrasound mechanical index values may cause microsphere cavitation or rupture and lead to ventricular arrhythmias. Additionally, end-systolic triggering with high mechanical indices has been reported to cause ventricular arrhythmias.
Lumason is not recommended for use at mechanical indices greater than 0.8.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Cardiopulmonary reactions [see Warnings and Precautions (5.1)]
- Hypersensitivity reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In completed clinical trials, a total of 6984 adult subjects (128 healthy volunteers and 6856 patients) received Lumason at cumulative doses ranging from 0.2 to 161 mL (mean 9.8 mL). Lumason was administered mainly as single or multiple injections; however, some subjects received infusion dosing. The majority (75%) of subjects received Lumason at cumulative doses of 10 mL or less. There were 64% men and 36% women, with an average age of 59 years (range 17 to 99 years). A total of 79% subjects were White; 4% were Black; 16% were Asian; <1% were Hispanic; and <1% were in other racial groups or race was not reported.
In the clinical trials, serious adverse reactions were observed in 2 subjects; one who experienced a hypersensitivity- type rash and presyncope and another who experienced anaphylactic shock shortly following Lumason administration.
The most commonly reported adverse reactions among patients (occurring among at least 0.2% of patients) are listed below (Table 1). Most adverse reactions were mild to moderate in intensity and resolved spontaneously.
*occurring in at least 0.2% of patients Table 1. Adverse Reactions in Adult Patients*
n = 6856Number (%) of Patients with Adverse Reactions 340 (5%) Headache 65 (1%) Nausea 37 (0.5%) Dysgeusia 29 (0.4%) Injection site pain 23 (0.3%) Feeling Hot 18 (0.3%) Chest discomfort 17 (0.2%) Chest pain 12 (0.2%) Dizziness 11 (0.2%) Injection Site Warmth 11 (0.2%) In completed clinical trials for echocardiography, a total of 12 pediatric patients received Lumason at a dose of 0.03 mL/kg. No adverse reactions were identified in pediatric patients [see Clinical Studies (14.1)].
6.2 Postmarketing Experience
In the international postmarketing clinical experience and clinical trials, serious adverse reactions have uncommonly been reported following administration of Lumason. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The serious adverse reactions include fatalities, especially in a pattern of symptoms suggestive of anaphylactoid/hypersensitivity reactions. Other serious reactions included arrhythmias and hypertensive episodes. These reactions typically occurred within 30 minutes of Lumason administration.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no data with Lumason use in pregnant women to inform any drug-associated risks. No adverse developmental outcomes were observed in animal reproduction studies with administration of sulfur hexafluoride lipid-type A microspheres in pregnant rats and rabbits during organogenesis at doses up to at least 10 and 20 times, respectively, the maximum human dose of 4.8 mL based on body surface area (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Lumason was administered intravenously to rats at doses of 0.2, 1, and 5 mL/kg (approximately 0.4, 2, and 10 times the recommended maximum human dose of 4.8 mL, respectively, based on body surface area); Lumason doses were administered daily for about 30 consecutive days, from two weeks before pairing until the end of organogenesis. Lumason was administered intravenously to rabbits at doses of 0.2, 1, and 5 mL/kg (approximately 0.8, 4, and 20 times the recommended maximum human dose, respectively, based on body surface area); Lumason doses were administered daily from gestation day 6 to day 19 inclusive. No significant findings on the fetus were observed.
8.2 Lactation
There are no data on the presence of Lumason in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Lumason and any potential adverse effects on the breastfed infant from Lumason or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness have been established for use in pediatric patients with suboptimal echocardiograms to opacify the left ventricular chamber and to improve delineation of the left endocardial border. Safety and effectiveness in pediatric patients are based on adequate and well-controlled studies in adults and are supported by a clinical study in 12 pediatric patients (mean age: 13.8 years) with extrapolation of efficacy to younger pediatric patients. No new adverse reactions were reported in the pediatric study [see Adverse Reactions (6.1) and Clinical Studies (14.1)]. Safety of intravenous use of Lumason was based on evaluation of published literature involving the use of Lumason in over 1400 pediatric patients (0 to 17 years).
Safety and effectiveness in pediatric patients has been established for use in ultrasonography of the liver for characterization of focal liver lesions from adequate and well controlled trials in adult patients and a clinical study of 44 pediatric patients [see Clinical Studies (14)]. Safety of intravenous use of Lumason was based on evaluation of published literature involving use of Lumason in over 1400 pediatric patients. Non-fatal anaphylaxis was reported in one pediatric patient.
Ultrasonography of the Urinary Tract
Safety and effectiveness in pediatric patients has been established for use in ultrasonography of the urinary tract for the evaluation of suspected or known vesicoureteral reflux from two published studies comprising a total of 411 pediatric patients [see Clinical Studies (14)]. Safety of intravesical use of Lumason was based on evaluation of published literature involving use of Lumason in over 6000 pediatric patients. No adverse reactions were reported.
8.5 Geriatric Use
Of the total number of 6856 adult patients in clinical studies of Lumason, 39% were 65 and over, while 11% were 75 and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly or younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
11 DESCRIPTION
Lumason (sulfur hexafluoride lipid-type A microspheres) for injectable suspension, for intravenous or intravesical use is used to prepare the ultrasound contrast agent. The single-patient use kit contains the following three items:
1) one clear glass 10 mL vial containing 25 mg of white lyophilized powder lipid-type A, 60.7 mg of sulfur hexafluoride gas and capped with a blue flip-cap
2) one prefilled syringe containing 5 mL Sodium Chloride 0.9% Injection, USP (Diluent)
Each vial is formulated as a 25 mg sterile, pyrogen-free lyophilized powder containing 24.56 mg of polyethylene glycol 4000, 0.19 mg of distearoylphosphatidyl-choline (DSPC), 0.19 mg of dipalmitoylphosphatidylglycerol sodium (DPPG-Na) and 0.04 mg of palmitic acid. The headspace of each vial contains 6.07 mg/mL (± 2 %) sulfur hexafluoride, SF6, or 60.7 mg per vial.
Each prefilled syringe with 5 mL of diluent 0.9% Sodium Chloride Injection is sterile, nonpyrogenic, preservative free containing 9 mg sodium chloride per mL.
Upon reconstitution with 5mL diluent, Lumason is a milky white, homogeneous suspension containing sulfur hexafluoride lipid-type A microspheres. The suspension is isotonic and has a pH of 4.5 to 7.5.
The sulfur hexafluoride lipid microspheres are composed of SF6 gas in the core surrounded by an outer shell monolayer of phospholipids consisting DSPC and DPPG-Na with palmitic acid as a stabilizer.
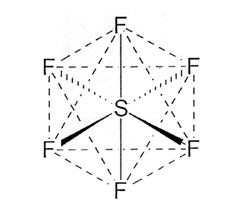
Sulfur hexafluoride has a molecular weight of 145.9 and the following chemical structure:
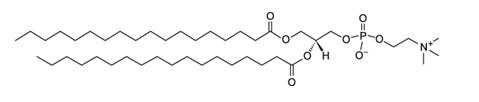
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC), with empirical formula C44H88NO8P, has a molecular weight of 790.6 and the following chemical structure:
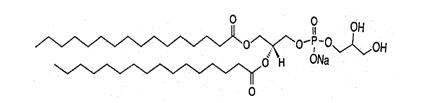
1,2-Dipalmitoyl-sn-glycero-3-phospho-rac-glycerol sodium (DPPG-Na), with empirical formula C38H74 NaO10P, has a molecular weight of 745 and the following chemical structure:
Each milliliter of reconstituted Lumason suspension contains 1.5 to 5.6 x 108 microspheres, 68 mcg SF6 (12 mcL), 0.038 mg DSPC, 0.038 mg DPPG-Na, 4.91 mg polyethylene glycol 4000 and 0.008 mg palmitic acid. The sulphur hexafluoride associated with the microspheres suspension is 45 mcg/mL. Fifteen to twenty three percent of the total lipids in the suspension are associated with the microspheres.
The sulfur hexafluoride lipid microsphere characteristics are listed in Table 2:
Table 2. Microsphere Characteristics Mean diameter range 1.5 – 2.5 μm Percent of microspheres ≤ 10 µm ≥ 99% Upper size limit 100.0% ≤ 20 µm -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Within the blood, the acoustic impedance of Lumason microspheres is lower than that of the surrounding non-aqueous tissue. Therefore, an ultrasound beam is reflected from the interface between the microspheres and the surrounding tissue. The reflected ultrasound signal provides a visual image that shows a contrast between the blood and the surrounding tissues.
For ultrasonography of the urinary tract in pediatric patients, the intravesically administered Lumason microspheres increase signal intensity of fluids within the urethra, bladder, ureters, and renal pelvis.
12.2 Pharmacodynamics
Lumason provides useful echocardiographic signal intensity for two minutes after intravenous injection. Lumason microspheres are destroyed and contrast enhancement decreases as the mechanical index increases (values of 0.8 or less are recommended).
For ultrasonography of the liver, Lumason provides dynamic patterns of differential signal intensity enhancement between focal liver lesions and liver parenchyma during the arterial, portal venous, and late phase of signal intensity enhancement of the microvasculature.
In ultrasonography of the urinary tract, Lumason facilitates the detection of reflux of fluid from the bladder into the ureters.
The effect of Lumason on pulmonary hemodynamics was studied in a prospective, open-label study of 36 patients scheduled for right heart catheterization, including 18 with mean pulmonary arterial pressure (MPAP) > 25 mmHg and 18 with MPAP ≤ 25 mmHg. No clinically important pulmonary hemodynamic changes were observed. This study did not assess the effect of Lumason on visualization of cardiac or pulmonary structures.
12.3 Pharmacokinetics
The pharmacokinetic of the SF6 gas component of Lumason was evaluated in 12 healthy adult subjects. After intravenous bolus injections of 0.03 mL/kg and 0.3 mL/kg of Lumason, corresponding to approximately 1 and 10 times the recommended doses, concentrations of SF6 in blood peaked within 1 to 2 minutes for both doses. The terminal half-life of SF6 in blood was approximately 10 minutes for the 0.3 mL/kg dose. The area-under-thecurve of SF6 was dose-proportional over the dose range studied.
In a study of healthy subjects, the mean values for the apparent steady-state volume of distribution of SF6 following intravenous administration, were 341 mcL and 710 mcL for Lumason doses of 0.03 mL/kg and 0.3 mL/kg, respectively. Preferential distribution to the lung is likely responsible for these values.
Following intravenous administration, the SF6 component of Lumason is eliminated via the lungs. In a clinical study that examined SF6 elimination twenty minutes following Lumason injection, the mean cumulative recovery of SF6 in expired air was 82 ± 20% (SD) at the 0.03 mL/kg dose and 88 ± 26% (SD) at the 0.3 mL/kg dose.
SF6 undergoes first pass elimination within the pulmonary circulation; approximately 40% to 50% of the SF6 content was eliminated in the expired air during the first minute following Lumason injection.
SF6 undergoes little or no biotransformation; following intravenous administration, 88% of an administered dose is recovered unchanged in expired air.
Pharmacokinetics in Specific Populations
In a study of patients with pulmonary impairment, blood concentrations of SF6 peaked at 1 to 4 minutes following Lumason administration. The cumulative recovery of SF6 in expired air was 102 ± 18% (mean ± standard deviation), and the terminal half-life of SF6 in blood was similar to that measured in healthy subjects.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies were performed to evaluate the carcinogenic potential of Lumason. No evidence of genotoxicity was found in the following studies conducted with Lumason: 1) a bacterial mutagenesis (Ames) assay, 2) an in vitro human lymphocyte chromosome aberration assay, and 3) an in vivo mouse micronucleus assay.
No impairment of fertility was observed in rats receiving Lumason at doses up to 8 times the human dose based on body surface area.
-
14 CLINICAL STUDIES
14.1 Echocardiography
A total of 191 patients with suspected cardiac disease and suboptimal non-contrast echocardiography received Lumason in three multi-center controlled clinical trials (76 patients in Study A, 62 patients in Study B, and 53 patients in Study C). Among these patients, there were 127 men and 64 women. The mean age was 59 years (range 22 to 96 years). The racial and ethnic representations were 79% White, 16% Black, 4% Hispanic, < 1% Asian, and < 1% other racial or ethnic groups. The mean weight was 204 lbs (range 92 to 405 lbs). Approximately 20% of the patients had a chronic pulmonary disorder and 30% had a history of heart failure. Of the 106 patients for whom a New York Heart Association (NYHA) classification of heart failure was assigned, 49% were Class I, 33% were Class II, and 18% were Class III. Patients with NYHA Class IV heart failure were not included in these studies.
In Studies A and B, each patient received four intravenous bolus injections of Lumason (0.5, 1, 2, and 4 mL), in randomized order. In Study C, each patient received two doses of Lumason (1 mL and 2 mL) in randomized order. All three studies assessed endocardial border delineation and left ventricular opacification. For each patient in each study, echocardiography with Lumason was compared to non-contrast (baseline) echocardiography. A recording of 2D echocardiography was obtained from 30 seconds prior to each injection to at least 15 minutes after dosing or until the disappearance of the contrast effect, whichever was longer. Contrast and non-contrast echocardiographic images for each patient were evaluated by two independent reviewers, who were blinded to clinical information and the Lumason dose. Evaluation of left ventricular endocardial border consisted of segment based assessment involving six endocardial segments and using two apical views (2- and 4 chamber views).
Endocardial Border Delineation and Duration of Useful Contrast Effect
In all three studies, administration of Lumason improved left ventricular endocardial border delineation. The majority of the patients who received a 2.0 mL dose of Lumason had improvement in endocardial border delineation manifested as visualization of at least two additional endocardial border segments. Table 3 demonstrates the improvement in endocardial border delineation following Lumason administration as a reduction in percentage of patients with inadequate border delineation in at least one pair of adjacent segments (combined 2-chamber and 4-chamber view). The results are shown by reader.
Table 3. Reduction in Percentage of Patients with Inadequate Border Delineation Reader Study A
N = 76Study B
N = 62Study C
N = 53Non-contrast Lumason Non-contrast Lumason Non-contrast Lumason A 60 (79%) 22 (33%) 31 (50%) 12 (19%) 12 (23%) 10 (19%) B 62 (82%) 29 (37%) 54 (87%) 6 (10%) 45 (85%) 20 (38%) Following the first appearance of contrast within the left ventricle the mean duration of useful contrast effect ranged from 1.7 to 3.1 minutes.
Left Ventricular Opacification
In all three studies, complete left ventricular opacification was observed in 52% to 80% of the patients following administration of a 2.0-mL dose of Lumason. The studies did not sufficiently assess the effect of Lumason upon measures of left ventricular ejection fraction and wall motion.
Twelve pediatric patients 9 to 17 years of age with suspected cardiac disease and suboptimal non-contrast echocardiography received Lumason in one prospective multicenter clinical trial. Patients received Lumason at a dose of 0.03 mL/kg (mean 1.83 mL). There were 7 female, 10 white, and 2 black patients.
For both the non-contrast and contrast-enhanced images, standard apical 4-, 2-, and 3-chamber views with harmonic imaging were acquired. Contrast and non-contrast images for each patient were evaluated by three independent reviewers, who were blinded to clinical information.
Endocardial Border Delineation
Evaluation of left ventricular endocardial border delineation consisted of segment-based assessment of the left ventricle divided into 17 endocardial segments. The delineation of each segment’s endocardial border was rated as inadequate, sufficient, or good. An exam was considered suboptimal if any of the patient’s apical views had 2 or more adjacent segments with inadequate delineation scores.
The majority of screened patients had adequate delineation of the left ventricular endocardial border without administration of contrast. The number of patients with inadequate left ventricular endocardial border delineation without contrast and after Lumason are shown for the 12 patients, by reader, in Table 4.
a Reader A had missing segment data with contrast echocardiography for one patient;
b Reader B had missing segment data with non-contrast echocardiography for one patient;
bb Reader B had missing segment data with contrast echocardiography for three patients;
c Reader C had missing segment data with contrast echocardiography for one patientTable 4. Number of Pediatric Patients with Inadequate Border Delineation with and without Lumason Reader A Reader B Reader C Non-contrast 12/12 11/11b 2/12 Lumason 1/11a 0/9bb 0/11c Left Ventricular Opacification
Complete left ventricular opacification was observed in all the patients by all 3 readers following administration of Lumason.
14.2 Ultrasonography of the Liver
A total of 499 patients with at least 1 focal liver lesion requiring characterization were evaluated in two studies (259 patients in Study A, 240 patients in Study B). Among these patients, there were 259 men and 240 women. The mean age was 56 years (range 19 to 93 years). The racial and ethnic representations were 74% White, 11% Black, 9% Hispanic, 5% Asian, and 1% other racial or ethnic groups. The mean weight was 80 kg (range 44 to 173 kg).
In both studies, prior to Lumason administration, gray scale and Doppler (color or power imaging) ultrasound examinations of the target lesion were performed using commercially available ultrasound equipment and using standard techniques. Each patient received an intravenous injection of 2.4 mL of Lumason (up to 2 injections were allowed, 91% patients received 1 injection). Following Lumason administration, ultrasound examination of the target lesion was carried out using contrast-specific imaging modes operating at MI ≤ 0.4. The probe was positioned to provide optimal visualization over the target lesion and was kept in the same position for at least 180 seconds.
Truth standard included: histology/surgery, contrast CT, contrast MRI, and/or 6 month follow-up.
For each study, the interpretation of images was conducted by three independent readers who were blinded to clinical data. Lesions were characterized as malignant or benign. Separate blinded readers assessed the truth standard images.
Results of both studies demonstrated an improvement in characterization of focal liver lesions using Lumason ultrasound compared to non-contrast ultrasound images. Table 5 summarizes the efficacy results by reader.
* Statistically significant improvement from non-contrast (p<0.05 based on McNemar’s test) Table 5. Diagnostic Performance of Lumason Ultrasound for Characterization of Focal Liver Lesions Study A: Sensitivity (patients with malignant lesions)
N=119Specificity (patients with benign lesions)
N=140Lumason
%Non-contrast
%Difference
(95% CI)Lumason
%Non-contrast
%Difference
(95% CI)Reader 1 87* 49 38 (30, 54) 71 63 8 (-4, 21) Reader 2 76* 35 41 (29, 52) 83* 54 29 (21, 44) Reader 3 92* 16 76 (67, 84) 73* 22 51 (40, 61) Study B: Sensitivity (patients with malignant lesions)
N=124Specificity (patients with benign lesions)
N=116Lumason
%Non-contrast
%Difference
(95% CI)Lumason
%Non-contrast
%Difference
(95% CI)Reader 4 65 53 12 (-1, 23) 72* 24 48 (35, 58) Reader 5 61* 41 20 (7, 32) 67* 7 60 (50, 70) Reader 6 47 66 -19 (-31, -7) 88* 59 29 (18, 40) In one published study, 44 patients with an indeterminate focal liver lesion (23 males, 21 females, age range: 4-18 years, median 11.5 years) were evaluated after intravenous bolus administration of 1.2 to 2.4 mL of Lumason. The findings of Lumason ultrasound images were compared to CT, MRI or histology. Specificity was 98% (43/44 patients).
14.3 Ultrasonography of the Urinary Tract
The efficacy of Lumason for the evaluation of pediatric patients with suspected or known vesicoureteral reflux was established in two published open-label single center studies (A and B). Patients received 1 mL of Lumason intravesically and underwent voiding urosonography (VUS). Patients were also evaluated with voiding cystourethrography (VCUG) as the reference standard. The presence or absence of urinary reflux with Lumason ultrasound was compared to the radiographic reference standard.
Study A evaluated 183 patients (94 male, 89 female; age 2 days - 44 months) with a total of 366 kidney-ureter units. The images were interpreted by one on-site reader, blinded to the reference standard. Out of 103 reference standard-positive images, Lumason VUS was positive in 89 units and falsely negative in 14 units. In 263 units with negative reference standard, the Lumason ultrasonography was negative in 226 and falsely positive in 37.
Study B evaluated 228 patients (123 male, 105 female; age 6 days -13 years) with a total of 463 kidney-ureter units (some patients had more than 2 units). The images were interpreted independently by two on-site readers, blinded to the reference standard. Out of 71 reference standard positive images, Lumason ultrasonography was positive in 57 and falsely negative in 14. In 392 units with negative reference standard, Lumason ultrasonography was negative in 302 and falsely positive in 90.
-
16 HOW SUPPLIED/STORAGE
AND HANDLING
Lumason (sulfur hexafluoride lipid-type A microspheres) for injectable suspension is supplied as a single patient- use kit as follows:
- One Lumason vial of 25 mg lipid-type A white lyophilized powder with headspace fill of 60.7 mg of sulfur hexafluoride
- One prefilled syringe containing 5mL of Sodium Chloride 0.9% Injection, USP (Diluent)
- One Mini-Spike
Each kit is packaged in a clear plastic container.
(NDC: 0270-7099-73) 5 Kits per carton -
17 PATIENT COUNSELING INFORMATION
Advise patients to inform their healthcare provider if they develop any symptoms of hypersensitivity after LUMASON administration including rash, wheezing, or shortness of breath.
Manufactured for:
Bracco Diagnostics Inc., Monroe Township, NJ 08831
by
BRACCO Suisse SA
Plan-les-Ouates Geneve, Switzerland (Lumason lyophilized powder vial-25 mg lipid-type A/60.7 mg sulfur hexafluoride gas)Vetter Pharma-Fertigung GmbH & Co. KG
88212 Ravensburg, Germany (Sodium Chloride 0.9% Injection, USP)B. Braun Melsungen AG
34212 Melsungen, Germany (Mini-Spike)This product is covered by US Patent No. 5,686,060

- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LUMASON
sulfur hexafluoride kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0270-7099 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0270-7099-73 5 in 1 BOX 01/15/2020 1 1 in 1 KIT; Type 1: Convenience Kit of Co-Package Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 25 mg Part 2 1 SYRINGE 5 mL Part 1 of 2 LUMASON
sulfur hexafluoride injection, powder, lyophilized, for suspensionProduct Information Route of Administration INTRAVESICAL, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SULFUR HEXAFLUORIDE (UNII: WS7LR3I1D6) (SULFUR HEXAFLUORIDE - UNII:WS7LR3I1D6) SULFUR HEXAFLUORIDE 60.7 mg in 1 mg DISTEAROYLPHOSPHATIDYLCHOLINE, DL- (UNII: EAG959U971) (DISTEAROYLPHOSPHATIDYLCHOLINE, DL- - UNII:EAG959U971) DISTEAROYLPHOSPHATIDYLCHOLINE, DL- .19 mg in 1 mg SODIUM 1,2-DIPALMITOYL-SN-GLYCERO-3-PHOSPHO-(1'-RAC-GLYCEROL) (UNII: 841B886EJ7) (SODIUM 1,2-DIPALMITOYL-SN-GLYCERO-3-PHOSPHO-(1'-RAC-GLYCEROL) - UNII:841B886EJ7) SODIUM 1,2-DIPALMITOYL-SN-GLYCERO-3-PHOSPHO-(1'-RAC-GLYCEROL) .19 mg in 1 mg Inactive Ingredients Ingredient Name Strength POLYETHYLENE GLYCOL 4000 (UNII: 4R4HFI6D95) 24.56 mg in 1 mg PALMITIC ACID (UNII: 2V16EO95H1) .04 mg in 1 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 25 mg in 1 VIAL; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203684 01/15/2020 Part 2 of 2 DILUENT
sodium chloride 0.9% injection,usp injectionProduct Information Route of Administration INTRAVESICAL, INTRAVENOUS Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) .9 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 5 mL in 1 SYRINGE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203684 01/15/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203684 01/15/2020 Labeler - BRACCO DIAGNOSTICS INC (849234661) Registrant - BRACCO DIAGNOSTICS INC (849234661) Establishment Name Address ID/FEI Business Operations Bracco Suisse SA 485635705 MANUFACTURE(0270-7099) , ANALYSIS(0270-7099) Establishment Name Address ID/FEI Business Operations AIRGAS USA LLC 831996595 API MANUFACTURE(0270-7099)





Trademark Results [Lumason]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LUMASON 88284789 5834181 Live/Registered |
Bracco Diagnostics Inc. 2019-01-31 |
 LUMASON 85929727 4786320 Live/Registered |
Bracco Diagnostics Inc. 2013-05-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.