UMECLIDINIUM ELLIPTA- umeclidinium aerosol, powder
Drug Labeling and Warnings
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Umeclidinium ELLIPTA safely and effectively. See full prescribing information for Umeclidinium ELLIPTA.
Umeclidinium ELLIPTA inhalation powder, for oral inhalation use
Initial U.S. Approval: 2013INDICATIONS AND USAGE
Umeclidinium ELLIPTA is an anticholinergic indicated for the maintenance treatment of patients with chronic obstructive pulmonary disease (COPD). (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Inhalation powder: 62.5 mcg of umeclidinium per actuation. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Do not initiate in acutely deteriorating COPD. Do not use to treat acute symptoms. (5.1)
- If paradoxical bronchospasm occurs, discontinue Umeclidinium ELLIPTA and institute alternative therapy. (5.2)
- Worsening of narrow-angle glaucoma may occur. Use with caution in patients with narrow-angle glaucoma and instruct patients to contact a healthcare provider immediately if symptoms occur. (5.4)
- Worsening of urinary retention may occur. Use with caution in patients with prostatic hyperplasia or bladder-neck obstruction and instruct patients to contact a healthcare provider immediately if symptoms occur. (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥2% and more common than placebo) include nasopharyngitis, upper respiratory tract infection, cough, arthralgia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Prasco Laboratories at 1-866-525-0688 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Anticholinergics: May interact additively with concomitantly used anticholinergic medications. Avoid administration of Umeclidinium ELLIPTA with other anticholinergic-containing drugs. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Deterioration of Disease and Acute Episodes
5.2 Paradoxical Bronchospasm
5.3 Hypersensitivity Reactions, including Anaphylaxis
5.4 Worsening of Narrow-Angle Glaucoma
5.5 Worsening of Urinary Retention
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Anticholinergics
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Dose-Ranging Trials
14.2 Maintenance Treatment: Confirmatory Trials
14.3 Maintenance Treatment: Combination with an ICS/LABA Trials
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
The recommended dosage of Umeclidinium ELLIPTA for maintenance treatment of COPD is 1 actuation (umeclidinium 62.5 mcg) once daily by oral inhalation.
- Umeclidinium ELLIPTA should be used at the same time every day. Do not use Umeclidinium ELLIPTA more than 1 time every 24 hours.
- No dosage adjustment is required for geriatric patients, patients with renal impairment, or patients with moderate hepatic impairment [see Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Deterioration of Disease and Acute Episodes
Umeclidinium ELLIPTA should not be initiated in patients during rapidly deteriorating or potentially life-threatening episodes of COPD. Umeclidinium ELLIPTA has not been studied in subjects with acutely deteriorating COPD. The initiation of Umeclidinium ELLIPTA in this setting is not appropriate.
Umeclidinium ELLIPTA should not be used for the relief of acute symptoms, i.e., as rescue therapy for the treatment of acute episodes of bronchospasm. Umeclidinium ELLIPTA has not been studied in the relief of acute symptoms and extra doses should not be used for that purpose. Acute symptoms should be treated with an inhaled, short-acting beta2-agonist.
If Umeclidinium ELLIPTA no longer controls symptoms of bronchoconstriction; the patient’s inhaled, short-acting beta2-agonist becomes less effective; or the patient needs more short-acting beta2-agonist than usual, these may be markers of deterioration of disease. In this setting re-evaluate the patient and the COPD treatment regimen at once. Increasing the daily dose of Umeclidinium ELLIPTA beyond the recommended dose is not appropriate in this situation.
5.2 Paradoxical Bronchospasm
As with other inhaled therapies, Umeclidinium ELLIPTA can produce paradoxical bronchospasm, which may be life threatening. If paradoxical bronchospasm occurs following dosing with Umeclidinium ELLIPTA, it should be treated immediately with an inhaled, short-acting bronchodilator; Umeclidinium ELLIPTA should be discontinued immediately; and alternative therapy should be instituted.
5.3 Hypersensitivity Reactions, including Anaphylaxis
Hypersensitivity reactions such as anaphylaxis, angioedema, pruritus, rash, and urticaria may occur after administration of Umeclidinium ELLIPTA. Discontinue Umeclidinium ELLIPTA if such reactions occur. There have been reports of anaphylactic reactions in patients with severe milk protein allergy after inhalation of other powder medications containing lactose; therefore, patients with severe milk protein allergy should not use Umeclidinium ELLIPTA [see Contraindications (4), Adverse Reactions (6.2)].
5.4 Worsening of Narrow-Angle Glaucoma
Umeclidinium ELLIPTA should be used with caution in patients with narrow-angle glaucoma. Prescribers and patients should also be alert for signs and symptoms of acute narrow-angle glaucoma (e.g., eye pain or discomfort, blurred vision, visual halos or colored images in association with red eyes from conjunctival congestion and corneal edema). Instruct patients to consult a healthcare provider immediately if any of these signs or symptoms develop.
5.5 Worsening of Urinary Retention
Umeclidinium ELLIPTA, like all therapies containing an anticholinergic, should be used with caution in patients with urinary retention. Prescribers and patients should be alert for signs and symptoms of urinary retention (e.g., difficulty passing urine, painful urination), especially in patients with prostatic hyperplasia or bladder-neck obstruction. Instruct patients to consult a healthcare provider immediately if any of these signs or symptoms develop.
-
6 ADVERSE REACTIONS
The following adverse reactions are described in greater detail in other sections:
- Paradoxical bronchospasm [see Warnings and Precautions (5.2)]
- Worsening of narrow-angle glaucoma [see Warnings and Precautions (5.4)]
- Worsening of urinary retention [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the 8 clinical trials conducted to support initial approval of Umeclidinium ELLIPTA, a total of 1,663 subjects with COPD (mean age: 62.7 years; 89% white; 65% male across all treatments, including placebo) received at least 1 inhalation dose of umeclidinium at doses of 62.5 or 125 mcg. In the 4 randomized, double-blind, placebo- or active-controlled, efficacy clinical trials, 1,185 subjects received umeclidinium for up to 24 weeks, of which 487 subjects received the recommended dose of umeclidinium 62.5 mcg. In a 12-month, randomized, double-blind, placebo-controlled, long-term safety trial, 227 subjects received umeclidinium 125 mcg for up to 52 weeks [see Clinical Studies (14)].
The incidence of adverse reactions associated with umeclidinium ELLIPTA in Table 1 is based upon 2 placebo-controlled trials: one 24-week trial (Trial 1) and one 12-week trial (Trial 2) [see Clinical Studies (14.2)].
Table 1. Adverse Reactions with Umeclidinium ELLIPTA with ≥1% Incidence and More Common than Placebo in Subjects with Chronic Obstructive Pulmonary Disease Adverse Reaction
Umeclidinium ELLIPTA
(n = 487)
%
Placebo
(n = 348)
%
Infections and infestations
Nasopharyngitis
8%
7%
Upper respiratory tract infection
5%
4%
Pharyngitis
1%
<1%
Viral upper respiratory tract infection
1%
<1%
Respiratory, thoracic, and mediastinal disorders
Cough
3%
2%
Musculoskeletal and connective tissue disorders
Arthralgia
2%
1%
Myalgia
1%
<1%
Gastrointestinal disorders
Abdominal pain upper
1%
<1%
Toothache
1%
<1%
Injury, poisoning, and procedural complications
Contusion
1%
<1%
Cardiac disorders
Tachycardia
1%
<1%
Other adverse reactions with umeclidinium ELLIPTA observed with an incidence <1% but more common than placebo included atrial fibrillation.
In a long-term safety trial (Trial 3), 336 subjects (n = 227 umeclidinium 125 mcg, n = 109 placebo) were treated for up to 52 weeks with umeclidinium 125 mcg or placebo. The demographic and baseline characteristics of the long-term safety trial were similar to those of the efficacy trials described above. Adverse reactions that occurred with a frequency ≥1% in subjects receiving umeclidinium 125 mcg that exceeded that in placebo in this trial were: nasopharyngitis, upper respiratory tract infection, urinary tract infection, pharyngitis, pneumonia, lower respiratory tract infection, rhinitis, supraventricular tachycardia, supraventricular extrasystoles, sinus tachycardia, idioventricular rhythm, headache, dizziness, sinus headache, cough, back pain, arthralgia, pain in extremity, neck pain, myalgia, nausea, dyspepsia, diarrhea, rash, depression, and vertigo.
The safety and efficacy of umeclidinium ELLIPTA in combination with an inhaled corticosteroid/long-acting beta2-adrenergic agonist (ICS/LABA) were also evaluated in four 12‑week clinical trials (Trial 4, Trial 5, Trial 6, and Trial 7). A total of 1,637 subjects with COPD across four 12‑week, randomized, double-blind clinical trials received at least 1 dose of umeclidinium ELLIPTA (62.5 mcg) or placebo administered once daily in addition to background ICS/LABA (mean age: 64 years, 88% white, 65% male across all treatments). Two trials (Trials 4 and 5) evaluated umeclidinium ELLIPTA in combination with fluticasone furoate/vilanterol (FF/VI) 100 mcg/25 mcg administered once daily, and 2 trials (Trials 6 and 7) evaluated umeclidinium ELLIPTA administered once daily in combination with fluticasone propionate/salmeterol (FP/SAL) 250 mcg/50 mcg administered twice daily [see Clinical Studies (14.3)]. Adverse reactions that occurred with umeclidinium ELLIPTA in combination with an ICS/LABA were similar to those reported with umeclidinium ELLIPTA as monotherapy. In addition to the umeclidinium monotherapy adverse reactions reported above, adverse reactions occurring with umeclidinium ELLIPTA in combination with an ICS/LABA, at an incidence of ≥1% and exceeding ICS/LABA alone, were oropharyngeal pain and dysgeusia.
6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postapproval use of Umeclidinium ELLIPTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These events have been chosen for inclusion due to either their seriousness, frequency of reporting, or causal connection to Umeclidinium ELLIPTA or a combination of these factors.
Eye Disorders
Eye pain, glaucoma, vision blurred.
Immune System Disorders
Hypersensitivity reactions, including anaphylaxis, angioedema, pruritus, and urticaria.
Renal and Urinary Disorders
Dysuria, urinary retention.
Respiratory, Thoracic and Mediastinal Disorders
Dysphonia, oropharyngeal pain.
-
7 DRUG INTERACTIONS
7.1 Anticholinergics
There is potential for an additive interaction with concomitantly used anticholinergic medicines. Therefore, avoid coadministration of Umeclidinium ELLIPTA with other anticholinergic-containing drugs as this may lead to an increase in anticholinergic adverse effects [see Warnings and Precautions (5.4, 5.5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are insufficient data on the use of umeclidinium in pregnant women to inform a drug‑associated risk. Umeclidinium administered via inhalation or subcutaneously to pregnant rats and rabbits was not associated with adverse effect on embryofetal development at exposures approximately 50 and 200 times, respectively, the human exposure at the maximum recommended human daily inhaled dose (MRHDID). (See Data.)
The estimated risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: In separate embryofetal developmental studies, pregnant rats and rabbits received umeclidinium during the period of organogenesis at doses up to approximately 50 and 200 times the MRHDID, respectively (on an AUC basis at maternal inhalation doses up to 278 mcg/kg/day in rats and at maternal subcutaneous doses up to 180 mcg/kg/day in rabbits). No evidence of teratogenic effects was observed in either species.
In a perinatal and postnatal developmental study in rats, dams received umeclidinium during late gestation and lactation periods with no evidence of effects on offspring development at doses up to approximately 26 times the MRHDID (on an AUC basis at maternal subcutaneous doses up to 60 mcg/kg/day).
8.2 Lactation
Risk Summary
There is no information available on the presence of umeclidinium in human milk, the effects on the breastfed child, or the effects on milk production. Umeclidinium was detected in the plasma of offspring of lactating rats treated with umeclidinium suggesting its presence in maternal milk. (See Data.) The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Umeclidinium ELLIPTA and any potential adverse effects on the breastfed child from umeclidinium or from the underlying maternal condition.
Data
Subcutaneous administration of umeclidinium to lactating rats at greater than or equal to 60 mcg/kg/day resulted in a quantifiable level of umeclidinium in 2 of 54 pups, which may indicate transfer of umeclidinium in milk.
8.4 Pediatric Use
The safety and effectiveness of Umeclidinium ELLIPTA have not been established in pediatric patients. Umeclidinium ELLIPTA is not indicated for use in pediatric patients.
8.5 Geriatric Use
Based on available data, no adjustment of the dosage of Umeclidinium ELLIPTA in geriatric patients is necessary, but greater sensitivity in some older individuals cannot be ruled out.
Clinical trials of umeclidinium ELLIPTA included 810 subjects aged 65 years and older, and, of those, 183 subjects were aged 75 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger subjects.
8.6 Hepatic Impairment
Patients with moderate hepatic impairment (Child-Pugh score of 7-9) showed no relevant increases in Cmax or AUC, nor did protein binding differ between subjects with moderate hepatic impairment and their healthy controls. Studies in subjects with severe hepatic impairment have not been performed [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Patients with severe renal impairment (CrCl <30 mL/min) showed no relevant increases in Cmax or AUC, nor did protein binding differ between subjects with severe renal impairment and their healthy controls. No dosage adjustment is required in patients with renal impairment [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Umeclidinium ELLIPTA is an inhalation powder drug product for delivery of umeclidinium (an anticholinergic) to patients by oral inhalation.
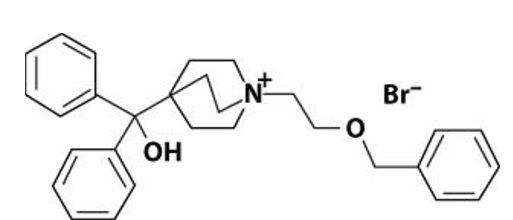
Umeclidinium bromide has the chemical name 1-[2-(benzyloxy)ethyl]-4-(hydroxydiphenylmethyl)-1-azoniabicyclo[2.2.2]octane bromide and the following chemical structure:
Umeclidinium bromide is a white powder with a molecular weight of 508.5, and the empirical formula is C29H34NO2Br (as a quaternary ammonium bromide compound). It is slightly soluble in water.
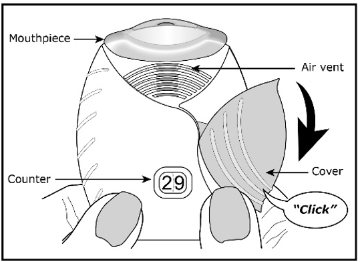
Umeclidinium ELLIPTA is a light grey and light green plastic inhaler containing a foil blister strip. Each blister on the strip contains a white powder blend of micronized umeclidinium bromide (74.2 mcg equivalent to 62.5 mcg of umeclidinium), magnesium stearate (75 mcg), and lactose monohydrate (to 12.5 mg). The lactose monohydrate contains milk proteins. After the inhaler is activated, the powder within the blister is exposed and ready for dispersion into the airstream created by the patient inhaling through the mouthpiece.
Under standardized in vitro test conditions, Umeclidinium ELLIPTA delivers 55 mcg of umeclidinium per dose when tested at a flow rate of 60 L/min for 4 seconds.
In adult subjects with obstructive lung disease and severely compromised lung function (COPD with forced expiratory volume in 1 second/forced vital capacity [FEV1/FVC] <70% and FEV1 <30% predicted or FEV1 <50% predicted plus chronic respiratory failure), mean peak inspiratory flow through the ELLIPTA inhaler was 67.5 L/min (range: 41.6 to 83.3 L/min).
The actual amount of drug delivered to the lung will depend on patient factors, such as inspiratory flow profile.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Umeclidinium is a long-acting muscarinic antagonist, which is often referred to as an anticholinergic. It has similar affinity to the subtypes of muscarinic receptors M1 to M5. In the airways, it exhibits pharmacological effects through inhibition of M3 receptors at the smooth muscle leading to bronchodilation. The competitive and reversible nature of antagonism was shown with human and animal origin receptors and isolated organ preparations. In preclinical in vitro as well as in vivo studies, prevention of methacholine- and acetylcholine-induced bronchoconstrictive effects was dose-dependent and lasted longer than 24 hours. The clinical relevance of these findings is unknown. The bronchodilation following inhalation of umeclidinium is predominantly a site-specific effect.
12.2 Pharmacodynamics
Cardiac Electrophysiology
QTc interval prolongation was studied in a double-blind, multiple-dose, placebo- and positive‑controlled, crossover study in 86 healthy subjects. Following repeat doses of umeclidinium 500 mcg once daily (8 times the recommended dosage) for 10 days, umeclidinium does not prolong QTc to any clinically relevant extent.
12.3 Pharmacokinetics
Linear pharmacokinetics was observed for umeclidinium (62.5 to 500 mcg).
Absorption
Umeclidinium plasma levels may not predict therapeutic effect. Following inhaled administration of umeclidinium in healthy subjects, Cmax occurred at 5 to 15 minutes. Umeclidinium is mostly absorbed from the lung after inhaled doses with minimum contribution from oral absorption. Following repeat dosing of inhaled Umeclidinium ELLIPTA, steady state was achieved within 14 days with up to 1.8-fold accumulation.
Distribution
Following intravenous administration to healthy subjects, the mean volume of distribution was 86 L. In vitro plasma protein binding in human plasma was on average 89%.
Elimination
Metabolism: In vitro data showed that umeclidinium is primarily metabolized by the enzyme cytochrome P450 2D6 (CYP2D6) and is a substrate for the P-glycoprotein (P-gp) transporter. The primary metabolic routes for umeclidinium are oxidative (hydroxylation, O-dealkylation) followed by conjugation (e.g., glucuronidation), resulting in a range of metabolites with either reduced pharmacological activity or for which the pharmacological activity has not been established. Systemic exposure to the metabolites is low.
Excretion: The effective half-life after once-daily orally inhaled dosing is 11 hours. Following intravenous dosing with radiolabeled umeclidinium, mass balance showed 58% of the radiolabel in the feces and 22% in the urine. The excretion of the drug-related material in the feces following intravenous dosing indicated elimination in the bile. Following oral dosing to healthy male subjects, radiolabel recovered in feces was 92% of the total dose and that in urine was <1% of the total dose, suggesting negligible oral absorption.
Specific Populations
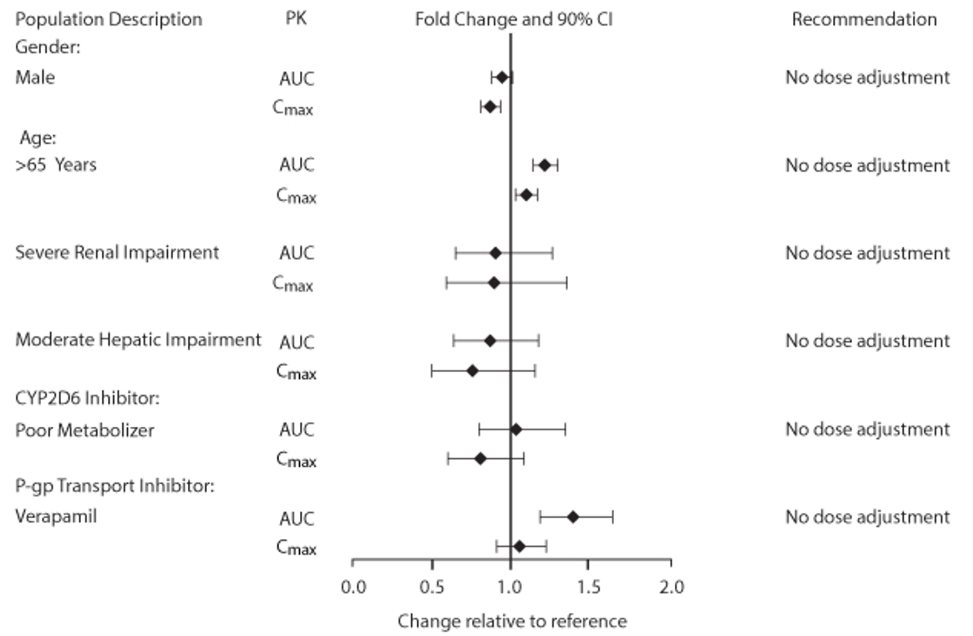
Population pharmacokinetic analysis showed no evidence of a clinically significant effect of age (40 to 93 years) (Figure 1), gender (69% male) (Figure 1), inhaled corticosteroid use (48%), or weight (34 to 161 kg) on systemic exposure of umeclidinium. In addition, there was no evidence of a clinically significant effect of race.
Patients with Hepatic Impairment: The impact of hepatic impairment on the pharmacokinetics of Umeclidinium ELLIPTA has been evaluated in subjects with moderate hepatic impairment (Child‑Pugh score of 7-9). There was no evidence of an increase in systemic exposure to umeclidinium (Cmax and AUC) (Figure 1). There was no evidence of altered protein binding in subjects with moderate hepatic impairment compared with healthy subjects. Umeclidinium ELLIPTA has not been evaluated in subjects with severe hepatic impairment.
Patients with Renal Impairment: The pharmacokinetics of Umeclidinium ELLIPTA has been evaluated in subjects with severe renal impairment (CrCl <30 mL/min). There was no evidence of an increase in systemic exposure to umeclidinium (Cmax and AUC) (Figure 1). There was no evidence of altered protein binding in subjects with severe renal impairment compared with healthy subjects.
Figure 1. Impact of Intrinsic and Extrinsic Factors on the Systemic Exposure of Umeclidinium
Drug Interaction Studies
Umeclidinium and P-glycoprotein Transporter: Umeclidinium is a substrate of P-gp. The effect of the moderate P-gp transporter inhibitor verapamil (240 mg once daily) on the steady-state pharmacokinetics of umeclidinium was assessed in healthy subjects. No effect on umeclidinium Cmax was observed; however, an approximately 1.4-fold increase in umeclidinium AUC was observed (Figure 1).
Umeclidinium and Cytochrome P450 2D6: In vitro metabolism of umeclidinium is mediated primarily by CYP2D6. However, no clinically meaningful difference in systemic exposure to umeclidinium (500 mcg) (8 times the approved dose) was observed following repeat daily inhaled dosing to normal (ultrarapid, extensive, and intermediate metabolizers) and CYP2D6 poor metabolizer subjects (Figure 1).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Umeclidinium produced no treatment-related increases in the incidence of tumors in 2-year inhalation studies in rats and mice at inhaled doses up to 137 and 295/200 mcg/kg/day (male/female), respectively (approximately 20 and 25/20 times, respectively, the MRHDID for adults on an AUC basis).
Umeclidinium tested negative in the following genotoxicity assays: the in vitro Ames assay, in vitro mouse lymphoma assay, and in vivo rat bone marrow micronucleus assay.
No evidence of impairment of fertility was observed in male and female rats at subcutaneous doses up to 180 mcg/kg/day and at inhaled doses up to 294 mcg/kg/day, respectively (approximately 100 and 50 times, respectively, the MRHDID for adults on an AUC basis).
-
14 CLINICAL STUDIES
The safety and efficacy of umeclidinium 62.5 mcg were evaluated in 3 dose-ranging trials, 2 placebo-controlled clinical trials (one 12-week trial and one 24-week trial), and a 12-month long‑term safety trial. The efficacy of umeclidinium ELLIPTA is based primarily on the dose‑ranging trials in 624 subjects and the 2 placebo-controlled confirmatory trials in 1,738 subjects with COPD, including chronic bronchitis and/or emphysema. [See Clinical Studies (14.1, 14.2).]
The safety and efficacy of umeclidinium ELLIPTA in combination with an ICS/LABA were also evaluated in four 12-week clinical trials. The efficacy of umeclidinium ELLIPTA in combination with an ICS/LABA is based on 1,637 subjects with COPD. [See Clinical Studies (14.3).]
Evidence of efficacy for umeclidinium ELLIPTA on COPD exacerbations was established by the efficacy of the umeclidinium component as part of a fixed-dose combination with an ICS/LABA, as assessed in a 12-month trial in 10,355 subjects. [See Clinical Studies (14.3).]
14.1 Dose-Ranging Trials
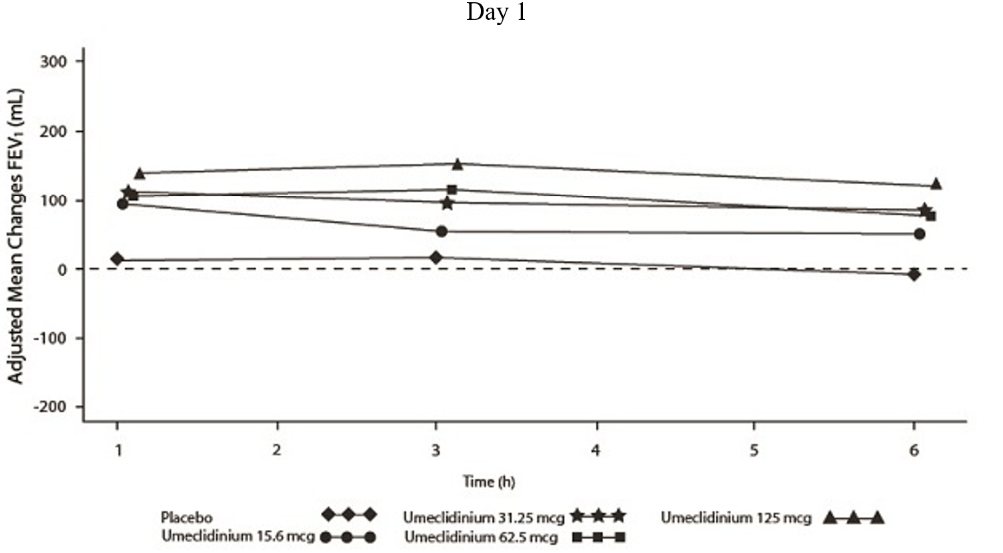
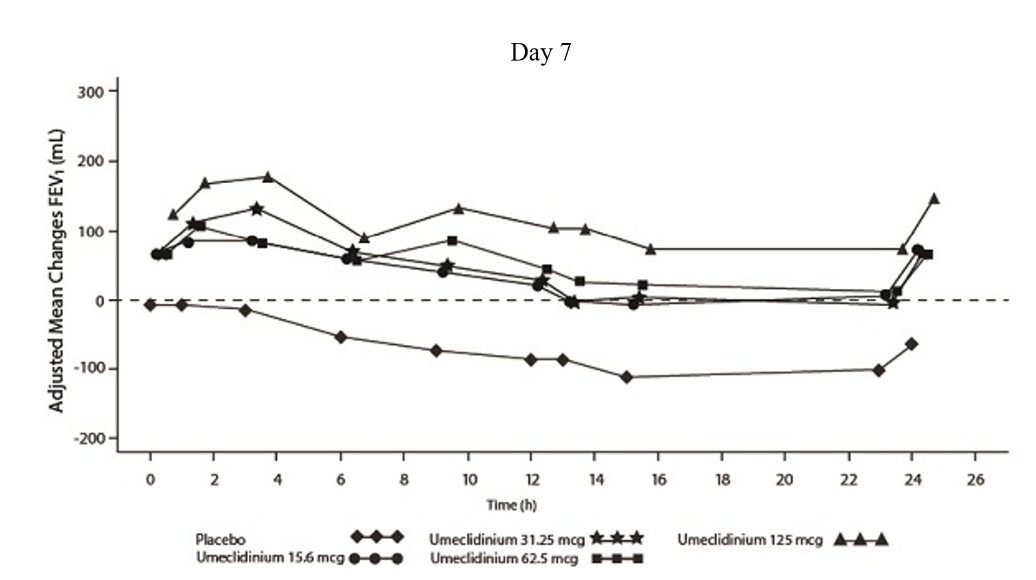
Dose selection for umeclidinium in COPD was supported by a 7-day, randomized, double-blind, placebo-controlled, crossover trial evaluating 4 doses of umeclidinium (15.6 to 125 mcg) or placebo dosed once daily in the morning in 163 subjects with COPD. A dose ordering was observed, with the 62.5- and 125-mcg doses demonstrating larger improvements in FEV1 over 24 hours compared with the lower doses of 15.6 and 31.25 mcg (Figure 2).
The differences in trough FEV1 from baseline after 7 days for placebo and the 15.6-, 31.25-, 62.5-, and 125-mcg doses were -74 mL (95% CI: -118, -31), 38 mL (95% CI: -6, 83), 27 mL (95% CI: -18, 72), 49 mL (95% CI: 6, 93), and 109 mL (95% CI: 65, 152), respectively. Two additional dose-ranging trials in subjects with COPD demonstrated minimal additional benefit at doses above 125 mcg. The dose-ranging results supported the evaluation of 2 doses of umeclidinium, 62.5 and 125 mcg, in the confirmatory COPD trials to further assess dose response.
Evaluations of dosing interval by comparing once- and twice-daily dosing supported selection of a once-daily dosing interval for further evaluation in the confirmatory COPD trials.
Figure 2. Adjusted Mean Change from Baseline in Postdose Serial FEV1 (mL) on Days 1 and 7
14.2 Maintenance Treatment: Confirmatory Trials
Lung Function
The clinical development program for umeclidinium ELLIPTA included 2 randomized, double‑blind, placebo-controlled, parallel-group trials in subjects with COPD designed to evaluate the efficacy of umeclidinium ELLIPTA on lung function. Trial 1 (NCT01313650) was a 24-week placebo‑controlled trial, and Trial 2 (NCT01387230) was a 12-week placebo‑controlled trial. These trials treated subjects that had a clinical diagnosis of COPD, were 40 years of age or older, had a history of smoking ≥10 pack-years, had a post-albuterol FEV1 ≤70% of predicted normal values, had a ratio of FEV1/FVC of <0.7, and had a Modified Medical Research Council (mMRC) score ≥2. Subjects in Trial 1 had a mean age of 63 years and an average smoking history of 46 pack-years, with 50% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 47% (range: 13% to 74%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (range: 0.20 to 0.74), and the mean percent reversibility was 15% (range: -35% to 109%). The majority of subjects (72%) reported not having a COPD exacerbation in the prior 12 months. Baseline demographics and lung function for subjects in Trial 2 were similar to those in Trial 1.
Trial 1 evaluated umeclidinium 62.5 mcg and placebo. The primary endpoint was change from baseline in trough (predose) FEV1 at Day 169 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 168) compared with placebo. Umeclidinium ELLIPTA 62.5 mcg demonstrated a larger increase in mean change from baseline in trough (predose) FEV1 relative to placebo (Table 2). Similar results were obtained from Trial 2.
Table 2. Least Squares Mean Change from Baseline in Trough FEV1 (mL) at Day 169 in the Intent-to-Treat Population (Trial 1) n = Number in intent-to-treat population. Treatment
n
Trough FEV1 (mL) at Day 169
Difference from Placebo
(95% CI)
n = 280
Umeclidinium ELLIPTA
n = 418
115 (76, 155)
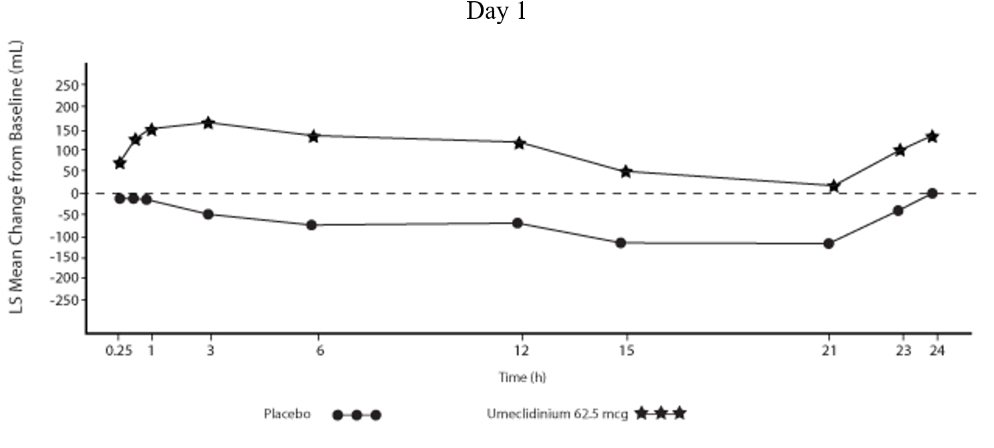
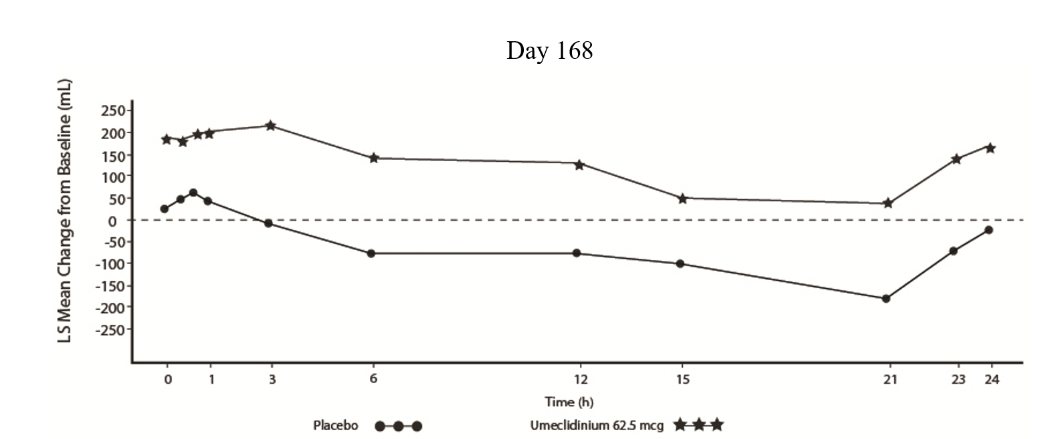
Serial spirometric evaluations throughout the 24-hour dosing interval were performed in a subset of subjects (n = 54, umeclidinium 62.5 mcg; n = 36, placebo) at Days 1, 84, and 168 in Trial 1, and for all patients at Days 1 and 84 in Trial 2. Results from Trial 1 at Day 1 and Day 168 are shown in Figure 3.
Figure 3. Least Squares (LS) Mean Change from Baseline in FEV1 (mL) over Time (0‑24 h) on Days 1 and 168 (Trial 1 Subset Population)
In Trial 1, the mean peak FEV1 (over the first 6 hours relative to baseline) at Day 1 and at Day 168 for the group receiving umeclidinium 62.5 mcg compared with placebo was 126 and 130 mL, respectively.
Health-Related Quality of Life
Health-related quality of life was measured using St. George’s Respiratory Questionnaire (SGRQ). Umeclidinium demonstrated an improvement in mean SGRQ total score compared with placebo treatment at Day 168: -4.69 (95% CI: -7.07,-2.31). The proportion of patients with a clinically meaningful decrease (defined as a decrease of at least 4 units from baseline) at Week 24 was greater for umeclidinium ELLIPTA 62.5 mcg (42%; 172/410) compared with placebo (31%; 86/274).
14.3 Maintenance Treatment: Combination with an ICS/LABA Trials
Lung Function
The efficacy of umeclidinium ELLIPTA in combination with an ICS/LABA was evaluated in 4 randomized, double-blind, parallel-group trials in subjects with COPD. These trials, all of similar study design, were of 12-weeks’ treatment duration. Subjects were randomized to umeclidinium ELLIPTA 62.5 mcg + ICS/LABA or placebo + ICS/LABA. Entry criteria for subjects enrolled in these trials were similar to those described above in Section 14.2. The primary endpoint for these trials was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84). Baseline FEV1 was measured while subjects were on background ICS/LABA.
Combination with Fluticasone Furoate + Vilanterol
Trial 4 (NCT01957163) and Trial 5 (NCT02119286) randomized subjects to umeclidinium ELLIPTA 62.5 mcg + FF/VI 100 mcg/25 mcg administered once daily or placebo + FF/VI 100 mcg/25 mcg administered once daily. Trial population demographics and results for Trials 4 and 5 were similar; therefore, only Trial 4 results are presented below.
Subjects in Trial 4 across all treatment arms had a mean age of 64 years and an average smoking history of 50 pack-years, with 42% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 45% (range: 13% to 76%), the mean postbronchodilator FEV1/FVC ratio was 0.48 (range: 0.22 to 0.70), and the mean percent reversibility was 14% (range: -20% to 71%). The majority of subjects (85%) reported not having a COPD exacerbation in the prior 12 months.
The primary endpoint was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84) compared with placebo (Umeclidinium ELLIPTA + FF/VI vs. placebo + FF/VI). Umeclidinium ELLIPTA + FF/VI demonstrated a larger mean change from baseline in trough (predose) FEV1 relative to placebo + FF/VI (Table 3).
Table 3. Least Squares Mean Change from Baseline in Trough FEV1 (mL) at Day 85 in the Intent-to-Treat Population (Trial 4) FF/VI = fluticasone furoate/vilanterol. n = Number in intent-to-treat population. Treatment
n
Trough FEV1 (mL) at Day 85
Difference from Placebo + FF/VI
(95% CI)
n = 206
Umeclidinium ELLIPTA + FF/VI
n = 206
124 (93, 154)
Combination with Fluticasone Propionate + Salmeterol
Trial 6 (NCT01772134) and Trial 7 (NCT01772147) randomized subjects to umeclidinium ELLIPTA 62.5 mcg + FP/SAL 250 mcg/50 mcg or placebo + FP/SAL 250 mcg/50 mcg. The treatments with umeclidinium ELLIPTA and placebo were administered once daily, while the FP/SAL treatment was administered twice daily. Trial population demographics and results for Trials 6 and 7 were similar; therefore, only Trial 6 results are presented below.
Subjects in Trial 6 across all treatment arms had a mean age of 63 years and an average smoking history of 50 pack-years, with 54% identified as current smokers. At screening, the mean postbronchodilator percent predicted FEV1 was 47% (range: 12% to 70%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (range: 0.22 to 0.69), and the mean percent reversibility was 16% (range: -36% to 79%). The majority of subjects (79%) reported not having a COPD exacerbation in the prior 12 months.
The primary endpoint was change from baseline in trough (predose) FEV1 at Day 85 (defined as the mean of the FEV1 values obtained at 23 and 24 hours after the previous dose on Day 84) compared with placebo (Umeclidinium ELLIPTA + FP/SAL vs. placebo + FP/SAL). Umeclidinium ELLIPTA + FP/SAL demonstrated a larger mean change from baseline in trough (predose) FEV1 relative to placebo + FP/SAL (Table 4).
Table 4. Least Squares Mean Change from Baseline in Trough FEV1 (mL) at Day 85 in the Intent-to-Treat Population (Trial 6) FP/SAL = fluticasone propionate/salmeterol. n = Number in intent-to-treat population. Treatment
n
Trough FEV1 (mL) at Day 85
Difference from Placebo + FP/SAL
(95% CI)
n = 205
Umeclidinium ELLIPTA+FP/SAL
n = 204
147 (107, 187)
Exacerbations
In Trial 8 (NCT02164513), a total of 10,355 subjects with COPD with a history of 1 or more moderate or severe exacerbations in the prior 12 months were randomized (2:2:1) to receive fluticasone furoate/umeclidinium/vilanterol 100 mcg/62.5 mcg/25 mcg (n = 4,151), fluticasone furoate/vilanterol 100 mcg/25 mcg (n = 4,133), or umeclidinium/vilanterol 62.5 mcg/25 mcg (n = 2,070) administered once daily in a 12-month trial. The population demographics across all treatments were: mean age of 65 years, 77% white, 66% male, and an average smoking history of 46.6 pack-years, with 35% identified as current smokers. At trial entry, the most common COPD medications were ICS + anticholinergic + LABA (34%), ICS + LABA (26%), anticholinergic + LABA (8%), and anticholinergic (7%). The mean postbronchodilator percent predicted FEV1 was 46% (standard deviation: 15%), the mean postbronchodilator FEV1/FVC ratio was 0.47 (standard deviation: 0.12), and the mean percent reversibility was 10% (range:
-59% to 125%).The primary endpoint was annual rate of on-treatment moderate and severe exacerbations in subjects treated with fluticasone furoate/umeclidinium/vilanterol compared with the fixed-dose combinations of fluticasone furoate/vilanterol and umeclidinium/vilanterol. Exacerbations were defined as worsening of 2 or more major symptoms (dyspnea, sputum volume, and sputum purulence) or worsening of any 1 major symptom together with any 1 of the following minor symptoms: sore throat, colds (nasal discharge and/or nasal congestion), fever without other cause, and increased cough or wheeze for at least 2 consecutive days. Exacerbations were considered to be of moderate severity if treatment with systemic corticosteroids and/or antibiotics was required and were considered to be severe if resulted in hospitalization or death.
Evidence of efficacy for Umeclidinium ELLIPTA on COPD exacerbations was established by the efficacy of the umeclidinium component of fluticasone furoate/umeclidinium/vilanterol in Trial 8. Treatment with fluticasone furoate/umeclidinium/vilanterol statistically significantly reduced the on-treatment annual rate of moderate/severe exacerbations by 15% compared with fluticasone furoate/vilanterol (Table 5). A reduction in risk of on-treatment moderate/severe exacerbation (as measured by time to first) was also observed for the same comparison.
Table 5. Moderate and Severe Chronic Obstructive Pulmonary Disease Exacerbations (Trial 8) FF/UMEC/VI = Fluticasone furoate/umeclidinium/vilanterol 100 mcg/62.5 mcg/25 mcg, FF/VI = Fluticasone furoate/vilanterol 100 mcg/25 mcg, UMEC/VI = Umeclidinium/vilanterol 62.5 mcg/25 mcg. a On-treatment analyses excluded exacerbation data collected after discontinuation of study treatment. Treatment
n
Mean Annual Rate
(exacerbations/year)
FF/UMEC/VI Rate Ratio vs. Comparator
(95% CI)
% Reduction in Exacerbation Rate
(95% CI)
P Value
FF/UMEC/VI
4,145
0.91
FF/VI
4,133
1.07
0.85
(0.80, 0.90)
15
(10, 20)
P<0.001
UMEC/VI
2,069
1.21
0.75
(0.70, 0.81)
25
(19, 30)
P<0.001
-
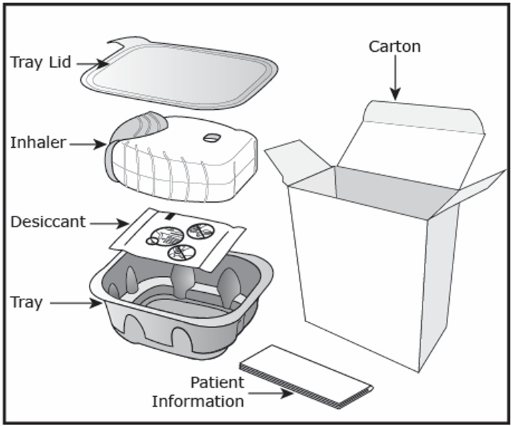
16 HOW SUPPLIED/STORAGE AND HANDLING
Umeclidinium ELLIPTA is supplied as a disposable light grey and light green plastic inhaler containing a foil strip with 30 blisters (NDC: 66993-189-97).
The inhaler is packaged in a moisture-protective foil tray with a desiccant and a peelable lid.
Store at room temperature between 68°F and 77°F (20°C and 25°C); excursions permitted from 59°F to 86°F (15°C to 30°C) [See USP Controlled Room Temperature]. Store in a dry place away from direct heat or sunlight. Keep out of reach of children.
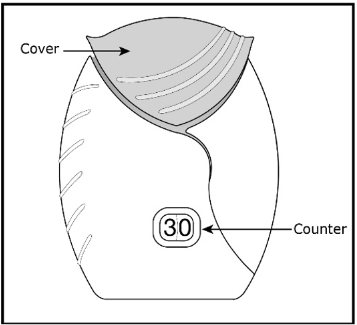
Umeclidinium ELLIPTA should be stored inside the unopened moisture-protective foil tray and only removed from the tray immediately before initial use. Discard Umeclidinium ELLIPTA 6 weeks after opening the foil tray or when the counter reads “0” (after all blisters have been used), whichever comes first. The inhaler is not reusable. Do not attempt to take the inhaler apart.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Not for Acute Symptoms
Inform patients that Umeclidinium ELLIPTA is not meant to relieve acute symptoms of COPD and extra doses should not be used for that purpose. Advise patients to treat acute symptoms with an inhaled, short-acting beta2-agonist such as albuterol. Provide patients with such medication and instruct them in how it should be used.
Instruct patients to seek medical attention immediately if they experience any of the following:
- Decreasing effectiveness of inhaled, short-acting beta2-agonists
- Need for more inhalations than usual of inhaled, short-acting beta2-agonists
- Significant decrease in lung function as outlined by the physician
Tell patients they should not stop therapy with Umeclidinium ELLIPTA without physician/provider guidance since symptoms may recur after discontinuation. [See Warnings and Precautions (5.1).]
Paradoxical Bronchospasm
As with other inhaled medicines, Umeclidinium ELLIPTA can cause paradoxical bronchospasm. If paradoxical bronchospasm occurs, instruct patients to discontinue Umeclidinium ELLIPTA and contact their healthcare provider right away. [See Warnings and Precautions (5.2).]
Worsening of Narrow-Angle Glaucoma
Instruct patients to be alert for signs and symptoms of acute narrow-angle glaucoma (e.g., eye pain or discomfort, blurred vision, visual halos or colored images in association with red eyes from conjunctival congestion and corneal edema). Instruct patients to consult a physician immediately if any of these signs or symptoms develop. [See Warnings and Precautions (5.4).]
Worsening of Urinary Retention
Instruct patients to be alert for signs and symptoms of urinary retention (e.g., difficulty passing urine, painful urination). Instruct patients to consult a physician immediately if any of these signs or symptoms develop. [See Warnings and Precautions (5.5).]
ELLIPTA is a trademark owned by or licensed to the GSK group of companies.
Manufactured for:
Prasco Laboratories
Mason, OH 45040 USA
Manufactured by:
GlaxoSmithKline
Durham, NC 27701
UMC-PS:1PI
-
PATIENT INFORMATION
Umeclidinium ELLIPTA (Ue-ME-kli-DIN-ee-um e-LIP-ta)
inhalation powder
for oral inhalation use
What is Umeclidinium ELLIPTA?
- Umeclidinium ELLIPTA is an anticholinergic medicine (umeclidinium).
- Anticholinergic medicines such as umeclidinium help the muscles around the airways in your lungs stay relaxed to prevent symptoms such as wheezing, cough, chest tightness, and shortness of breath. These symptoms can happen when the muscles around the airways tighten. This makes it hard to breathe.
- Umeclidinium ELLIPTA is not used to relieve sudden breathing problems and will not replace a rescue inhaler.
- Umeclidinium ELLIPTA is a prescription medicine used long term (chronic) to treat people with chronic obstructive pulmonary disease (COPD). COPD is a chronic lung disease that includes chronic bronchitis, emphysema, or both.
- Umeclidinium ELLIPTA is used as 1 inhalation 1 time each day to improve symptoms of COPD for better breathing and to reduce the number of flare-ups (the worsening of your COPD symptoms for several days).
- Umeclidinium ELLIPTA should not be used in children. It is not known if Umeclidinium ELLIPTA is safe and effective in children.
Do not use Umeclidinium ELLIPTA if you:
- have a severe allergy to milk proteins. Ask your healthcare provider if you are not sure.
- are allergic to umeclidinium or any of the ingredients in Umeclidinium ELLIPTA. See the end of this Patient Information for a complete list of ingredients in Umeclidinium ELLIPTA.
Before using Umeclidinium ELLIPTA, tell your healthcare provider about all of your medical conditions, including if you:
- have heart problems.
- have eye problems such as glaucoma. Umeclidinium ELLIPTA may make your glaucoma worse.
- are allergic to milk proteins.
- have prostate or bladder problems, or problems passing urine. Umeclidinium ELLIPTA may make these problems worse.
- are pregnant or plan to become pregnant. It is not known if Umeclidinium ELLIPTA may harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if the medicine in Umeclidinium ELLIPTA passes into your breast milk and if it can harm your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Umeclidinium ELLIPTA and certain other medicines may interact with each other. This may cause serious side effects.
Especially tell your healthcare provider if you take:
- anticholinergics (including tiotropium, ipratropium, aclidinium)
- atropine
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I use Umeclidinium ELLIPTA?
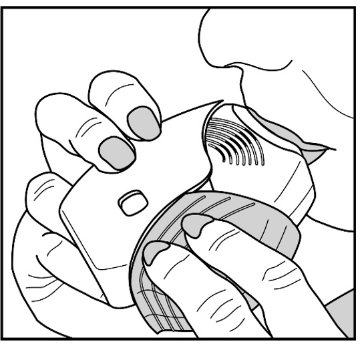
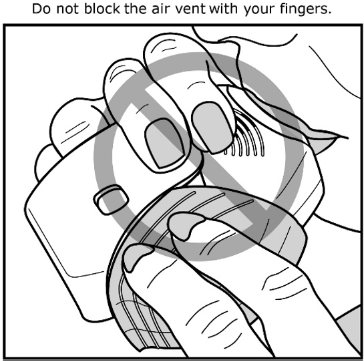
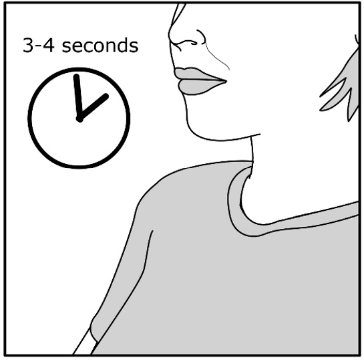
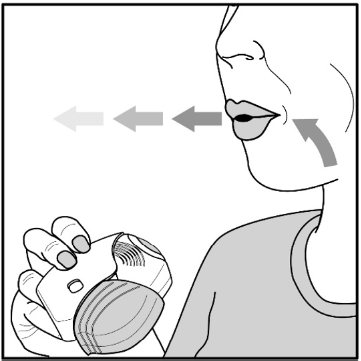
Read the step-by-step instructions for using Umeclidinium ELLIPTA at the end of this Patient Information.
- Do not use Umeclidinium ELLIPTA unless your healthcare provider has taught you how to use the inhaler and you understand how to use it correctly.
- Use Umeclidinium ELLIPTA exactly as your healthcare provider tells you to use it. Do not use Umeclidinium ELLIPTA more often than prescribed.
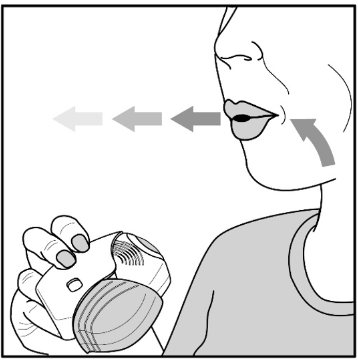
- Use 1 inhalation of Umeclidinium ELLIPTA 1 time each day. Use Umeclidinium ELLIPTA at the same time each day.
- If you miss a dose of Umeclidinium ELLIPTA, take it as soon as you remember. Do not take more than 1 inhalation per day. Take your next dose at your usual time. Do not take 2 doses at 1 time.
- If you take too much Umeclidinium ELLIPTA, call your healthcare provider or go to the nearest hospital emergency room right away if you have any unusual symptoms, such as worsening shortness of breath, chest pain, increased heart rate, or shakiness.
- Do not use other medicines that contain an anticholinergic for any reason. Ask your healthcare provider or pharmacist if any of your other medicines are anticholinergic medicines.
- Do not stop using Umeclidinium ELLIPTA unless told to do so by your healthcare provider because your symptoms might get worse. Your healthcare provider will change your medicines as needed.
- Talk to your healthcare provider right away if you stop using Umeclidinium ELLIPTA.
- Umeclidinium ELLIPTA does not relieve sudden symptoms of COPD and you should not take extra doses of Umeclidinium ELLIPTA to relieve these sudden symptoms. Always have a rescue inhaler with you to treat sudden symptoms. If you do not have a rescue inhaler, call your healthcare provider to have one prescribed for you.
-
Call your healthcare provider or get medical care right away if:
- o your breathing problems get worse.
- o you need to use your rescue inhaler more often than usual.
- o your rescue inhaler does not work as well to relieve your symptoms.
What are the possible side effects of Umeclidinium ELLIPTA?
Umeclidinium ELLIPTA can cause serious side effects, including:
- sudden breathing problems immediately after inhaling your medicine. If you have sudden breathing problems immediately after inhaling your medicine, stop using Umeclidinium ELLIPTA and call your healthcare provider right away.
- serious allergic reactions (anaphylaxis). Stop using Umeclidinium ELLIPTA and call your healthcare provider or go to the nearest emergency room right away if you get any of the following symptoms of a serious allergic reaction:
- o rash
- o hives
- o severe itching
- o swelling of your face, mouth, and tongue
- o breathing problems
- new or worsening eye problems including acute narrow-angle glaucoma. You should have regular eye exams while using Umeclidinium ELLIPTA. Acute narrow-angle glaucoma can cause permanent loss of vision if not treated. Symptoms of acute narrow-angle glaucoma may include:
- o eye pain or discomfort
- o nausea or vomiting
- o blurred vision
- o seeing halos or bright colors around lights
- o red eyes
If you have these symptoms, call your healthcare provider right away before taking another dose.
- urinary retention. People who take Umeclidinium ELLIPTA may develop new or worse urinary retention. Symptoms of urinary retention may include:
- o difficulty urinating
- o painful urination
- o urinating frequently
- o urination in a weak stream or drips
If you have these symptoms of urinary retention, stop taking Umeclidinium ELLIPTA, and call your healthcare provider right away before taking another dose.
Common side effects of Umeclidinium ELLIPTA include:
- upper respiratory tract infection
- stuffy or runny nose
- cough
- mouth and throat pain
- joint pain
- change in taste
- muscle pain
- tooth pain
- stomach pain
- bruising or dark areas of skin
- fast or irregular heartbeat
These are not all the possible side effects of Umeclidinium ELLIPTA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Umeclidinium ELLIPTA?
- Store Umeclidinium ELLIPTA at room temperature between 68°F and 77°F (20°C and 25°C). Keep in a dry place away from heat and sunlight.
- Store Umeclidinium ELLIPTA in the unopened tray and only open when ready for use.
- Safely throw away Umeclidinium ELLIPTA in the trash 6 weeks after you open the tray or when the counter reads “0”, whichever comes first. Write the date you open the tray on the label on the inhaler.
- Keep Umeclidinium ELLIPTA and all medicines out of the reach of children.
General information about the safe and effective use of Umeclidinium ELLIPTA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use Umeclidinium ELLIPTA for a condition for which it was not prescribed. Do not give Umeclidinium ELLIPTA to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about Umeclidinium ELLIPTA that is written for health professionals.
What are the ingredients in Umeclidinium ELLIPTA?
Active ingredient: umeclidinium
Inactive ingredients: lactose monohydrate (contains milk proteins), magnesium stearate
Manufactured for:
Prasco Laboratories
Mason, OH 45040 USA
Manufactured by:
GlaxoSmithKline
Durham, NC 27701
For more information about Umeclidinium ELLIPTA, call 1-866-525-0688.
ELLIPTA is a trademark owned by or licensed to the GSK group of companies.
UMC-PS: 1PIL
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: November 2025
-
This Instructions for Use has been approved by the U.S. Food and Drug Administration Revised: November 2025
-
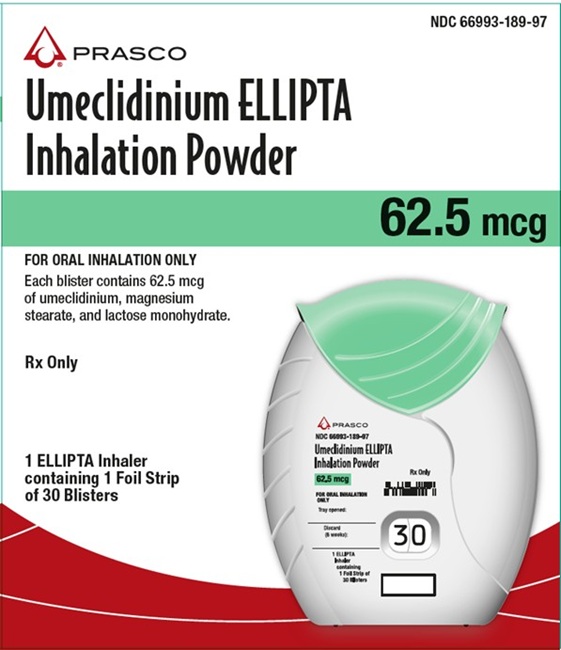
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 66993-189-97
Umeclidinium ELLIPTA Inhalation Powder
62.5 mcg
PRASCO
FOR ORAL INHALATION ONLY
Each blister contains 62.5 mcg of umeclidinium, magnesium stearate, and lactose monohydrate.
Rx Only
1 ELLIPTA Inhaler containing 1 Foil Strip of 30 Blisters
Made in Singapore
Rev. 11/25
62000000102497
-
INGREDIENTS AND APPEARANCE
UMECLIDINIUM ELLIPTA
umeclidinium aerosol, powderProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 66993-189 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength UMECLIDINIUM BROMIDE (UNII: 7AN603V4JV) (UMECLIDINIUM - UNII:GE2T1418SV) UMECLIDINIUM 62.5 ug Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 66993-189-97 1 in 1 CARTON 04/06/2026 1 1 in 1 TRAY 1 30 in 1 INHALER; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA authorized generic NDA205382 04/06/2026 Labeler - Prasco Laboratories (065969375)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.