diclofenac sodium- Diclofenac Sodium tablet, delayed release diclofenac sodium- Diclofenac Sodium tablet
Drug Labeling and Warnings
Drug Details [pdf]
- N/A - Section Title Not Found In Database
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
Diclofenac sodium, is a benzene-acetic acid derivative, designated chemically as 2-[(2,6-dichlorophenyl)amino] benzeneacetic acid, monosodium salt. The structural formula is:
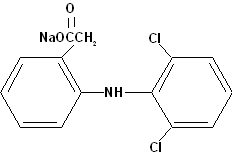
C14H10Cl2NNaO2 M.W. 318.14
Diclofenac sodium is a faintly yellowish white to light beige, virtually odorless, slightly hygroscopic crystalline powder. It is freely soluble in methanol, soluble in ethanol, sparingly soluble in water and practically insoluble in chloroform and in dilute acid. The n-octanol/water partition coefficient is 13.4 at pH 7.4 and 1545 at pH 5.2. Diclofenac sodium has a dissociation constant (pKa) of 4.0 ± 0.2 at 25°C in water.
Each enteric-coated tablet, for oral administration, contains 25 mg, 50 mg, or 75 mg of diclofenac sodium. In addition, each tablet contains the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, enteric-coating (black iron oxide, FD&C Blue No. 2, FD&C Red No. 40, FD&C Yellow No. 6, methacrylic acid copolymer, methylparaben, n-butyl alcohol, polysorbate 80, potassium sorbate, propylene glycol, propylparaben, sodium citrate, sodium lauryl sulfate, talc, titanium dioxide, triethyl citrate, and xanthan gum), lactose monohydrate, magnesium stearate, microcrystalline cellulose, pregelatinized starch, and stearic acid.
-
CLINICAL PHARMACOLOGY
Pharmacodynamics
Diclofenac is a nonsteroidal anti-inflammatory drug (NSAID). In pharmacologic studies, diclofenac has shown anti-inflammatory, analgesic, and antipyretic activity. As with other NSAIDs, its mode of action is not known; its ability to inhibit prostaglandin synthesis, however, may be involved in its anti-inflammatory activity.
Pharmacokinetics
Diclofenac sodium delayed-release tablets are in a pharmaceutic formulation that resists dissolution in the low pH of gastric fluid but allows a rapid release of drug in the higher pH-environment in the duodenum. Its pattern of drug release and absorption is illustrated below:
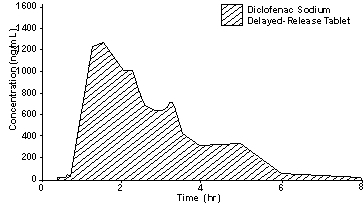
± 1 SD plasma diclofenac concentrations after a single dose of a 50 mg diclofenac sodium delayed-release tablet (N=38)
Absorption
When diclofenac sodium delayed-release tablets are administered orally after fasting, diclofenac is completely absorbed from the gastrointestinal tract. Of this, only 50% of the absorbed dose of diclofenac from diclofenac sodium is systemically available, due to first-pass metabolism. Peak plasma levels are achieved in 2 hours in fasting normal volunteers, with a range from 1 to 4 hours. The area-under-the plasma-concentration curve (AUC) is dose-proportional within the range of 25 mg to 150 mg. Peak plasma levels are less than dose-proportional and are approximately 1.0, 1.5, and 2 µg/mL for 25 mg, 50 mg and 75 mg doses, respectively. It should be noted that the administration of several individual diclofenac sodium tablets may not yield equivalent results in peak concentration as the administration of one tablet of a higher strength. This is probably due to the staggered gastric emptying of tablets into the duodenum. After repeated oral administration of diclofenac sodium 50 mg b.i.d., diclofenac did not accumulate in plasma.
When diclofenac sodium is taken with food, there is usually a delay in the onset of absorption of 1 to 4.5 hours, with delays as long as 10 hours in some patients, and a reduction in peak plasma levels of approximately 40%. The extent of absorption of diclofenac, however, is not significantly affected by food intake.
Distribution
Plasma concentrations of diclofenac decline from peak levels in a biexponential fashion, with the terminal phase having a half-life of approximately 2 hours. Clearance and volume of distribution are about 350 mL/min and 550 mL/kg, respectively. More than 99% of diclofenac is reversibly bound to human plasma albumin.
A 4-week study, comparing plasma level profiles of diclofenac (diclofenac sodium 50 mg b.i.d.) in younger (26 to 46 years) versus older (66 to 81 years) adults, did not show differences between age groups (10 patients per age group).
As with other NSAIDs, diclofenac diffuses into and out of the synovial fluid. Diffusion into the joint occurs when plasma levels are higher than those in the synovial fluid, after which the process reverses and synovial fluid levels are higher than plasma levels. It is not known whether diffusion into the joint plays a role in the effectiveness of diclofenac.
Metabolism and Elimination
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites. Approximately 65% of the dose is excreted in the urine, and approximately 35% in the bile.
Conjugates of unchanged diclofenac account for 5 to 10% of the dose excreted in the urine and for less than 5% excreted in the bile. Little or no unchanged unconjugated drug is excreted. Conjugates of the principal metabolite account for 20 to 30% of the dose excreted in the urine and for 10 to 20% of the dose excreted in the bile. Conjugates of three other metabolites together account for 10 to 20% of the dose excreted in the urine and for small amounts excreted in the bile. The elimination half-life values for these metabolites are shorter than those for the parent drug. Urinary excretion of an additional metabolite (half-life 80 hours) accounts for only 1.4% of the oral dose. The degree of accumulation of diclofenac metabolites is unknown. Some of the metabolites may have activity.
Patients with Renal and/or Hepatic Impairment
To date, no differences in the pharmacokinetics of diclofenac have been detected in studies of patients with renal (50 mg intravenously) or hepatic impairment (100 mg oral solution). In patients with renal impairment (N=5, creatinine clearance 3 to 42 mL/min), AUC values and elimination rates were comparable to those in healthy subjects. In patients with biopsy-confirmed cirrhosis or chronic active hepatitis (variably elevated transaminases and mildly elevated bilirubins, N=10), diclofenac concentrations and urinary elimination values were comparable to those in healthy subjects.
Clinical Studies
Osteoarthritis:
Diclofenac sodium was evaluated for the management of the signs and symptoms of osteoarthritis of the hip or knee in a total of 633 patients treated for up to 3 months in placebo and active-controlled clinical trials against aspirin (N=449), and naproxen (N=92). Diclofenac sodium was given both in variable (100 to 150 mg/day) and fixed (150 mg/day) dosing schedules on either b.i.d. or t.i.d. dosing regimens. In these trials, diclofenac sodium was found to be comparable to 2400 to 3600 mg/day of aspirin or 500 mg/day of naproxen. Diclofenac was effective when administered as either b.i.d. or t.i.d. dosing regimens.
Rheumatoid Arthritis:
Diclofenac sodium was evaluated for managing the signs and symptoms of rheumatoid arthritis in a total of 468 patients treated for up to 3 months in placebo- and active- controlled clinical trials against aspirin (N=290), and ibuprofen (N = 74). Diclofenac sodium was given in a fixed (150 or 200 mg/day) dosing schedule as either b.i.d. or t.i.d. dosing regimens. Diclofenac sodium was found to be comparable to 3600 to 4800 mg/day of aspirin, and 2400 mg/day of ibuprofen. Diclofenac sodium was used b.i.d. or t.i.d., administering 150 mg/day in most trials, but 50 mg q.i.d. (200 mg/day) was also studied.
Ankylosing Spondylitis:
Diclofenac sodium was evaluated for the management of the signs and symptoms of ankylosing spondylitis in a total of 132 patients in one active controlled clinical trial against indomethacin (N=130). Both diclofenac sodium and indomethacin patients were started on 25 mg t.i.d. and were permitted to increase the dose 25 mg per day each week to a maximum dose of 125 mg/day. Diclofenac sodium 75 to 125 mg/day was found to be comparable to indomethacin 75 to 125 mg/day.
G.I. Blood Loss/Endoscopy Data:
G.I. blood loss and endoscopy studies were performed with diclofenac sodium delayed-release [enteric-coated] tablets that, unlike immediate-release tablets, do not dissolve in the stomach where the endosopic lesions are primarily seen. A repeat-dose endoscopy study, in patients with rheumatoid arthritis or osteoarthritis treated with diclofenac sodium delayed-release tablets 75 mg b.i.d. (N=101), or naproxen (immediate-release tablets) 500 mg b.i.d. (N=103) for three months, resulted in a significantly smaller number of patients with an increase in endoscopy score from baseline and a significantly lower mean endoscopy score after treatment in the diclofenac sodium treated patients. Two repeat-dose endoscopic studies, in normal volunteers, showed that daily doses of diclofenac sodium delayed-release tablets 75 or 100 mg (N=6 and 14, respectively) for 1 week caused fewer gastric lesions, and those that did occur had lower scores than those observed following daily 500 mg doses of naproxen (immediate-release tablets). In healthy subjects, the daily administration of 150 mg of diclofenac sodium (N=8) for 3 weeks resulted in a mean fecal blood loss of less than that observed with 3 g of aspirin daily (N=8). In four repeat-dose studies, mean fecal blood loss with 150 mg of diclofenac was also less than that observed with 750 mg of naproxen (N=8 and 6) or 150 mg of indomethacin (N=8 and 6). The clinical significance of these findings is unknown since there is no evidence available to indicate that diclofenac sodium is less likely than other drugs of its class to cause serious gastrointestinal lesions when used in chronic therapy.
Individualization of Dosage
Diclofenac, like other NSAIDs, shows interindividual differences in both pharmacokinetics and clinical response (pharmacodynamics). Consequently, the recommended strategy for initiating therapy is to use a starting dose likely to be effective for the majority of patients and to adjust dosage thereafter based on observation of diclofenac’s beneficial and adverse effects.
In patients weighing less than 60 kg (132 lbs), or where the severity of the disease, concomitant medication, or other diseases warrant, the maximum recommended total daily dose of diclofenac should be reduced. Experience with other NSAIDs has shown that starting therapy with maximal doses in patients at increased risk due to renal or hepatic disease, low body weight (<60 kg), advanced age, a known ulcer diathesis, or known sensitivity to NSAID effects, is likely to increase frequency of adverse reactions and is not recommended (see PRECAUTIONS).
Osteoarthritis/Rheumatoid Arthritis/Ankylosing Spon-dylitis:
The usual starting dose of diclofenac sodium delayed-release tablets for patients with osteoarthritis, is 100 to 150 mg/day, using a b.i.d. or t.i.d. dosing regimen. In two variable-dose clinical trials in osteoarthritis, of 266 patients started on 100 mg/day, 176 chose to increase the dose to 150 mg/day. Dosages above 150 mg/day have not been studied in patients with osteoarthritis.
The usual starting dose of diclofenac sodium for most patients with rheumatoid arthritis is 150 mg/day, using a b.i.d. or t.i.d. dosing regimen. Patients requiring more relief of pain and inflammation may increase the dose to 200 mg/day. In clinical trials, patients receiving 200 mg/day were less likely to drop form the trial due to lack of efficacy than patients receiving 150 mg/day. Dosages above 225 mg/day are not recommended in patients with rheumatoid arthritis because of increased risk of adverse events.
The recommended dose of diclofenac sodium delayed-release tablets for patients with ankylosing spondylitis is 100 to 125 mg/day, using a q.i.d. dosing regimen (see DOSAGE AND ADMINISTRATION regarding the 125 mg/day dosage regimen). In a variable-dose clinical trial, of 132 patients started on 75 mg/day, 122 chose to increase the dose to 125 mg/day. Dosages above 125 mg/day have not been studied in patients with ankylosing spondylitis.
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Diclofenac sodium delayed-release tablets are contraindicated in patients with hypersensitivity to the product. Diclofenac should not be given to patients who have experienced asthma, urticaria, or other allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal, anaphylactic-like reactions to diclofenac have been reported in such patients.
-
WARNINGS
Gastrointestinal Effects
Peptic ulceration and gastrointestinal bleeding have been reported in patients receiving diclofenac. Physicians and patients should therefore remain alert for ulceration and bleeding in patients treated chronically with diclofenac even in the absence of previous G.I. tract symptoms. It is recommended that patients be maintained on the lowest dose of diclofenac possible consistent with achieving a satisfactory therapeutic response.
Risk of G.I. Ulcerations, Bleeding, and Perforation with NSAID Therapy
Serious gastrointestinal toxicity such as bleeding, ulceration, and perforation can occur at any time, with or without warning symptoms, in patients treated chronically with NSAID therapy. Although minor upper gastrointestinal problems, such as dyspepsia, are common, usually developing early in therapy, physicians should remain alert for ulceration and bleeding in patients treated chronically with NSAIDs even in the absence of previous G.I. tract symptoms. In patients observed in clinical trials of several months to 2 years duration, symptomatic upper G.I. ulcers, gross bleeding, or perforation appear to occur in approximately 1% of patients for 3 to 6 months, and in about 2 to 4% of patients treated for 1 year. Physicians should inform patients about the signs and/or symptoms of serious G.I. toxicity and what steps to take if they occur.
Studies to date have not identified any subset of patients not at risk of developing peptic ulceration and bleeding. Except for a prior history of serious G.I. events and other risk factors known to be associated with peptic ulcer disease, such as alcoholism, smoking, etc., no risk factors (e.g., age, sex) have been associated with increased risk. Elderly or debilitated patients seem to tolerate ulceration or bleeding less well than other individuals and most spontaneous reports of fatal G.I. events are in this population. Studies to date are inconclusive concerning the relative risk of various NSAIDs in causing such reactions. High doses of any NSAID probably carry a greater risk of these reactions, although controlled clinical trials showing this do not exist in most cases. In considering the use of relatively large doses (within the recommended dosage range), sufficient benefit should be anticipated to offset the potential increased risk of G.I. toxicity.
Hepatic Effects
As with other NSAIDs, elevations of one or more liver tests may occur during diclofenac therapy. These laboratory abnormalities may progress, may remain unchanged, or may be transient with continued therapy. Borderline elevations, (i.e., less than 3 times the ULN [=the Upper Limit of the Normal range]), or greater elevations of transaminases occurred in about 15% of diclofenac-treated patients. Of the hepatic enzymes, ALT (SGPT) is the one recommended for the monitoring of liver injury.
In clinical trials, meaningful elevations (i.e., more than 3 times the ULN) of AST (SGOT) (ALT was not measured in all studies) occurred in about 2% of approximately 5700 patients at some time during diclofenac sodium treatment. In a large, open, controlled trial, meaningful elevations of ALT and/or AST occurred in about 4% of 3700 patients treated for 2 to 6 months, including marked elevations (i.e., more than 8 times the ULN) in about 1% of the 3700 patients. In that open-label study, a higher incidence of borderline (less than 3 times the ULN), moderate (3 to 8 times the ULN), and marked (> 8 times the ULN) elevations of ALT or AST was observed in patients receiving diclofenac when compared to other NSAIDs. Transaminase elevations were seen more frequently in patients with osteoarthritis than in those with rheumatoid arthritis (see ADVERSE REACTIONS).
In addition to the enzyme elevations seen in clinical trials, rare cases of severe hepatic reactions, including jaundice and fatal fulminant hepatitis, have been reported.
Physicians should measure transaminases periodically in patients receiving long-term therapy with diclofenac, because severe hepatotoxicity may develop without a prodrome of distinguishing symptoms. The optimum times for making the first and subsequent transaminase measurements are not known. In the largest U.S. trial (open-label), that involved 3700 patients monitored first at 8 weeks and 1200 patients monitored again at 24 weeks, almost all meaningful elevations in transaminases were detected before patients became symptomatic. In 42 of the 51 patients in all trials who developed marked transaminase elevations, abnormal tests occurred during the first 2 months of therapy with diclofenac. Based on this experience, if diclofenac is used chronically, the first transaminase measurement should be made no later than 8 weeks after the start of diclofenac treatment. As with other NSAIDs, if abnormal liver tests persist or worsen, if clinical signs and/or symptoms consistent with liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.), diclofenac should be discontinued.
To minimize the possibility that hepatic injury will become severe between transaminase measurements, physicians should inform patients of the warning signs and symptoms of hepatotoxicity (e.g., nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness and “flu-like” symptoms), and the appropriate action patients should take if these signs and symptoms appear.
-
PRECAUTIONS
General
Allergic Reactions:
As with other NSAIDs, allergic reactions including anaphylaxis, have been reported with diclofenac. Specific allergic manifestations consisting of swelling of eyelids, lips, pharynx and larynx, urticaria, asthma, and bronchospasm, sometimes with a concomitant fall in blood pressure (severe at times) have been observed in clinical trials and/or the marketing experience with diclofenac. Anaphylaxis has rarely been reported from foreign sources; in U.S. clinical trials with diclofenac in over 6000 patients, 1 case of anaphylaxis was reported. In controlled clinical trials, allergic reactions have been observed at an incidence of 0.5%. These reactions can occur without prior exposure to the drug.
Fluid Retention and Edema:
Fluid retention and edema have been observed in some patients taking diclofenac. Therefore, as with other NSAIDs, diclofenac should be used with caution in patients with a history of cardiac decompensation, hypertension, or other conditions predisposing to fluid retention.
Renal Effects:
As a class, NSAIDs have been associated with renal papillary necrosis and other abnormal renal pathology in long-term administration to animals. In oral diclofenac studies in animals, some evidence of renal toxicity was noted. Isolated incidents of papillary necrosis were observed in a few animals at high doses (20 to 120 mg/kg) in several baboon subacute studies. In patients treated with diclofenac, rare cases of interstitial nephritis and papillary necrosis have been reported (see ADVERSE REACTIONS).
A second form of renal toxicity generally associated with NSAIDs, is seen in patients with conditions leading to a reduction in renal blood flow or blood volume, where renal prostaglandins have a supportive role in the maintenance of renal perfusion. In these patients, administration of a NSAID results in a dose-dependent decrease in prostaglandin synthesis and, secondarily, in a reduction of renal blood flow, which may precipitate overt renal failure. Patients at greatest risk of this reaction are those with impaired renal function, heart failure, liver dysfunction, those taking diuretics, and the elderly. Discontinuation of NSAID therapy is typically followed by recovery to the pretreatment state.
Cases of significant renal failure in patients receiving diclofenac have been reported from marketing experience, but were not observed in over 4000 patients in clinical trials during which serum creatinine and BUN values were followed serially. There were only 11 patients (0.3%) whose serum creatinine and concurrent serum BUN values were greater than 2.0 mg/dL and 40 mg/dL, respectively, while on diclofenac (mean rise in the 11 patients: creatinine 2.3 mg/dL and BUN 28.4 mg/dL).
Since diclofenac metabolites are eliminated primarily by the kidneys, patients with significantly impaired renal function should be more closely monitored than subjects with normal renal function.
Porphyria:
The use of diclofenac in patients with hepatic porphyria should be avoided. To date, one patient has been described in whom diclofenac probably triggered a clinical attack of porphyria. The postulated mechanism. demonstrated in rats, for causing such attacks by diclofenac, as well as some other NSAIDs, is through stimulation of the porphyrin precursor deltaaminolevulinic acid (ALA).
Information for Patients
Diclofenac, like other drugs of its class, is not free of side effects. The side effects of these drugs can cause discomfort and, rarely, there are more serious side effects, such as gastrointestinal bleeding and, more rarely, liver toxicity (see WARNINGS: Hepatic Effects), which may result in hospitalization and even fatal outcomes.
NSAIDs are often essential agents in the management of arthritis and have a major role in the management of pain but they also may be commonly employed for conditions that are less serious.
Physicians may wish to discuss with their patients the potential risks (see WARNINGS, PRECAUTIONS, and ADVERSE REACTIONS) and likely benefits of NSAID treatment, particularly when the drugs are used for less serious conditions where treatment without NSAIDs may represent an acceptable alternative to both the patient and physician.
Laboratory Tests
Because serious G.I. tract ulceration and bleeding can occur without warning symptoms, physicians should follow chronically treated patients for the signs and symptoms of ulceration and bleeding and should inform them of the importance of this follow-up (see WARNINGS: Risk of G.I. Ulcerations, Bleeding, and Perforation with NSAID Therapy). If diclofenac is used chronically, patients should also be instructed to report any signs and symptoms that might be due to hepatotoxicity of diclofenac; these symptoms may be evident between visits when periodic liver laboratory tests are performed (see WARNINGS: Hepatic Effects).
Drug Interactions
Aspirin:
Concomitant administration of diclofenac and aspirin is not recommended because diclofenac is displaced from its binding sites during the concomitant administration of aspirin, resulting in lower plasma concentrations, peak plasma levels, and AUC values.
Anticoagulants:
While studies have not shown diclofenac to interact with anti-coagulants of the warfarin type, caution should be exercised, nonetheless, since interactions have been seen with other NSAIDs. Because prostaglandins play an important role in hemostasis, and NSAIDs affect platelet function as well, concurrent therapy with all NSAIDs, including diclofenac, and warfarin requires close monitoring of patients to be certain that no change in their anticoagulant dosage is required.
Digoxon, Methotrexate, Cyclosporine:
Diclofenac, like other NSAIDs, may affect renal prostaglandins and increase the toxicity of certain drugs. Ingestion of diclofenac may increase serum concentrations of digoxin and methotrexate and increase cyclosporine's nephrotoxicity. Patients who begin taking diclofenac or who increase their diclofenac dose or any other NSAID while taking digoxin, methotrexate, or cyclosporine may develop toxicity characteristics of these drugs. They should be observed closely, particularly if renal function is impaired. In case of digoxin, serum levels should be monitored.
Lithium:
Diclofenac decreases lithium renal clearance and increases lithium plasma levels. In patients taking diclofenac and lithium concomitantly, lithium toxicity may develop.
Oral Hypoglycemics:
Diclofenac does not alter glucose metabolism in normal subjects nor does it alter the effects of oral hypoglycemic agents. There are rare reports, however, from marketing experiences of changes in effects of insulin or oral hypoglycemic agents in the presence of diclofenac that necessitated changes in the doses of such agents. Both hypo- and hyperglycemic effects have been reported. A direct causal relationship has not been established, but physicians should consider the possibility that diclofenac may alter a diabetic patient’s response to insulin or oral hypoglycemic agents.
Protein Binding
In vitro, diclofenac interferes minimally or not at all with the protein binding of salicylic acid (20% decrease in binding), tolbutamide, prednisolone (10% decrease in binding), or warfarin. Benzylpenicillin, ampicillin, oxacillin, chlortetracycline, doxycycline, cephalothin, erythromycin, and sulfamethoxazole have no influence in vitro on the protein binding of diclofenac in human serum.
Drug/Laboratory Test Interactions
Effect on Blood Coagulation:
Diclofenac increases platelet aggregation time but does not affect bleeding time, plasma thrombin clotting time, plasma fibrinogen, or factors V and VII to XII. Statistically significant changes in prothrombin and partial thromboplastin times have been reported in normal volunteers. The mean changes were observed to be less than 1 second in both instances, however, and are unlikely to be clinically important. Diclofenac is a prostaglandin synthetase inhibitor, however, and all drugs that inhibit prostaglandin synthesis interfere with platelet function to some degree; therefore, patients who may be adversely affected by such an action should be carefully observed.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies in rats given diclofenac sodium up to 2 mg/kg/day (12 mg/m2/day, approximately the human dose) have revealed no significant increases in tumor incidence. There was a slight increase in benign mammary fibroadenomas in mid-dose-treated (0.5 mg/kg/day or 3 mg/m2/day) female rats (high-dose females had excessive mortality), but the increase was not significant for this common rat tumor. A two-year carcinogenicity study conducted in mice employing diclofenac sodium at doses up to 0.3 mg/kg/day (0.9 mg/m2/day) in males and 1 mg/kg/day (3 mg/m2/day) in females did not reveal any oncogenic potential. Diclofenac sodium did not show mutagenic activity in in vitro point mutation assays in mammalian (mouse lymphoma) and microbial (yeast, Ames) test systems, and was nonmutagenic in several mammalian in vitro and in vivo tests, including dominant lethal and male germinal epithelial chromosomal studies in mice, and nucleus anomaly and chromosomal aberration studies in Chinese hamsters. Diclofenac sodium administered to male and female rats at 4 mg/kg/day did not affect fertility.
Teratogenic Effects
There are no adequate and well-controlled studies in pregnant women. Diclofenac should be used during pregnancy only if the benefits to the mother justify the potential risk to the fetus.
Pregnancy Category B:
Reproduction studies have been performed in mice given diclofenac sodium (up to 20 mg/kg/day or 60 mg/m2/day) and in rats and rabbits given diclofenac sodium (up to 10 mg/kg/day or 60 mg/m2/day for rats, and 80 mg/m2/day for rabbits), and have revealed no evidence of teratogenicity despite the induction of maternal toxicity and fetal toxicity. In rats, maternally toxic doses were associated with dystocia, prolonged gestation, reduced fetal weights and growth, and reduced fetal survival. Diclofenac has been shown to cross the placental barrier in mice and rats.
Labor and Delivery
The effects of diclofenac on labor and delivery in pregnant women are unknown. Because of the known effects of postaglandin-inhibiting drugs on the fetal cardiovascular system (closure of ductus arteriosus), use of diclofenac during late pregnancy should be avoided and, as with other nonsteriodal anti-inflammatory drugs, it is possible that diclofenac may inhibit uterine contraction.
Nursing Mothers
Diclofenac has been found in the milk of nursing mothers. As with other drugs that are excreted in milk, diclofenac is not recommended for use in nursing women.
Pediatric Use
Safety and effectiveness of diclofenac in pediatric patients have not been established.
Geriatic Use
Of the more than 6000 patients treated with diclofenac in U.S. trials, 31% were older than 65 years of age. No overall difference was observed between efficacy, adverse event or pharmacokinetic profiles of older and younger patients. As with any NSAID, the elderly are likely to tolerate adverse reactions less well than younger patients.
-
ADVERSE REACTIONS
Adverse reaction information is derived from blinded, controlled and open-label clinical trials as well as worldwide marketing experience. In the description below, rates of more common events represent clinical study results; rarer events are derived principally from marketing experience and publications, and accurate rate estimates are generally not possible.
The incidence of common adverse reactions (greater than 1%) is based upon controlled clinical trials in 1543 patients treated up to 13 weeks with diclofenac sodium delayed-release tablets. By far the most common adverse effects were gastrointestinal symptoms, most of them minor, occurring in about 20%, and leading to discontinuation in about 3%, of patients. Peptic ulcer or G.I. bleeding occurred in clinical trials in 0.6% (95%-confidence interval: 0.2% to 1%) of approximately 1800 patients during their first 3 months of diclofenac treatment and 1.6% (95%-confidence interval: 0.8% to 2.4%) of approximately 800 patients followed for 1 year.
Gastrointestinal symptoms were followed in frequency by central nervous system side effects such as headache (7%) and dizziness (3%).
Meaningful (exceeding 3 times the Upper Limit of Normal) elevations of ALT (SGPT) or AST (SGOT) occurred at an overall rate of approximately 2% during the first 2 months of diclofenac sodium treatment. Unlike aspirin-related elevations, which occur more frequently in patients with rheumatoid arthritis, these elevations were more frequently observed in patients with osteoarthritis (2.6%) than in patients with rheumatoid arthritis (0.7%). Marked elevations (exceeding 8 times the ULN) were seen in 1% of patients treated for 2 to 6 months (see WARNINGS: Hepatic Effects).
The following adverse reactions were reported in patients treated with diclofenac:
Incidence Greater Than 1%–Causal Relationship Probable
(All derived from clinical trials)
Digestive:
Diarrhea*, indigestion*, nausea*, constipation*, flatulence, liver test abnormalities*, PUB, i.e., peptic ulcer, with or without bleeding and/or perforation, or bleeding without ulcer (see above and also WARNINGS).
Incidence Less Than 1% - Causal Relationship Probable
The following reactions have been reported in patients taking diclofenac under circumstances that do not permit a clear attribution of the reaction to diclofenac. These reactions are being included as alerting information for physicians. Adverse reactions reported only in worldwide marketing experience or in the literature, not seen in clinical trials, are considered rare and are italicized.
Body as a Whole:
Malaise, swelling of lips and tongue, photosensitivity, anaphylaxis, anaphylactoid reactions.
Digestive:
Vomiting, jaundice, melena, aphthous stomatitis, dry mouth and mucous membranes, bloody diarrhea, hepatitis, hepatic necrosis, appetite change, pancreatitis with or without concomitant hepatitis, colitis.
Hemic and Lymphatic:
Hemoglobin decrease, leukopenia, thrombocytopenia, hemolytic anemia, aplastic anemia, agranulocytosis, purpura, allergic purpura.
Nervous System:
Insomnia, drowsiness, depression, diplopia, anxiety, irritability, aseptic meningitis.
Incidence Less Than 1% - Causal Relationship Unknown
(Adverse reactions reported only in worldwide marketing experience or in the literature, not seen in clinical trials, are considered rare and are italicized.)
Cardiovascular:
Palpitations, flushing, tachycardia, premature ventricular contractions, myocardial infarction.
-
OVERDOSAGE
Worldwide reports on overdosage with diclofenac cover 66 cases. In approximately one-half of these reports of overdosage, concomitant medications were also taken. The highest dose of diclofenac was 5 g in a 17-year-old male who suffered loss of consciousness, increased intracranial pressure, aspiration pneumonitis, and died 2 days after overdose. The next highest doses of diclofenac were 4 g and 3.75 g. The 24-year-old female who took 4 g and the 28- and 42-year-old females, each of whom took 3.75 g, did not develop any clinically significant signs or symptoms. However, there was a report of a 17-year-old female who experienced vomiting and drowsiness after an overdose of 2.37 g of diclofenac.
Animal LD50 values show a wide range of susceptibilities to acute overdosage, with primates being more resistant to acute toxicity than rodents (LD50 in mg/kg–rats, 55; dogs, 500; monkeys, 3200).
In case of acute overdosage it is recommended that the stomach be emptied by vomiting or lavage. Forced diuresis may theoretically be beneficial because the drug is excreted in the urine. The effect of dialysis or hemoperfusion in the elimination of diclofenac (99% protein-bound, see CLINICAL PHARMACOLOGY) remains unproven. In addition to supportive measures, the use of oral activated charcoal may help to reduce the absorption of diclofenac.
-
DOSAGE AND ADMINISTRATION
Diclofenac sodium may be administered as 25 mg, 50 mg, or 75 mg delayed-release tablets. Regardless of the indication, the dosage of diclofenac should be individualized to the lowest effective dose to minimize adverse effects (see CLINICAL PHARMACOLOGY: Individualization of Dosage)
Osteoarthritis:
The recommended dosage is 100 to 150 mg/day in divided doses, 50 mg b.i.d. or t.i.d. or 75 mg b.i.d. Dosages above 150 mg/day have not been studied in patients with osteoarthritis.
-
HOW SUPPLIED
Diclofenac Sodium Delayed-Release Tablets USP
25 mg white, round, enteric-coated tablets
Tablets Identified 54 140
NDC: 0054-8223-25: Unit dose, 10 tablets per strip, 10 strips per shelf pack, ten shelf packs per shipper.
NDC: 0054-4223-25: Bottles of 100 tablets.
50 mg white, round, enteric-coated tablets
Tablets Identified 54 592
NDC: 0054-8221-25: Unit dose, 10 tablets per strip, 10 strips per shelf pack, ten shelf packs per shipper.
NDC: 0054-4221-25: Bottles of 100 tablets.
NDC: 0054-4221-31: Bottles of 1000 tablets.
75 mg white, round, enteric-coated tablets
Tablets Identiied 54 839
NDC: 0054-8222-25: Unit dose, 10 tablets per strip, 10 strips per shelf pack, ten shelf packs per shipper.
NDC: 0054-4222-25: Bottles of 100 tablets.
NDC: 0054-4222-31: Bottles of 1000 tablets.
Do not store above 30°C (86°F).
Protect from moisture.
Dispense in a tight, light-resistant container as defined in the USP/NF.
4048700//03
© RLI, 2005
-
INGREDIENTS AND APPEARANCE
DICLOFENAC SODIUM
diclofenac sodium tablet, delayed releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0054-8221 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength diclofenac sodium (UNII: QTG126297Q) 50 mg Inactive Ingredients Ingredient Name Strength black iron oxide () colloidal silicon dioxide () croscarmellose sodium () FD&C Blue No. 2 () FD&C Red No. 40 () FD&C Yellow No. 6 () lactose monohydrate () magnesium stearate (UNII: 70097M6I30) methacrylic acid copolymer () methylparaben () microcrystalline cellulose () n-butyl alcohol (UNII: 8PJ61P6TS3) polysorbate 80 () potassium sorbate () pregelatinized starch () propylene glycol (UNII: 6DC9Q167V3) propylparaben (UNII: Z8IX2SC1OH) sodium citrate (UNII: 1Q73Q2JULR) sodium lauryl sulfate (UNII: 368GB5141J) stearic acid (UNII: 4ELV7Z65AP) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) triethyl citrate () xanthan gum () Product Characteristics Color WHITE (WHITE) Score no score Shape ROUND (ROUND) Size 8mm Flavor Imprint Code 54;592 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0054-8221-25 10 in 1 PACKAGE 1 100 in 1 BLISTER PACK 2 NDC: 0054-4221-25 100 in 1 BOTTLE, PLASTIC 3 NDC: 0054-4221-31 1000 in 1 BOTTLE, PLASTIC DICLOFENAC SODIUM
diclofenac sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0054-4223 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength diclofenac sodium (UNII: QTG126297Q) 25 mg Inactive Ingredients Ingredient Name Strength black iron oxide () colloidal silicon dioxide () croscarmellose sodium () FD&C Blue No. 2 () FD&C Red No. 40 () FD&C Yellow No. 6 () lactose monhydrate () magnesium stearate (UNII: 70097M6I30) methacrylic acid copolymer () methylparaben () microcrystalline cellulose () n-butyl alcohol (UNII: 8PJ61P6TS3) polysorbate 80 () potassium sorbate () pregelatinized starch () propylene glycol (UNII: 6DC9Q167V3) propylparaben (UNII: Z8IX2SC1OH) sodium citrate (UNII: 1Q73Q2JULR) sodium lauryl sulfate (UNII: 368GB5141J) stearic acid (UNII: 4ELV7Z65AP) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) triethyl citrate () xanthan gum () Product Characteristics Color WHITE (WHITE) Score no score Shape ROUND (ROUND) Size 6mm Flavor Imprint Code 54;140 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0054-4223-25 100 in 1 BOTTLE, PLASTIC 2 NDC: 0054-8223-25 10 in 1 PACKAGE 2 100 in 1 BLISTER PACK DICLOFENAC SODIUM
diclofenac sodium tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0054-8222 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength diclofenac sodium (UNII: QTG126297Q) 75 mg Inactive Ingredients Ingredient Name Strength black iron oxide () colloidal silicon dioxide () croscarmellose sodium () FD&C Blue No. 2 () FD&C Red No. 40 () FD&C Yellow No. 6 () lactose monohydrate () magnesium stearate (UNII: 70097M6I30) methacrylic acid copolymer () methylparaben () microcrystalline cellulose () n-butyl alcohol (UNII: 8PJ61P6TS3) polysorbate 80 () potassium sorbate () pregelatinized starch () propylene glycol (UNII: 6DC9Q167V3) propylparaben (UNII: Z8IX2SC1OH) sodium citrate (UNII: 1Q73Q2JULR) sodium lauryl sulfate (UNII: 368GB5141J) stearic acid (UNII: 4ELV7Z65AP) talc (UNII: 7SEV7J4R1U) titanium dioxide (UNII: 15FIX9V2JP) triethyl citrate () xanthan gum () Product Characteristics Color WHITE (WHITE) Score no score Shape ROUND (ROUND) Size 10mm Flavor Imprint Code 54;839 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0054-8222-25 10 in 1 PACKAGE 1 100 in 1 BLISTER PACK 2 NDC: 0054-4222-25 100 in 1 BOTTLE, PLASTIC 3 NDC: 0054-4222-31 1000 in 1 BOTTLE, PLASTIC Labeler - Boehringer Ingelheim Roxane Laboratories
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.