quinidine gluconate- Quinidine Gluconate tablet, extended release
Drug Labeling and Warnings
Drug Details [pdf]
- N/A - Section Title Not Found In Database
-
DESCRIPTION
Quinidine is an antimalarial schizonticide and an antiarrhythmic agent with Class Ia activity; it is the d-isomer of quinine, and its molecular weight is 324.43. Quinidine gluconate is the gluconate salt of quinidine; its chemical name is cinchonan-9-ol, 6’-methoxy-, (9S)-, mono-D-gluconate. The structural formula is represented below:
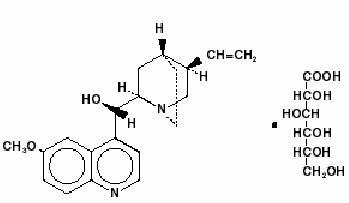
C20H24N2O2C6H12O7 M.W. 520.58
Quinidine gluconate contains 62.3% of the anhydrous quinidine alkaloid, whereas quinidine sulfate contains 82.86%. In prescribing Quinidine Gluconate Extended-release Tablets, this factor should be considered.
Each tablet, for oral administration, contains 324 mg of quinidine gluconate (202 mg of quinidine base) in a matrix to provide extended-release.
Quinidine Gluconate Extended-release Tablets contain the following inactive ingredients: carnauba wax, ethylcellulose, magnesium stearate and povidone.
This product meets USP Drug Release Test 1.
-
CLINICAL PHARMACOLOGY
Pharmacokinetics and Metabolism
The absolute bioavailability of quinidine from quinidine gluconate is 70 to 80%. Relative to a solution of quinidine sulfate, the bioavailability of quinidine from quinidine gluconate is reported to be 1.03. The less-than-complete bioavailability is thought to be due to first-pass elimination by the liver. Peak serum levels generally appear 3 to 5 hours after dosing; when the drug is taken with food, absorption is increased in both rate (27%) and extent (17%). The rate and extent of absorption of quinidine from quinidine gluconate are not significantly affected by the coadministration of an aluminum-hydroxide antacid. The rate of absorption of quinidine following the ingestion of grapefruit juice may be decreased.
The volume of distribution of quinidine is 2 to 3 L/kg in healthy young adults, but this may be reduced to as little as 0.5 L/kg in patients with congestive heart failure, or increased to 3 to 5 L/kg in patients with cirrhosis of the liver. At concentrations of 2 to 5 mg/L (6.5 to 16.2 µmol/L), the fraction of quinidine bound to plasma proteins (mainly to α1-acid glycoprotein and to albumin) is 80 to 88% in adults and older children, but it is lower in pregnant women, and in infants and neonates it may be as low as 50 to 70%. Because α1-acid glycoprotein levels are increased in response to stress, serum levels of total quinidine may be greatly increased in settings such as acute myocardial infarction, even though the serum content of unbound (active) drug may remain normal. Protein binding is also increased in chronic renal failure, but binding abruptly descends toward or below normal when heparin is administered for hemodialysis.
Quinidine clearance typically proceeds at 3 to 5 mL/min/kg in adults, but clearance in children may be twice or three times as rapid. The elimination half-life is 6 to 8 hours in adults and 3 to 4 hours in children. Quinidine clearance is unaffected by hepatic cirrhosis, so the increased volume of distribution seen in cirrhosis leads to a proportionate increase in the elimination half-life.
Most quinidine is eliminated hepatically via the action of cytochrome P450IIIA4; there are several different hydroxylated metabolites, and some of these have antiarrhythmic activity.
The most important of quinidine’s metabolites is 3-hydroxy-quinidine (3HQ), serum levels of which can approach those of quinidine in patients receiving conventional doses of quinidine gluconate. The volume of distribution of 3HQ appears to be larger than that of quinidine, and the elimination half-life of 3HQ is about 12 hours.
As measured by antiarrhythmic effects on animals, by QTc prolongation in human volunteers, or by various in vitro techniques, 3HQ has at least half the antiarrhythmic activity of the parent compound, so it may be responsible for a substantial fraction of the effect of quinidine gluconate in chronic use.
When the urine pH is less than 7, about 20% of administered quinidine appears unchanged in the urine, but this fraction drops to as little as 5% when the urine is more alkaline. Renal clearance involves both glomerular filtration and active tubular secretion, moderated by (pH-dependent) tubular reabsorption. The net renal clearance is about 1 mL/min/kg in healthy adults.
When renal function is taken into account, quinidine clearance is apparently independent of patient age.
Assays of serum quinidine levels are widely available, but the results of modern assays may not be consistent with results cited in the older medical literature. The serum levels of quinidine cited in this package insert are those derived from specific assays, using either benzene extraction or (preferably) reverse-phase high-pressure liquid chromatography. In matched samples, older assays might unpredictably have given results that were as much as two or three times higher. A typical “therapeutic” concentration range is 2 to 6 mg/L (6.2 to 18.5 µmol/L).
Mechanisms of Action
In patients with malaria, quinidine acts primarily as an intra-erythrocytic schizonticide, with little effect upon sporozites or upon pre-erythrocytic parasites. Quinidine is gametocidal to Plasmodium vivax and P. malariae, but not to P. falciparum.
In cardiac muscle and in Purkinje fibers, quinidine depresses the rapid inward depolarizing sodium current, thereby slowing phase-0 depolarization and reducing the amplitude of the action potential without affecting the resting potential. In normal Purkinje fibers, it reduces the slope of phase-4 depolarization, shifting the threshold voltage upward toward zero. The result is slowed conduction and reduced automaticity in all parts of the heart, with increase of the effective refractory period relative to the duration of the action potential in the atria, ventricles, and Purkinje tissues. Quinidine also raises the fibrillation thresholds of the atria and ventricles, and it raises the ventricular defibrillation threshold as well. Quinidine’s actions fall into Class Ia in the Vaughn-Williams classification.
By slowing conduction and prolonging the effective refractory period, quinidine can interrupt or prevent reentrant arrhythmias and arrhythmias due to increased automaticity, including atrial flutter, atrial fibrillation, and paroxysmal supraventricular tachycardia.
In patients with the sick sinus syndrome, quinidine can cause marked sinus node depression and bradycardia. In most patients, however, use of quinidine is associated with an increase in the sinus rate.
Like other antiarrhythmic drugs with Class Ia activity, quinidine prolongs the QT interval in a dose-related fashion. This may lead to increased ventricular automaticity and polymorphic ventricular tachycardias, including torsades de pointes (seeWARNINGS).
In addition, quinidine has anticholinergic activity, it has negative inotropic activity, and it acts peripherally as an α-adrenergic antagonist (that is, as a vasodilator).
-
CLINICAL EFFECTS
Maintenance of Sinus Rhythm After Conversion From Atrial Fibrillation
In six clinical trials (published between 1970 and 1984) with a total of 808 patients, quinidine (418 patients) was compared to nontreatment (258 patients) or placebo (132 patients) for the maintenance of sinus rhythm after cardioversion from chronic atrial fibrillation. Quinidine was consistently more efficacious in maintaining sinus rhythm, but a meta-analysis found that mortality in the quinidine-exposed patients (2.9%) was significantly greater than mortality in the patients who had not been treated with active drug (0.8%).
Suppression of atrial fibrillation with quinidine has theoretical patient benefits (e.g., improved exercise tolerance; reduction in hospitalization for cardioversion; lack of arrhythmia-related palpitations, dyspnea and chest pain; reduced incidence of systemic embolism and/or stroke), but these benefits have never been demonstrated in clinical trials. Some of these benefits (e.g., reduction in stroke incidence) may be achievable by other means (anticoagulation).
By slowing the atrial rate in atrial flutter/fibrillation, quinidine can decrease the degree of atrioventricular block and cause an increase, sometimes marked, in the rate at which supraventricular impulses are successfully conducted by the atrioventricular node, with a resultant paradoxical increase in ventricular rate (seeWARNINGS).
Non-Life-Threatening Ventricular Arrhythmias
In studies of patients with a variety of ventricular arrhythmias (mainly frequent ventricular premature beats and non-sustained ventricular tachycardia, quinidine (total n=502) has been compared with flecainide (n=141), mexiletine (n=246), propafenone (n=53), and tocainide (n=67). In each of these studies, the mortality in the quinidine group was numerically greater than the mortality in the comparator group. When the studies were combined in a meta-analysis, quinidine was associated with a statistically significant threefold relative risk of death.
At therapeutic doses, quinidine’s only consistent effect upon the surface electrocardiogram is an increase in the QT interval. This prolongation can be monitored as a guide to safety, and it may provide better guidance than serum drug levels (seeWARNINGS).
-
INDICATIONS AND USAGE
Conversion of Atrial Fibrillation/ Flutter
In patients with symptomatic atrial fibrillation/flutter whose symptoms are not adequately controlled by measures that reduce the rate of ventricular response, quinidine gluconate is indicated as a means of restoring normal sinus rhythm. If this use of quinidine gluconate does not restore sinus rhythm within a reasonable time (seeDOSAGE AND ADMINISTRATION), then quinidine gluconate should be discontinued.
Reduction of Frequency of Relapse into Atrial Fibrillation/Flutter
Chronic therapy with quinidine gluconate is indicated for some patients at high risk of symptomatic atrial fibrillation/flutter, generally patients who have had previous episodes of atrial fibrillation/flutter that were so frequent and poorly tolerated as to outweigh, in the judgment of the physician and the patient, the risks of prophylactic therapy with quinidine gluconate. The increased risk of death should specifically be considered. Quinidine gluconate should be used only after alternative measures (e.g., use of other drugs to control the ventricular rate) have been found to be inadequate.
In patients with histories of frequent symptomatic episodes of atrial fibrillation/flutter, the goal of therapy should be an increase in the average time between episodes. In most patients, the tachyarrhythmia will recur during therapy, and a single recurrence should not be interpreted as therapeutic failure.
Suppression of Ventricular Arrhythmias
Quinidine gluconate is also indicated for the suppression of recurrent documented ventricular arrhythmias, such as sustained ventricular tachycardia, that in the judgment of the physician are life-threatening. Because of the proarrhythmic effects of quinidine, its use with ventricular arrhythmias of lesser severity is generally not recommended, and treatment of patients with asymptomatic ventricular premature contractions should be avoided. Where possible, therapy should be guided by the results of programmed electrical stimulation and/or Holter monitoring with exercise.
Antiarrhythmic drugs (including quinidine gluconate) have not been shown to enhance survival in patients with ventricular arrhythmias.
-
CONTRAINDICATIONS
Quinidine is contraindicated in patients who are known to be allergic to it, or who have developed thrombocytopenic purpura during prior therapy with quinidine or quinine.
In the absence of a functioning artificial pacemaker, quinidine is also contraindicated in any patient whose cardiac rhythm is dependent upon a junctional or idioventricular pacemaker, including patients in complete atrioventricular block.
Quinidine is also contraindicated in patients who, like those with myasthenia gravis, might be adversely affected by an anticholinergic agent.
-
WARNINGS
Mortality
In many trials of antiarrhythmic therapy for non-life-threatening arrhythmias, active antiarrhythmic therapy has resulted in increased mortality; the risk of active therapy is probably greatest in patients with structural heart disease. In the case of quinidine used to prevent or defer recurrence of atrial flutter/fibrillation, the best available data come from a meta-analysis described under CLINICAL PHARMACOLOGY/Clinical Effects above. In the patients studied in the trials there analyzed, the mortality associated with the use of quinidine was more than three times as great as the mortality associated with the use of placebo. Another meta-analysis, also described under CLINICAL PHARMACOLOGY/Clinical Effects, showed that in patients with various non-life-threatening ventricular arrhythmias, the mortality associated with the use of quinidine was consistently greater than that associated with the use of any of a variety of alternative antiarrhythmics. Proarrhythmic Effects
Like many other drugs (including all other Class Ia antiarrhythmics), quinidine prolongs the QTc interval, and this can lead to torsades de pointes, a life-threatening ventricular arrhythmia (seeOVERDOSAGE). The risk of torsades is increased by bradycardia, hypokalemia, hypomagnesemia or high serum levels of quinidine, but it may appear in the absence of any of these risk factors. The best predictor of this arrhythmia appears to be the length of QTc interval, and quinidine should be used with extreme care in patients who have preexisting long-QT syndromes, who have histories of torsades de pointes of any cause, or who have previously responded to quinidine (or other drugs that prolong ventricular repolarization) with marked lengthening of the QTc interval. Estimation of the incidence of torsades in patients with therapeutic levels of quinidine is not possible from the available data.
Other ventricular arrhythmias that have been reported with quinidine include frequent extrasystoles, ventricular tachycardia, ventricular flutter, and ventricular fibrillation.
Paradoxical Increase in Ventricular Rate in Atrial Flutter/Fibrillation
When quinidine is administered to patients with atrial flutter/fibrillation, the desired pharmacologic reversion to sinus rhythm may (rarely) be preceded by a slowing of the atrial rate with a consequent increase in the rate of beats conducted to the ventricles. The resulting ventricular rate may be very high (greater than 200 beats per minute) and poorly tolerated. This hazard may be decreased if partial atrioventricular block is achieved prior to initiation of quinidine therapy, using conduction-reducing drugs such as digitalis, verapamil, diltiazem, or a ß-receptor blocking agent.
Exacerbated Bradycardia in Sick Sinus Syndrome
In patients with the sick sinus syndrome, quinidine has been associated with marked sinus node depression and bradycardia.
Pharmacokinetic Considerations
Renal or hepatic dysfunction causes the elimination of quinidine to be slowed, while congestive heart failure causes a reduction in quinidine’s apparent volume of distribution. Any of these conditions can lead to quinidine toxicity if dosage is not appropriately reduced. In addition, interactions with coadministered drugs can alter the serum concentration and activity of quinidine, leading either to toxicity or to lack of efficacy if the dose of quinidine is not appropriately modified. (SeePRECAUTIONS, Drug and Diet Interactions.)
-
PRECAUTIONS
Heart Block
In patients without implanted pacemakers who are at high risk of complete atrioventricular block (e.g., those with digitalis intoxication, second degree atrioventricular block, or severe intraventricular conduction defects), quinidine should be used only with caution.
Drug and Diet Interactions
Altered Pharmacokinetics of Quinidine
Diltiazem significantly decreases the clearance and increases the t1/2 of quinidine, but quinidine does not alter the kinetics of diltiazem.
Drugs that alkalinize the urine (carbonic-anhydrase inhibitors, sodium bicarbonate, thiazide diuretics) reduce renal elimination of quinidine.
By pharmacokinetic mechanisms that are not well understood, quinidine levels are increased by coadministration of amiodarone or cimetidine. Very rarely, and again by mechanisms not understood, quinidine levels are decreased by coadministration of nifedipine.
Hepatic elimination of quinidine may be accelerated by coadministration of drugs (phenobarbital, phenytoin, rifampin) that induce production of cytochrome P450IIIA4.
Perhaps because of competition for the P450IIIA4 metabolic pathway, quinidine levels rise when ketoconazole is coadministered.
Coadministration of propranolol usually does not affect quinidine pharmacokinetics, but in some studies the ß-blocker appeared to cause increases in the peak serum levels of quinidine, decreases in quinidine’s volume of distribution, and decreases in total quinidine clearance. The effects (if any) of coadministration of other ß-blockers on quinidine pharmacokinetics have not been adequately studied.
Hepatic clearance of quinidine is significantly reduced during coadministration of verapamil, with corresponding increases in serum levels and half-life.
Grapefruit juice
Grapefruit juice inhibits P450 3A4-mediated metabolism of quinidine to 3-hydroxyquinidine. Although the clinical significance of this interaction is unknown, grapefruit juice should be avoided.
Dietary salt
The rate and extent of quinidine absorption may be affected by changes in dietary salt intake; a decrease in dietary salt intake may lead to an increase in plasma quinidine concentrations.
Altered Pharmacokinetics of Other Drugs
Quinidine slows the elimination of digoxin and simultaneously reduces digoxin’s apparent volume of distribution. As a result, serum digoxin levels may be as much as doubled. When quinidine and digoxin are coadministered, digoxin doses usually need to be reduced. Serum levels of digitoxin are also raised when quinidine is coadministered, although the effect appears to be smaller.
By a mechanism that is not understood, quinidine potentiates the anticoagulatory action of warfarin, and the anticoagulant dosage may need to be reduced.
Cytochrome P450IID6 is an enzyme critical to the metabolism of many drugs, notably including mexiletine, some phenothiazines, and most polycyclic antidepressants. Constitutional deficiency of cytochrome P450IID6 is found in less than 1% of Orientals, in about 2% of American blacks, and in about 8% of American whites. Testing with debrisoquine is sometimes used to distinguish the P450IID6- deficient “poor metabolizers” from the majority-phenotype “extensive metabolizers”.
When drugs whose metabolism is P450IID6-dependent are given to poor metabolizers, the serum levels achieved are higher, sometimes much higher, than the serum levels achieved when identical doses are given to extensive metabolizers. To obtain similar clinical benefit without toxicity, doses given to poor metabolizers may need to be greatly reduced. In the case of prodrugs whose actions are actually mediated by P450IID6- produced metabolites (for example, codeine and hydrocodone, whose analgesic and antitussive effects appear to be mediated by morphine and hydromorphone, respectively), it may not be possible to achieve the desired clinical benefits in poor metabolizers.
Quinidine is not metabolized by cytochrome P450IID6, but therapeutic serum levels of quinidine inhibit the action of cytochrome P450IID6, effectively converting extensive metabolizers into poor metabolizers. Caution must be exercised whenever quinidine is prescribed together with drugs metabolized by cytochrome P450IID6.
Perhaps by competing for pathways of renal clearance, coadministration of quinidine causes an increase in serum levels of procainamide.
Serum levels of haloperidol are increased when quinidine is coadministered.
Presumably because both drugs are metabolized by cytochrome P450IIIA4, coadministration of quinidine causes variable slowing of the metabolism of nifedipine. Interactions with other dihydropyridine calcium channel blockers have not been reported, but these agents (including felodipine, nicardipine, and nimodipine) are all dependent upon P450IIIA4 for metabolism, so similar interactions with quinidine should be anticipated.
Altered Pharmacodynamics of Other Drugs
Quinidine’s anticholinergic, vasodilating, and negative inotropic actions may be additive to those of other drugs with these effects, and antagonistic to those of drugs with cholinergic, vasoconstricting, and positive inotropic effects. For example, when quinidine and verapamil are coadministered in doses that are each well tolerated as monotherapy, hypotension attributable to additive peripheral α-blockade is sometimes reported.
Quinidine potentiates the actions of depolarizing (succinylcholine, decamethonium) and nondepolarizing ( d-tubocurarine, pancuronium) neuromuscular blocking agents. These phenomena are not well understood, but they are observed in animal models as well as in humans. In addition, in vitro addition of quinidine to the serum of pregnant women reduces the activity of pseudocholinesterase, an enzyme that is essential to the metabolism of succinylcholine.
Non-interactions of Quinidine With Other Drugs
Quinidine has no clinically significant effect on the pharmacokinetics of diltiazem, flecainide, mephenytoin, metoprolol, propafenone, propranolol, quinine, timolol, or tocainide.
Conversely, the pharmacokinetics of quinidine are not significantly affected by caffeine, ciprofloxacin, digoxin, felodipine, omeprazole, or quinine. Quinidine’s pharmacokinetics are also unaffected by cigarette smoking.
Information for Patients
Before prescribing quinidine gluconate as prophylaxis against recurrence of atrial fibrillation, the physician should inform the patient of the risks and benefits to be expected (seeCLINICAL PHARMACOLOGY). Discussion should include the facts:
-
that the goal of therapy will be a reduction (probably not to zero) in the frequency of episodes of atrial fibrillation; and
-
that reduced frequency of fibrillatory episodes may be expected, if achieved, to bring symptomatic benefit; but
-
that no data are available to show that reduced frequency of fibrillatory episodes will reduce the risks of irreversible harm through stroke or death; and in fact
-
that such data as are available suggest that treatment with quinidine gluconate is likely to increase the patient’s risk of death.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Animal studies to evaluate quinidine’s carcinogenic or mutagenic potential have not been performed. Similarly, there are no animal data as to quinidine’s potential to impair fertility.
Pregnancy
Pregnancy Category C. Animal reproductive studies have not been conducted with quinidine. There are no adequate and well-controlled studies in pregnant women. Quinidine should be given to a pregnant woman only if clearly needed.
In one neonate whose mother had received quinidine throughout her pregnancy, the serum level of quinidine was equal to that of the mother, with no apparent ill effect. The level of quinidine in amniotic fluid was about three times higher than that found in serum.
Labor and Delivery
Quinine is said to be oxytocic in humans, but there are no adequate data as to quinidine’s effects (if any) on human labor and delivery.
Nursing Mothers
Quinidine is present in human milk at levels slightly lower than those in maternal serum; a human infant ingesting such milk should (scaling directly by weight) be expected to develop serum quinidine levels at least an order of magnitude lower than those of the mother. On the other hand, the pharmacokinetics and pharmacodynamics of quinidine in human infants have not been adequately studied, and neonates’ reduced protein binding of quinidine may increase their risk of toxicity at low total serum levels. Administration of quinidine should (if possible) be avoided in lactating women who continue to nurse.
Geriatric Use
Safety and efficacy of quinidine in elderly patients have not been systematically studied.
Pediatric Use
In antimalarial trials, quinidine was as safe and effective in pediatric patients as in adults. Notwithstanding the known pharmacokinetic differences between children and adults (seePharmacokinetics and Metabolism), children in these trials received the same doses (on a mg/kg basis) as adults.
Safety and effectiveness of antiarrhythmic use in pediatric patients have not been established.
-
-
ADVERSE REACTIONS
Quinidine preparations have been used for many years, but there are only sparse data from which to estimate the incidence of various adverse reactions. The adverse reactions most frequently reported have consistently been gastrointestinal, including diarrhea, nausea, vomiting, and heartburn/esophagitis.
In the reported study that was closest in character to the predominant approved use of quinidine gluconate, 86 adult outpatients with atrial fibrillation were followed for six months while they received slow-release quinidine bisulfate tablets, 600 mg (approximately 400 mg of quinidine base) twice daily. The incidences of adverse experiences reported more than once were as shown in the table below. The most serious quinidine-associated adverse reactions are described above underWARNINGS.
ADVERSE EXPERIENCES REPORTED MORE THAN ONCE IN 86 PATIENTS WITH ATRIAL FIBRILLATION Incidence (%) diarrhea 21 (24%) fever 5 (6%) rash 5 (6%) arrhythmia 3 (3%) abnormal electrocardiogram 3 (3%) nausea/vomiting 3 (3%) dizziness 3 (3%) headache 3 (3%) asthenia 2 (2%) cerebral ischemia 2 (2%) Vomiting and diarrhea can occur as isolated reactions to therapeutic levels of quinidine, but they may also be the first signs of cinchonism, a syndrome that may also include tinnitus, reversible high-frequency hearing loss, deafness, vertigo, blurred vision, diplopia, photophobia, headache, confusion, and delirium. Cinchonism is most often a sign of chronic quinidine toxicity, but it may appear in sensitive patients after a single moderate dose.
A few cases of hepatotoxicity, including granulomatous hepatitis, have been reported in patients receiving quinidine. All of these have appeared during the first few weeks of therapy, and most (not all) have remitted once quinidine was withdrawn.
Autoimmune and inflammatory syndromes associated with quinidine therapy have included fever, urticaria, flushing, exfoliative rash, bronchospasm, psoriasiform rash, pruritus and lymphadenopathy, hemolytic anemia, vasculitis, thrombocytopenic purpura, uveitis, angioedema, agranulocytosis, the sicca syndrome, arthralgia, myalgia, elevation in serum levels of skeletal-muscle enzymes, a disorder resembling systemic lupus erythematosus, and pneumonitis.
Convulsions, apprehension, and ataxia have been reported, but it is not clear that these were not simply the results of hypotension and consequent cerebral hypoperfusion. There are many reports of syncope. Acute psychotic reactions have been reported to follow the first dose of quinidine, but these reactions appear to be extremely rare.
Other adverse reactions occasionally reported include depression, mydriasis, disturbed color perception, night blindness, scotomata, optic neuritis, visual field loss, photosensitivity, and abnormalities of pigmentation.
-
OVERDOSAGE
Overdoses with various oral formulations of quinidine have been well described. Death has been described after a 5-gram ingestion by a toddler, while an adolescent was reported to survive after ingesting 8 grams of quinidine.
The most important ill effects of acute quinidine overdoses are ventricular arrhythmias and hypotension. Other signs and symptoms of overdose may include vomiting, diarrhea, tinnitus, high-frequency hearing loss, vertigo, blurred vision, diplopia, photophobia, headache, confusion, and delirium.
Arrhythmias
Serum quinidine levels can be conveniently assayed and monitored, but the electrocardiographic QTc interval is a better predictor of quinidine-induced ventricular arrhythmias.
The necessary treatment of hemodynamically unstable polymorphic ventricular tachycardia (including torsades de pointes) is withdrawal of treatment with quinidine and either immediate cardioversion or, if a cardiac pacemaker is in place or immediately available, immediate overdrive pacing. After pacing or cardioversion, further management must be guided by the length of the QTc interval.
Quinidine-associated ventricular tachyarrhythmias with normal underlying QTc intervals have not been adequately studied. Because of the theoretical possibility of QT-prolonging effects that might be additive to those of quinidine, other antiarrhythmics with Class I (disopyramide, procainamide) or Class III activities should (if possible) be avoided. Similarly, although the use of bretylium in quinidine overdose has not been reported, it is reasonable to expect that the α-blocking properties of bretylium might be additive to those of quinidine, resulting in problematic hypotension.
If the post-cardioversion QTc interval is prolonged, then the precardioversion polymorphic ventricular tachycardia was (by definition) torsades de pointes. In this case, lidocaine and bretylium are unlikely to be of value, and other Class I antiarrhythmics (disopyramide, procainamide) are likely to exacerbate the situation. Factors contributing to QTc prolongation (especially hypokalemia and hypomagnesemia) should be sought out and (if possible) aggressively corrected. Prevention of recurrent torsades may require sustained overdrive pacing or the cautious administration of isoproterenol (30-150 ng/kg/min).
Hypotension
Quinidine-induced hypotension that is not due to an arrhythmia is likely to be a consequence of quinidine-related α-blockade and vasorelaxation. Simple repletion of central volume (Trendelenburg positioning, saline infusion) may be sufficient therapy; other interventions reported to have been beneficial in this setting are those that increase peripheral vascular resistance, including α-agonist catecholamines (norepinephrine, metaraminol) and the Military Anti-Shock Trousers.
Treatment
To obtain up-to-date information about the treatment of overdose, a good resource is your certified Regional Poison-Control Center. Telephone numbers of certified poison-control centers are listed in the Physicians’ Desk Reference (PDR). In managing overdose, consider the possibilities of multiple-drug overdoses, drug-drug interactions, and unusual drug kinetics in your patient.
Accelerated Removal
Adequate studies of orally-administered activated charcoal in human overdoses of quinidine have not been reported, but there are animal data showing significant enhancement of systemic elimination following this intervention, and there is at least one human case report in which the elimination half-life of quinidine in the serum was apparently shortened by repeated gastric lavage. Activated charcoal should be avoided if an ileus is present; the conventional dose is 1 gram/kg, administered every 2 to 6 hours as a slurry with 8 mL/kg of tap water.
Although renal elimination of quinidine might theoretically be accelerated by maneuvers to acidify the urine, such maneuvers are potentially hazardous and of no demonstrated benefit.
Quinidine is not usefully removed from the circulation by dialysis.
Following quinidine overdose, drugs that delay elimination of quinidine (cimetidine, carbonic-anhydrase inhibitors, diltiazem, thiazide diuretics) should be withdrawn unless absolutely required.
-
DOSAGE AND ADMINISTRATION
The dose of quinidine delivered by quinidine gluconate extended-release tablets may be titrated by breaking a tablet in half. If tablets are crushed or chewed, their extended-release properties will be lost.
The dosage of quinidine varies considerably depending upon the general condition and the cardiovascular state of the patient.
Conversion of Atrial Fibrillation/Flutter to Sinus Rhythm
Especially in patients with known structural heart disease or other risk factors for toxicity, initiation or dose-adjustment of treatment with quinidine gluconate should generally be performed in a setting where facilities and personnel for monitoring and resuscitation are continuously available. Patients with symptomatic atrial fibrillation/flutter should be treated with quinidine gluconate only after ventricular rate control (e.g., with digitalis or ß-blockers) has failed to provide satisfactory control of symptoms.
Adequate trials have not identified an optimal regimen of quinidine gluconate for conversion of atrial fibrillation/flutter to sinus rhythm. In one reported regimen, the patient first receives two tablets (648 mg; 403 mg of quinidine base) of quinidine gluconate every eight hours. If this regimen has not resulted in conversion after 3 or 4 doses, then the dose is cautiously increased. If, at any point during administration, the QRS complex widens to 130% of its pre-treatment duration; the QTc interval widens to 130% of its pre-treatment duration and is then longer than 500 ms; P waves disappear; or the patient develops significant tachycardia, symptomatic bradycardia, or hypotension, then quinidine gluconate is discontinued, and other means of conversion (e.g., direct-current cardioversion) are considered.
In another regimen sometimes used, the patient receives one tablet (324 mg; 202 mg of quinidine base) every eight hours for two days; then two tablets every twelve hours for two days; and finally two tablets every eight hours for up to four days. The four-day stretch may come at one of the lower doses if, in the judgment of the physician, the lower dose is the highest one that will be tolerated. The criteria for discontinuation of treatment with quinidine gluconate are the same as in the other regimen.
Reduction in the Frequency of Relapse into Atrial Fibrillation/Flutter
In a patient with a history of frequent symptomatic episodes of atrial fibrillation/flutter, the goal of therapy with quinidine gluconate should be an increase in the average time between episodes. In most patients, the tachyarrhythmia will recur during therapy with quinidine gluconate, and a single recurrence should not be interpreted as therapeutic failure.
Especially in patients with known structural heart disease or other risk factors for toxicity, initiation or dose-adjustment of treatment with quinidine gluconate should generally be performed in a setting where facilities and personnel for monitoring and resuscitation are continuously available. Monitoring should be continued for two or three days after initiation of the regimen on which the patient will be discharged.
Therapy with quinidine gluconate should begin with one tablet (324 mg; 202 mg of quinidine base) every eight or twelve hours. If this regimen is well tolerated, if the serum quinidine level is still well within the laboratory’s therapeutic range, and if the average time between arrhythmic episodes has not been satisfactorily increased, then the dose may be cautiously raised. The total daily dosage should be reduced if the QRS complex widens to 130% of its pre-treatment duration; the QTc interval widens to 130% of its pre-treatment duration and is then longer than 500 ms; P waves disappear; or the patient develops significant tachycardia, symptomatic bradycardia, or hypotension.
Suppression of Life-Threatening Ventricular Arrhythmias
Dosing regimens for the use of quinidine gluconate in suppressing life-threatening ventricular arrhythmias have not been adequately studied. Described regimens have generally been similar to the regimen described just above for the prophylaxis of symptomatic atrial fibrillation/flutter. Where possible, therapy should be guided by the results of programmed electrical stimulation and/or Holter monitoring with exercise.
-
HOW SUPPLIED
Quinidine Gluconate Extended-release Tablets USP 324 mg are 13/32”, unscored, round, off-white tablets imprinted DAN and 5538 supplied in bottles of 100, 250, and 500.
Dispense in a well-closed, light-resistant container with child-resistant closure.
Store at 20°-25°C (68°-77°F). [See USP controlled room temperature.]
Watson Laboratories, Inc.
Corona, CA 92880 USA -
INGREDIENTS AND APPEARANCE
QUINIDINE GLUCONATE
quinidine gluconate tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0591-5538 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Quinidine Gluconate (UNII: R6875N380F) (Quinidine - UNII:ITX08688JL) 324 mg Inactive Ingredients Ingredient Name Strength Carnauba wax () Ethylcellulose () Magnesium stearate (UNII: 70097M6I30) Povidone () Product Characteristics Color WHITE (Off-white) Score no score Shape ROUND Size 10mm Flavor Imprint Code DAN;5538 Contains Coating false Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0591-5538-01 100 in 1 BOTTLE 2 NDC: 0591-5538-25 250 in 1 BOTTLE 3 NDC: 0591-5538-05 500 in 1 BOTTLE Labeler - Watson Laboratories, Inc.
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.