tramadol hydrochloride- Tramadol Hydrochloride tablet, film coated
Drug Labeling and Warnings
Drug Details [pdf]
- N/A - Section Title Not Found In Database
-
DESCRIPTION
Tramadol hydrochloride tablet is a centrally acting analgesic. The chemical name for tramadol hydrochloride is (±) cis-2-[(dimethylamino)methyl]-1-(3-methoxyphenyl) cyclohexanol hydrochloride. Its structural formula is:
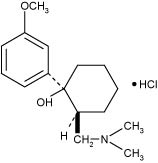
The molecular formula of tramadol hydrochloride is C16H25NO2HCl and its molecular weight is 299.8.
Tramadol hydrochloride is a white, bitter, crystalline and odorless powder. It is readily soluble in water and ethanol and has a pKa of 9.41. The n-octanol/water log partition coefficient (logP) is 1.35 at pH 7. Each tramadol hydrochloride tablet intended for oral administration contains 50 mg of tramadol hydrochloride.
In addition, it also contains the following inactive ingredients:
hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, pregelatinized starch, sodium starch glycolate and titanium dioxide. -
CLINICAL PHARMACOLOGY
Pharmacodynamics
Tramadol hydrochloride is a centrally acting synthetic opioid analgesic. Although its mode of action is not completely understood, from animal tests, at least two complementary mechanisms appear applicable: binding of parent and M1 metabolite to μ-opioid receptors and weak inhibition of reuptake of nor-epinephrine and serotonin.
Opioid activity is due to both low affinity binding of the parent compound and higher affinity binding of the Ο-demethylated metabolite M1 to μ-opioid receptors. In animal models, M1 is up to 6 times more potent than tramadol in producing analgesia and 200 times more potent in μ-opioid binding. Tramadol-induced analgesia is only partially antagonized by the opiate antagonist naloxone in several animal tests. The relative contribution of both tramadol and M1 to human analgesia is dependent upon the plasma concentrations of each compound (see CLINICAL PHARMACOLOGY, Pharmacokinetics).
Tramadol has been shown to inhibit reuptake of norepinephrine and serotonin in vitro, as have some other opioid analgesics. These mechanisms may contribute independently to the overall analgesic profile of tramadol hydrochloride. Analgesia in humans begins approximately within one hour after administration and reaches a peak in approximately two to three hours.
Apart from analgesia, tramadol hydrochloride administration may produce a constellation of symptoms (including dizziness, somnolence, nausea, constipation, sweating and pruritus) similar to that of other opioids. In contrast to morphine, tramadol has not been shown to cause histamine release. At therapeutic doses, tramadol hydrochloride has no effect on heart rate, left-ventricular function or cardiac index. Orthostatic hypotension has been observed.
Pharmacokinetics
The analgesic activity of tramadol hydrochloride is due to both parent drug and the M1 metabolite (see CLINICAL PHARMACOLOGY, Pharmacodynamics). Tramadol is administered as a racemate and both the [-] and [+] forms of both tramadol and M1 are detected in the circulation. Tramadol is well absorbed orally with an absolute bioavailability of 75%. Tramadol has a volume of distribution of approximately 2.7 L/kg and is only 20% bound to plasma proteins. Tramadol is extensively metabolized by a number of pathways, including CYP2D6 and CYP3A4, as well as by conjugation of parent and metabolites. One metabolite, M1, is pharmacologically active in animal models. The formation of M1 is dependent upon CYP2D6 and as such is subject to inhibition, which may affect the therapeutic response (see PRECAUTIONS - Drug Interactions). Tramadol and its metabolites are excreted primarily in the urine with observed plasma half-lives of 6.3 and 7.4 hours for tramadol and M1, respectively. Linear pharmacokinetics have been observed following multiple doses of 50 and 100 mg to steady-state.
Absorption:
Racemic tramadol is rapidly and almost completely absorbed after oral administration. The mean absolute bioavailability of a 100 mg oral dose is approximately 75%. The mean peak plasma concentration of racemic tramadol and M1 occurs at two and three hours, respectively, after administration in healthy adults. In general, both enantiomers of tramadol and M1 follow a parallel time course in the body following single and multiple doses although small differences (∼10%) exist in the absolute amount of each enantiomer present.
Steady-state plasma concentrations of both tramadol and M1 are achieved within two days with q.i.d. dosing. There is no evidence of self-induction (see Figure 1 and table 1 below).
Figure 1: Mean Tramadol and M1 Plasma Concentration Profiles after a Single 100 mg Oral Dose and after Twenty-Nine 100 mg Oral Doses of Tramadol HCl given q.i.d.:
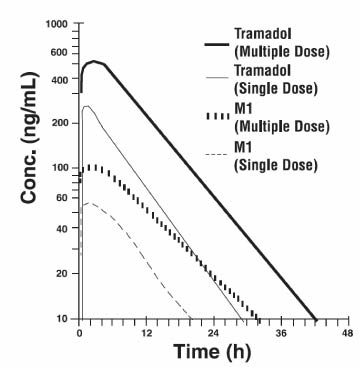
Table 1 Mean (%CV) Pharmacokinetic Parameter for Racemic Tramadol and M1 Metabolite - * SD = Single dose, MD = Multiple dose, p.o. = Oral administration, i.v. = Intravenous administration, q.i.d. = Four times daily
- † F represents the oral bioavailability of tramadol
- ‡ Not applicable
- § Not measured
Population/Dosage Regimen* Parent Drug/
MetabolitePeak Conc.
(ng/ml)Time to
Peak (hrs)Clearance/F†
(mL/min/Kg)t1/2 (hrs) Healthy Adults,
100 mg qid,
MD p.o.Tramadol 592 (30) 2.3 (61) 5.90 (25) 6.7 (15) M1 110 (29) 2.4 (46) ‡ 7.0 (14) Healthy Adults,
100 mg
SD p.o.Tramadol 308 (25) 1.6 (63) 8.50 (31) 5.6 (20) M1 55.0 (36) 3.0 (51) ‡ 6.7 (16) Geriatric,
(> 75 yrs)
50 mg SD p.o.Tramadol 208 (31) 2.1 (19) 6.89 (25) 7.0 (23) M1 § § ‡ § Hepatic Impaired,
50 mg
SD p.o.Tramadol 217 (11) 1.9 (16) 4.23 (56) 13.3 (11) M1 19.4 (12) 9.8 (20) ‡ 18.5 (15) Renal Impaired,
Clcr10-30 mL/min
100 mg SD i.v.Tramadol ‡ ‡ 4.23 (54) 10.6 (31) M1 ‡ ‡ ‡ 11.5 (40) Renal Impaired,
Clcr<5 mL/min
100 mg SD i.v.Tramadol ‡ ‡ 3.73 (17) 11.0 (29) M1 ‡ ‡ ‡ 16.9 (18) Food Effects:
Oral administration of tramadol hydrochloride with food does not significantly affect its rate or extent of absorption, therefore, tramadol hydrochloride can be administered without regard to food.
Distribution:
The volume of distribution of tramadol was 2.6 and 2.9 liters/kg in male and female subjects, respectively, following a 100 mg intravenous dose. The binding of tramadol to human plasma proteins is approximately 20% and binding also appears to be independent of concentration up to 10 mcg/mL. Saturation of plasma protein binding occurs only at concentrations outside the clinically relevant range.
Metabolism:
Tramadol is extensively metabolized after oral administration. Approximately 30% of the dose is excreted in the urine as unchanged drug, whereas 60% of the dose is excreted as metabolites. The remainder is excreted either as unidentified or as unextractable metabolites. The major metabolic pathways appear to be N- and O-demethylation and glucuronidation or sulfation in the liver. One metabolite ( O-desmethyltramadol, denoted M1) is pharmacologically active in animal models. Formation of M1 is dependent on the CYP2D6 and as such is subject to inhibition, which may affect the therapeutic response (see PRECAUTIONS - Drug Interactions).
Approximately 7% of the population has reduced activity of the CYP2D6 isoenzyme of cytochrome P-450. These individuals are “poor metabolizers” of debrisoquine, dextromethorphan, tricyclic anti-depressants, among other drugs. Based on a population PK analysis of Phase I studies in healthy subjects, concentrations of tramadol were approximately 20% higher in “poor metabolizers” versus “extensive metabolizers”, while M1 concentrations were 40% lower. Concomitant therapy with inhibitors of CYP2D6 such as fluoxetine, paroxetine and quinidine could result in significant drug interactions. In vitro drug interaction studies in human liver microsomes indicate that inhibitors of CYP2D6 such as fluoxetine and its metabolite norfluoxetine, amitriptyline and quinidine inhibit the metabolism of tramadol to various degrees, suggesting that concomitant administration of these compounds could result in increases in tramadol concentrations and decreased concentrations of M1. The full pharmacological impact of these alterations in terms of either efficacy or safety is unknown. Concomitant use of SEROTONIN re-uptake INHIBITORS and MAO INHIBITORS may enhance the risk of adverse events, including seizure (see WARNINGS) and serotonin syndrome.
Elimination:
Tramadol is eliminated primarily through metabolism by the liver and the metabolites are eliminated primarily by the kidneys. The mean terminal plasma elimination half-lives of racemic tramadol and racemic M1 are 6.3 ±1.4 and 7.4 ±1.4 hours, respectively. The plasma elimination half-life of racemic tramadol increased from approximately six hours to seven hours upon multiple dosing.
Special Populations
Renal:
Impaired renal function results in a decreased rate and extent of excretion of tramadol and its active metabolite, M1. In patients with creatinine clearances of less than 30 mL/min, adjustment of the dosing regimen is recommended (see DOSAGE AND ADMINISTRATION). The total amount of tramadol and M1 removed during a 4-hour dialysis period is less than 7% of the administered dose.
Hepatic:
Metabolism of tramadol and M1 is reduced in patients with advanced cirrhosis of the liver, resulting in both a larger area under the concentration time curve for tramadol and longer tramadol and M1 elimination half-lives (13 hrs. for tramadol and 19 hrs. for M1). In cirrhotic patients, adjustment of the dosing regimen is recommended (see DOSAGE AND ADMINISTRATION).
Geriatric:
Healthy elderly subjects aged 65 to 75 years have plasma tramadol concentrations and elimination half-lives comparable to those observed in healthy subjects less than 65 years of age. In subjects over 75 years, maximum serum concentrations are elevated (208 vs.162 ng/mL) and the elimination half-life is prolonged (7 vs. 6 hours) compared to subjects 65 to 75 years of age. Adjustment of the daily dose is recommended for patients older than 75 years (see DOSAGE AND ADMINISTRATION).
Gender:
The absolute bioavailability of tramadol was 73% in males and 79% in females. The plasma clearance was 6.4 mL/min/kg in males and 5.7 mL/min/kg in females following a 100 mg IV dose of tramadol. Following a single oral dose, and after adjusting for body weight, females had a 12% higher peak tramadol concentration and a 35% higher area under the concentration-time curve compared to males. The clinical significance of this difference is unknown.
Clinical Studies
Tramadol hydrochloride has been given in single oral doses of 50, 75 and 100 mg to patients with pain following surgical procedures and pain following oral surgery (extraction of impacted molars).
In single-dose models of pain following oral surgery, pain relief was demonstrated in some patients at doses of 50 mg and 75 mg. A dose of 100 mg tramadol hydrochloride tended to provide analgesia superior to codeine sulfate 60 mg, but it was not as effective as the combination of aspirin 650 mg with codeine phosphate 60 mg.
Tramadol hydrochloride has been studied in three long-term controlled trials involving a total of 820 patients, with 530 patients receiving tramadol hydrochloride. Patients with a variety of chronic painful conditions were studied in double-blind trials of one to three months duration. Average daily doses of approximately 250 mg of tramadol hydrochloride in divided doses were generally comparable to five doses of acetaminophen 300 mg with codeine phosphate 30 mg daily, five doses of aspirin 325 mg with codeine phosphate 30 mg daily, or two to three doses of acetaminophen 500 mg with oxycodone hydrochloride 5 mg daily.
Titration Trials
In a randomized, blinded clinical study with 129 to 132 patients per group, a 10-day titration to a daily tramadol hydrochloride dose of 200 mg (50 mg q.i.d.), attained in 50 mg increments every 3 days, was found to result in fewer discontinuations due to dizziness or vertigo than titration over only 4 days or no titration.
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Tramadol hydrochloride should not be administered to patients who have previously demonstrated hypersensitivity to tramadol, any other component of this product or opioids. Tramadol hydrochloride is contraindicated in any situation where opioids are contraindicated, including acute intoxication with any of the following: alcohol, hypnotics, narcotics, centrally acting analgesics, opioids or psychotropic drugs. Tramadol hydrochloride may worsen central nervous system and respiratory depression in these patients.
-
WARNINGS
Seizure Risk
Seizures have been reported in patients receiving tramadol hydrochloride within the recommended dosage range. Spontaneous post-marketing reports indicate that seizure risk is increased with doses of tramadol hydrochloride above the recommended range. Concomitant use of tramadol hydrochloride increases the seizure risk in patients taking:
- Selective serotonin reuptake inhibitors (SSRI antidepressants or anoretics),
- Tricyclic antidepressants (TCAs), and other tricyclic compounds (e.g., cyclobenzaprine, promethazine, etc.), or
- Other opioids.
Administration of tramadol hydrochloride may enhance the seizure risk in patients taking:
- MAO inhibitors (see also WARNINGS- Use with MAO inhibitors),
- Neuroleptics, or
- Other drugs that reduce the seizure threshold.
Risk of convulsions may also increase in patients with epilepsy, those with a history of seizures, or in patients with a recognized risk for seizure (such as head trauma, metabolic disorders, alcohol and drug withdrawal, CNS infections). In tramadol hydrochloride overdose, naloxone administration may increase the risk of seizure.
Anaphylactoid Reactions
Serious and rarely fatal anaphylactoid reactions have been reported in patients receiving therapy with tramadol hydrochloride. When these events do occur it is often following the first dose. Other reported allergic reactions include pruritus, hives, bronchospasm, angioedema, toxic epidermal necrolysis and Stevens-Johnson syndrome. Patients with a history of anaphylactoid reactions to codeine and other opioids may be at increased risk and therefore should not receive tramadol hydrochloride (see CONTRAINDICATIONS).
Respiratory Depression
Administer tramadol hydrochloride cautiously in patients at risk for respiratory depression. In these patients alternative non-opioid analgesics should be considered. When large doses of tramadol hydrochloride are administered with anesthetic medications or alcohol, respiratory depression may result. Respiratory depression should be treated as an overdose. If naloxone is to be administered, use cautiously because it may precipitate seizures (see WARNINGS , Seizure Risk and OVERDOSAGE).
Interaction with Central Nervous System (CNS) Depressants
Tramadol hydrochloride should be used with caution and in reduced dosages when administered to patients receiving CNS depressants such as alcohol, opioids, anesthetic agents, narcotics, phenothiazines, tranquilizers or sedative hypnotics. Tramadol hydrochloride increases the risk of CNS and respiratory depression in these patients.
Increased Intracranial Pressure or Head Trauma
Tramadol hydrochloride should be used with caution in patients with increased intracranial pressure or head injury. The respiratory depressant effects of opioids include carbon dioxide retention and secondary elevation of cerebrospinal fluid pressure, and may be markedly exaggerated in these patients. Additionally, pupillary changes (miosis) from tramadol may obscure the existence, extent, or course of intracranial pathology. Clinicians should also maintain a high index of suspicion for adverse drug reaction when evaluating altered mental status in these patients if they are receiving tramadol hydrochloride. (See Respiratory Depression)
Use in Ambulatory Patients
Tramadol hydrochloride may impair the mental and or physical abilities required for the performance of potentially hazardous tasks such as driving a car or operating machinery. The patient using this drug should be cautioned accordingly.
Use with MAO Inhibitors and serotonin re-uptake inhibitors
Use tramadol hydrochloride with great caution in patients taking monoamine oxidase inhibitors. Animal studies have shown increased deaths with combined administration. Concomitant use of tramadol hydrochloride with MAO inhibitors or SSRI's increases the risk of adverse events, including seizure and serotonin syndrome.
Withdrawal
Withdrawal symptoms may occur if tramadol hydrochloride tablets are discontinued abruptly (See DRUG ABUSE and DEPENDENCE). These symptoms may include: anxiety, sweating, insomnia, rigors, pain, nausea, tremors, diarrhea, upper respiratory symptoms, piloerection, and rarely hallucinations. Other symptoms that have been seen less frequently with tramadol hydrochloride tablets discontinuation include: panic attacks, severe anxiety, and paresthesias. Clinical experience suggests that withdrawal symptoms may be avoided by tapering tramadol hydrochloride tablets at the time of discontinuation.
Physical Dependence and Abuse
Tramadol hydrochloride may induce psychic and physical dependence of the morphine-type (μ-opioid) (See DRUG ABUSE and DEPENDENCE). Tramadol hydrochloride should not be used in opioid-dependent patients. Tramadol hydrochloride has been shown to reinitiate physical dependence in some patients that have been previously dependent on other opioids. Dependence and abuse, including drug-seeking behavior and taking illicit actions to obtain the drug, are not limited to those patients with prior history of opioid dependence.
Risk of Overdosage
Serious potential consequences of overdosage with tramadol hydrochloride are central nervous system depression, respiratory depression and death. In treating an overdose, primary attention should be given to maintaining adequate ventilation along with general supportive treatment (See OVERDOSAGE).
-
PRECAUTIONS
Acute Abdominal Conditions
The administration of tramadol hydrochloride may complicate the clinical assessment of patients with acute abdominal conditions.
Use in Renal and Hepatic Disease
Impaired renal function results in a decreased rate and extent of excretion of tramadol and its active metabolite, M1. In patients with creatinine clearances of less than 30 mL/min, dosing reduction is recommended (see DOSAGE AND ADMINISTRATION).
Metabolism of tramadol and M1 is reduced in patients with advanced cirrhosis of the liver. In cirrhotic patients, dosing reduction is recommended (see DOSAGE AND ADMINISTRATION).
With the prolonged half-life in these conditions, achievement of steady-state is delayed, so that it may take several days for elevated plasma concentrations to develop.
Information for Patients
- Tramadol hydrochloride tablets may impair mental or physical abilities required for the performance of potentially hazardous tasks such as driving a car or operating machinery
- Tramadol hydrochloride tablets should not be taken with alcohol containing beverages.
- Tramadol hydrochloride tablets should be used with caution when taking medications such as tranquilizers, hypnotics or other opiate containing analgesics.
- The patient should be instructed to inform the physician if they are pregnant, think they might become pregnant, or are trying to become pregnant (see PRECAUTIONS: Labor and Delivery).
- The patient should understand the single-dose and 24-hour dose limit and the time interval between doses, since exceeding these recommendations can result in respiratory depression, seizures and death.
Drug Interactions
In vitro studies indicate that tramadol is unlikely to inhibit the CYP3A4-mediated metabolism of other drugs when tramadol is administered concomitantly at therapeutic doses. Tramadol does not appear to induce its own metabolism in humans, since observed maximal plasma concentrations after multiple oral doses are higher than expected based on single-dose data. Tramadol is a mild inducer of selected drug metabolism pathways measured in animals.
Use with Carbamazepine
Patients taking carbamazepine may have a significantly reduced analgesic effect of tramadol hydrochloride. Because carbamazepine increases tramadol metabolism and because of the seizure risk associated with tramadol, concomitant administration of tramadol hydrochloride and carbamazepine is not recommended.
Use with Quinidine
Tramadol is metabolized to M1 by the CYP2D6. Quinidine is a selective inhibitor of that isoenzyme, so that concomitant administration of quinidine and tramadol hydrochloride results in increased concentrations of tramadol and reduced concentrations of M1. The clinical consequences of these findings are unknown. In vitro drug interaction studies in human liver microsomes indicate that tramadol has no effect on quinidine metabolism.
Use with Inhibitors of CYP2D6
In vitro drug interaction studies in human liver microsomes indicate that concomitant administration with inhibitors of CYP2D6 such as fluoxetine, paroxetine, and amitriptyline could result in some inhibition of the metabolism of tramadol.
Use with Cimetidine
Concomitant administration of tramadol hydrochloride with cimetidine does not result in clinically significant changes in tramadol pharmacokinetics. Therefore, no alteration of the tramadol hydrochloride dosage regimen is recommended.
Use with MAO Inhibitors
Interactions with MAO Inhibitors, due to interference with detoxification mechanisms, have been reported for some centrally acting drugs (see WARNINGS, Use with MAO Inhibitors).
Carcinogenesis, Mutagenesis, and Impairment of Fertility
A slight, but statistically significant, increase in two common murine tumors, pulmonary and hepatic, was observed in a mouse carcinogenicity study, particularly in aged mice. Mice were dosed orally up to 30 mg/kg (90 mg/m2 or 0.36 times the maximum daily human dosage of 246 mg/m2) for approximately two years, although the study was not done with the Maximum Tolerated Dose. This finding is not believed to suggest risk in humans. No such finding occurred in a rat carcinogenicity study (dosing orally up to 30 mg/kg, 180 mg/m2, or 0.73 times the maximum daily human dosage).
Tramadol was not mutagenic in the following assays: Ames Salmonella microsomal activation test, CHO/HPRT mammalian cell assay, mouse lymphoma assay (in the absence of metabolic activation), dominant lethal mutation tests in mice, chromosome aberration test in Chinese hamsters, and bone marrow micronucleus tests in mice and Chinese hamsters. Weakly mutagenic results occurred in the presence of metabolic activation in the mouse lymphoma assay and micronucleus test in rats. Overall, the weight of evidence from these tests indicates that tramadol does not pose a genotoxic risk to humans.
No effects on fertility were observed for tramadol at oral dose levels up to 50 mg/kg (300 mg/m2) in male rats and 75 mg/kg (450 mg/m2) in female rats. These dosages are 1.2 and 1.8 times the maximum daily human dosage of 246 mg/m2, respectively.
Pregnancy, Teratogenic Effects: Pregnancy Category C
Tramadol has been shown to be embryotoxic and fetotoxic in mice, (120 mg/kg or 360 mg/m2), rats (≥ 25 mg/kg or 150 mg/m2) and rabbits (≥75 mg/kg or 900 mg/m2) at maternally toxic dosages but was not teratogenic at these dose levels. These dosages on a mg/m2 basis are 1.4, ≥ 0.6, and ≥ 3.6 times the maximum daily human dosage (246 mg/m2) for mouse, rat and rabbit, respectively.
No drug-related teratogenic effects were observed in progeny of mice (up to 140 mg/kg or 420 mg/m2), rats (up to 80 mg/kg or 480 mg/m2) or rabbits (up to 300 mg/kg or 3600 mg/m2) treated with tramadol by various routes. Embryo and fetal toxicity consisted primarily of decreased fetal weights, skeletal ossification and increased supernumerary ribs at maternally toxic dose levels. Transient delays in developmental or behavioral parameters were also seen in pups from rat dams allowed to deliver. Embryo and fetal lethality were reported only in one rabbit study at 300 mg/kg (3600 mg/m2), a dose that would cause extreme maternal toxicity in the rabbit. The dosages listed for mouse, rat and rabbit are 1.7, 1.9 and 14.6 times the maximum daily human dosage (246 mg/m2), respectively.
Non-teratogenic Effects
Tramadol was evaluated in peri- and post-natal studies in rats. Progeny of dams receiving oral (gavage) dose levels of 50 mg/kg (300 mg/m2 or 1.2 times the maximum daily human tramadol dosage) or greater had decreased weights, and pup survival was decreased early in lactation at 80 mg/kg (480 mg/m2 or 1.9 and higher the maximum daily human dose).
There are no adequate and well-controlled studies in pregnant women. Tramadol hydrochloride should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Neonatal seizures, neonatal withdrawal syndrome, fetal death and still birth have been reported during post-marketing.
Labor and Delivery
Tramadol hydrochloride should not be used in pregnant women prior to or during labor unless the potential benefits outweigh the risks. Safe use in pregnancy has not been established. Chronic use during pregnancy may lead to physical dependence and post-partum withdrawal symptoms in the newborn (See DRUG ABUSE AND DEPENDENCE). Tramadol has been shown to cross the placenta. The mean ratio of serum tramadol in the umbilical veins compared to maternal veins was 0.83 for 40 women given tramadol during labor.
The effect of tramadol hydrochloride, if any, on the later growth, development, and functional maturation of the child is unknown.
Nursing Mothers
Tramadol hydrochloride is not recommended for obstetrical preoperative medication or for post-delivery analgesia in nursing mothers because its safety in infants and newborns has not been studied. Following a single IV 100 mg dose of tramadol, the cumulative excretion in breast milk within 16 hours postdose was 100 mcg of tramadol (0.1% of the maternal dose) and 27 mcg of M1.
Pediatric Use
The safety and efficacy of tramadol hydrochloride in patients under 16 years of age have not been established. The use of tramadol hydrochloride in the pediatric population is not recommended.
Geriatric Use
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function and of concomitant disease or other drug therapy. In patients over 75 years of age, daily doses in excess of 300 mg are not recommended (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
A total of 455 elderly (65 years of age or older) subjects were exposed to tramadol hydrochloride in controlled clinical trials. Of those, 145 subjects were 75 years of age and older.
In studies including geriatric patients, treatment-limiting adverse events were higher in subjects over 75 years of age compared to those under 65 years of age. Specifically, 30% of those over 75 years of age had gastrointestinal treatment-limiting adverse events compared to 17% of those under 65 years of age. Constipation resulted in discontinuation of treatment in 10% of those over 75.
-
ADVERSE REACTIONS
Tramadol hydrochloride was administered to 550 patients during the double-blind or open-label extension periods in U.S. studies of chronic nonmalignant pain. Of these patients, 375 were 65 years old or older. Table 2 reports the cumulative incidence rate of adverse reactions by 7, 30 and 90 days for the most frequent reactions (5% or more by 7 days). The most frequently reported events were in the central nervous system and gastrointestinal system. Although the reactions listed in the table are felt to be probably related to tramadol hydrochloride administration, the reported rates also include some events that may have been due to underlying disease or concomitant medication. The overall incidence rates of adverse experiences in these trials were similar for tramadol hydrochloride and the active control groups, acetaminophen 300 mg with codeine phosphate 30 mg, and aspirin 325 mg with codeine phosphate 30 mg however the rates of withdrawals due to adverse events appeared to be higher in the tramadol hydrochloride groups.
Table 2 Cumulative Incidence of Adverse Reactions for Tramadol Hydrochloride in Chronic Trials of Nonmalignant Pain (N=427) - * “CNS Stimulation” is a composite of nervousness, anxiety, agitation, tremor, spasticity,
euphoria, emotional lability and hallucinations.Up to
7 DaysUp to
30 DaysUp to
90 DaysDizziness/Vertigo 26% 31% 33% Nausea 24% 34% 40% Constipation 24% 38% 46% Headache 18% 26% 32% Somnolence 16% 23% 25% Vomiting 9% 13% 17% Pruritus 8% 10% 11% "CNS Stimulation"* 7% 11% 14% Asthenia 6% 11% 12% Sweating 6% 7% 9% Dyspepsia 5% 9% 13% Dry Mouth 5% 9% 10% Diarreah 5% 6% 10% Incidence 1% to less than 5%, possibly causally related: the following lists adverse reactions that occurred with an incidence of 1% to less than 5% in clinical trials, and for which the possibility of a causal relationship with tramadol hydrochloride exists.
Body as a Whole: Malaise.
Cardiovascular: Vasodilation.
Central Nervous System: Anxiety, Confusion, Coordination disturbance, Euphoria, Miosis, Nervousness, Sleep disorder.
Gastrointestinal: Abdominal pain, Anorexia, Flatulence.
Musculoskeletal: Hypertonia.
Skin: Rash.
Special Senses: Visual disturbance.
Urogenital: Menopausal symptoms, Urinary frequency, Urinary retention.Incidence less than 1%, possibly causally related: the following lists adverse reactions that occurred with an incidence of less than 1% in clinical trials and/or reported in post-marketing experience.
Body as a Whole: Accidental injury, Allergic reaction, Anaphylaxis, Death, Suicidal tendency, Weight loss, Serotonin syndrome (mental status change, hyperreflexia, fever, shivering, tremor, agitation, diaphoresis, seizures and coma)
Cardiovascular: Orthostatic hypotension, Syncope, Tachycardia.
Central Nervous System: Abnormal gait, Amnesia, Cognitive dysfunction, Depression, Difficulty in concentration, Hallucinations, Paresthesia, Seizure (see WARNINGS), Tremor.
Respiratory: Dyspnea.
Skin: Stevens-Johnson syndrome/Toxic epidermal necrolysis, Urticaria, Vesicles.
Special Senses: Dysgeusia.
Urogenital: Dysuria, Menstrual disorder.Other adverse experiences, causal relationship unknown: A variety of other adverse events were reported infrequently in patients taking tramadol hydrochloride during clinical trials and/or reported in post-marketing experience. A causal relationship between tramadol hydrochloride and these events has not been determined. However, the most significant events are listed below as alerting information to the physician.
Cardiovascular: Abnormal ECG, Hypertension, Hypotension, Myocardial ischemia, Palpitations, Pulmonary edema, Pulmonary embolism.
Central Nervous System: Migraine, Speech disorders.
Gastrointestinal: Gastrointestinal bleeding, Hepatitis, Stomatitis, Liver failure.
Laboratory Abnormalities: Creatinine increase, Elevated liver enzymes, Hemoglobin decrease, Proteinuria.
Sensory: Cataracts, Deafness, Tinnitus. - * “CNS Stimulation” is a composite of nervousness, anxiety, agitation, tremor, spasticity,
-
DRUG ABUSE AND DEPENDENCE
Tramadol hydrochloride may induce psychic and physical dependence of the morphine-type (μ-opioid) (See WARNINGS). Dependence and abuse, including drug-seeking behavior and taking illicit actions to obtain the drug are not limited to those patients with prior history of opioid dependence. The risk in patients with substance abuse has been observed to be higher. Tramadol hydrochloride is associated with craving and tolerance development. Withdrawal symptoms may occur if tramadol hydrochloride is discontinued abruptly. These symptoms may include: anxiety, sweating, insomnia, rigors, pain, nausea, tremors, diarrhea, upper respiratory symptoms, piloerection, and rarely hallucinations. Other symptoms that have been seen less frequently with tramadol hydrochloride discontinuation include: panic attacks, severe anxiety, and paresthesias. Clinical experience suggests that withdrawal symptoms may be relieved by reinstitution of opioid therapy followed by a gradual, tapered dose reduction of the medication combined with symptomatic support.
-
OVERDOSAGE
Serious potential consequences of overdosage are respiratory depression, lethargy, coma, seizure, cardiac arrest and death (See WARNINGS). Fatalities have been reported in post marketing in association with both intentional and unintentional overdose with tramadol hydrochloride. In treating an overdose, primary attention should be given to maintaining adequate ventilation along with general supportive treatment. While naloxone will reverse some, but not all, symptoms caused by overdosage with tramadol hydrochloride the risk of seizures is also increased with naloxone administration. In animals convulsions following the administration of toxic doses of tramadol could be suppressed with barbiturates or benzodiazepines but were increased with naloxone. Naloxone administration did not change the lethality of an overdose in mice. Hemodialysis is not expected to be helpful in an overdose because it removes less than 7% of the administered dose in a 4-hour dialysis period.
-
DOSAGE & ADMINISTRATION
Adults (17 years of age and over)
For patients with moderate to moderately severe chronic pain not requiring rapid onset of analgesic effect, the tolerability of tramadol hydrochloride can be improved by initiating therapy with a titration regimen: The total daily dose may be increased by 50 mg as tolerated every 3 days to reach 200 mg/day (50 mg q.i.d.). After titration, tramadol hydrochloride 50 mg to 100 mg can be administered as needed for pain relief every four to six hours, not to exceed 400 mg per day.For the subset of patients for whom rapid onset of analgesic effect is required and for whom the benefits outweigh the risk of discontinuation due to adverse events associated with higher initial doses, tramadol hydrochloride 50 mg to 100 mg can be administered as needed for pain relief every four to six hours, not to exceed 400 mg per day..
Individualization of Dose
Good pain management practice dictates that the dose be individualized according to patient need using the lowest beneficial dose. Studies with tramadol in adults have shown that starting at the lowest possible dose and titrating upwards will result in fewer discontinuations and increased tolerability.
- In all patients with creatinine clearance less than 30 mL/min, it is recommended that the dosing interval of tramadol hydrochloride be increased to 12 hours, with a maximum daily dose of 200 mg. Since only 7% of an administered dose is removed by hemodialysis, dialysis patients can receive their regular dose on the day of dialysis.
- The recommended dose for adult patients with cirrhosis is 50 mg every 12 hours.
- In general, dose selection for an elderly patient over 65 years old should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function and of concomitant disease or other drug therapy. For elderly patients over 75 years old, total dose should not exceed 300 mg/day.
-
HOW SUPPLIED
Tramadol hydrochloride tablets 50 mg are supplied as unscored, white, round film coated tablets debossed "cor" over “127”.
They are supplied as follows:
Bottles of 100 (NDC: 65162-127-10)
Bottles of 500 (NDC: 65162-127-50)
Bottles of 1000 (NDC: 65162-127-11)
Store at 20° to 25°C (68° to 77°F). [see USP Controlled Room Temperature]
Dispense in a tight container as defined in the USP.
Manufactured by:
Corepharma LLC
Middlesex, NJ 08846Distributed by:
Akyma Pharmaceuticals LLC
Glasgow, KY 42141
June 2006
MF # 153-06 -
INGREDIENTS AND APPEARANCE
TRAMADOL HYDROCHLORIDE
tramadol hydrochloride tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 65162-127 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TRAMADOL HYDROCHLORIDE (UNII: 9N7R477WCK) (Tramadol - UNII:39J1LGJ30J) 50 mg Inactive Ingredients Ingredient Name Strength hypromellose () lactose monohydrate () magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose () polyethylene glycol () polysorbate 80 () pregelatinized starch () sodium starch glycolate () titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color WHITE (White) Score no score Shape ROUND (round) Size 9mm Flavor Imprint Code cor;127 Contains Coating true Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 65162-127-10 100 in 1 BOTTLE, PLASTIC 2 NDC: 65162-127-50 500 in 1 BOTTLE, PLASTIC 3 NDC: 65162-127-11 1000 in 1 BOTTLE, PLASTIC Labeler - COREPHARMA LLC.
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.