OCALIVA- obeticholic acid tablet, film coated
Ocaliva by
Drug Labeling and Warnings
Ocaliva by is a Prescription medication manufactured, distributed, or labeled by Intercept Pharmaceuticals Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OCALIVA® safely and effectively. See full prescribing information for OCALIVA.
OCALIVA® (obeticholic acid) tablets, for oral use
Initial U.S. Approval: 2016WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
See full prescribing information for complete boxed warning
- In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended. (5.1)
- The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event. (2.2)
INDICATIONS AND USAGE
OCALIVA, a farnesoid X receptor (FXR) agonist, is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA.
This indication is approved under accelerated approval based on a reduction in alkaline phosphatase (ALP). An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. (1)
DOSAGE AND ADMINISTRATION
Important Dosage and Administration Instructions (2.1)
- Prior to starting OCALIVA in patients with suspected cirrhosis, use the nomogram to calculate Child-Pugh classification (A, B, or C) and determine the appropriate starting dosage.
Parameter Points Scored for Observed Findings 1 point 2 points 3 points Encephalopathy grade None 1 or 2 3 or 4 Ascites Absent Slight Moderate Serum bilirubin (mg/dL) < 2 2 to 3 > 3 Serum albumin (g/dL) > 3.5 2.8 to 3.5 < 2.8 International Normalized Ratio (INR) < 1.7 1.7 to 2.3 > 2.3 Child-Pugh Class is obtained by adding the points from all 5 parameters to derive a total score, which can range from 5 to 15 points.
Total Score: 5-6 points =A, 7-9 points =B, 10-15 points =C- Routinely monitor patients during OCALIVA treatment for biochemical response, tolerability, progression of PBC disease, and re-evaluate Child-Pugh classification to determine if dosage adjustment is needed.
- Reduce the dosing frequency from once daily to once weekly as appropriate for patients who progress to advanced disease (i.e., from Child-Pugh Class A to Child-Pugh Class B or C).
Recommended Dosage Regimen (2.2)
The recommended starting dosage and titration regimen of OCALIVA, for patients who have not achieved an adequate biochemical response to an appropriate dosage of UDCA for at least 1 year or who are intolerant to UDCA, is dependent on disease stage.
Staging/ Classification Non-Cirrhotic or Compensated Child-Pugh Class A Child-Pugh Class B or C or Patients with a Prior Decompensation Eventa Starting OCALIVA Dosage for first 3 months 5 mg once daily 5 mg once weekly OCALIVA Dosage Titration after first 3 months, for patients who have not achieved an adequate reduction in ALP and/or total bilirubin and who are tolerating OCALIVAb 10 mg once daily 5 mg twice weekly (at least 3 days apart)
Titrate to 10 mg twice weekly (at least 3 days apart) based on response and tolerabilityMaximum OCALIVA Dosage 10 mg once daily 10 mg twice weekly (at least 3 days apart) aGastroesophageal variceal bleeding, new or worsening jaundice, spontaneous bacterial peritonitis, etc.
bPrior to dosage adjustment, re-calculate the Child-Pugh classification. (2.1)Monitoring for Safety, Treatment Discontinuation (2.3)
- Routinely monitor all patients for progression of PBC disease.
- Reduce the dosing frequency for patients who progress from Child-Pugh Class A to Child-Pugh Class B or C. (2.2)
- Closely monitor patients at an increased risk of hepatic decompensation.
- Interrupt treatment in patients with laboratory or clinical evidence of worsening liver function indicating risk of decompensation, and monitor liver function.
- Consider discontinuing OCALIVA in patients who experience clinically significant liver-related adverse reactions.
Management of Patients with Intolerable Pruritus
- See full prescribing information for management options. (2.4)
Administration Instructions (2.5)
- Take with or without food.
- For patients taking bile acid binding resins, take OCALIVA at least 4 hours before or 4 hours after taking a bile acid binding resin, or at as great an interval as possible. (7.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 5 mg, 10 mg (3)
CONTRAINDICATIONS
Patients with complete biliary obstruction (4)
WARNINGS AND PRECAUTIONS
- Hepatic Decompensation and Failure in Incorrectly Dosed PBC Patients with Child-Pugh Class B or C or Decompensated Cirrhosis: Routinely monitor patients for progression of PBC disease, including liver-related complications, with laboratory and clinical assessments. Dosage adjustment, interruption, or discontinuation may be required. Discontinue in patients who develop complete biliary obstruction. (2.3, 4, 5.1)
- Severe Pruritus: Management strategies include the addition of bile acid binding resins or antihistamines; OCALIVA dosage reduction and/or temporary dosing interruption. (2.4, 5.3)
- Reduction in HDL-C: Monitor for changes in serum lipid levels during treatment. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (≥ 5%) are: pruritus, fatigue, abdominal pain and discomfort, rash, oropharyngeal pain, dizziness, constipation, arthralgia, thyroid function abnormality, and eczema. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Intercept Pharmaceuticals at 1-844-782-ICPT or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Warfarin: Potential for decreased INR; monitor INR and adjust the dosage of warfarin, as needed, to maintain the target INR range. (7.2)
- CYP1A2 Substrates with Narrow Therapeutic Index (e.g., theophylline and tizanidine): Potential for increased exposure to CYP1A2 substrates; monitor drug concentrations of CYP1A2 substrates with narrow therapeutic index. (7.3)
- Inhibitors of Bile Salt Efflux Pump (e.g., cyclosporine): Avoid use. If concomitant use is necessary, monitor serum transaminases and bilirubin. (7.4)
USE IN SPECIFIC POPULATIONS
Hepatic Impairment: Dosage adjustment is required in patients with Child-Pugh-Class B and C or a prior decompensation event. (2.2, 8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Instructions
2.2 Recommended Dosage Regimen
2.3 Monitoring to Assess Safety, Treatment Interruption or Discontinuation
2.4 Management of Patients with Intolerable Pruritus on OCALIVA
2.5 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Decompensation and Failure in Incorrectly Dosed PBC Patients with Child-Pugh Class B or C or Decompensated Cirrhosis
5.2 Liver-Related Adverse Reactions
5.3 Severe Pruritus
5.4 Reduction in HDL-C
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Bile Acid Binding Resins
7.2 Warfarin
7.3 CYP1A2 Substrates with Narrow Therapeutic Index
7.4 Inhibitors of Bile Salt Efflux Pump
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATIC DECOMPENSATION AND FAILURE IN INCORRECTLY DOSED PBC PATIENTS WITH CHILD-PUGH CLASS B OR C OR DECOMPENSATED CIRRHOSIS
- In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in patients with primary biliary cholangitis (PBC) with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than recommended [see Warnings and Precautions (5.1)].
- The recommended starting dosage of OCALIVA is 5 mg once weekly for patients with Child-Pugh Class B or C hepatic impairment or a prior decompensation event [see Dosage and Administration (2.2)].
-
1 INDICATIONS AND USAGE
OCALIVA® is indicated for the treatment of primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults with an inadequate response to UDCA, or as monotherapy in adults unable to tolerate UDCA.
This indication is approved under accelerated approval based on a reduction in alkaline phosphatase (ALP) [see Clinical Studies (14)]. An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Instructions
- Prior to the initiation of OCALIVA in patients with suspected cirrhosis, use the nomogram (see Table 1) to calculate the patient's score to determine their Child-Pugh classification (A, B, or C) and determine the appropriate starting dosage (see Table 2) [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
Table 1: Child-Pugh Nomogram Parameter Points Scored for Observed Findings 1 point 2 points 3 points Encephalopathy grade None 1 or 2 3 or 4 Ascites Absent Slight Moderate Serum bilirubin (mg/dL) < 2 2 to 3 > 3 Serum albumin (g/dL) > 3.5 2.8 to 3.5 < 2.8 International Normalized Ratio (INR) < 1.7 1.7 to 2.3 > 2.3 Child-Pugh Class is obtained by adding the points from all 5 parameters to derive a total score, which can range from 5 to 15 points.
Child-Pugh Class A: 5 to 6 points
Child-Pugh Class B: 7 to 9 points
Child-Pugh Class C: 10 to 15 points- Routinely monitor patients during OCALIVA treatment for biochemical response, tolerability, progression of PBC disease, and re-evaluate Child-Pugh classification to determine if dosage adjustment is needed.
- Reduce the dosing frequency from once daily to once weekly as appropriate for patients who progress to advanced disease (i.e., from Child-Pugh Class A to Child-Pugh Class B or C) [see Dosage and Administration (2.2)].
2.2 Recommended Dosage Regimen
The recommended starting dose and titration dosage regimen of OCALIVA for patients who have not achieved an adequate biochemical response to an appropriate dosage of UDCA for at least 1 year or are intolerant to UDCA [see Clinical Studies (14)] is dependent upon disease stage, as shown in Table 2:
- Non-cirrhotic patients or compensated cirrhotic patients with no or mild hepatic impairment (Child-Pugh Class A) are dosed once daily.
- Cirrhotic patients with moderate or severe hepatic impairment (Child-Pugh Class B or C) or patients who have previously experienced a decompensation event are dosed initially once weekly and not more than twice weekly.
Table 2: Dosage Regimen by Disease Stage Staging / Classification Non-Cirrhotic or Compensated Child-Pugh Class A Child-Pugh Class B or C or Patients with a Prior Decompensation Eventa Starting OCALIVA Dosage for first 3 months 5 mg once daily 5 mg once weekly OCALIVA Dosage Titration after first 3 months, for patients who have not achieved an adequate reduction in ALP and/or total bilirubin and who are tolerating OCALIVAb 10 mg once daily [see Clinical Pharmacology (12.2), Clinical Studies (14)] 5 mg twice weekly (at least 3 days apart)
Titrate to 10 mg twice weekly (at least 3 days apart) based on response and tolerability [see Use in Specific Populations (8.6)]Maximum OCALIVA Dosage 10 mg once daily 10 mg twice weekly (at least 3 days apart) aGastroesophageal variceal bleeding, new or worsening jaundice, spontaneous bacterial peritonitis, etc.
bPrior to dosage adjustment, re-calculate the Child-Pugh classification [see Dosage and Administration (2.1)].2.3 Monitoring to Assess Safety, Treatment Interruption or Discontinuation
Routinely monitor patients during OCALIVA treatment for progression of PBC disease with laboratory and clinical assessments to determine whether dosage adjustment is needed. Reduce the dosing frequency for patients who progress from Child-Pugh Class A to Child-Pugh Class B or C (see Table 2 above). Close monitoring is recommended for patients at an increased risk of hepatic decompensation, including those with laboratory evidence of worsening liver function (i.e., total bilirubin, INR, albumin) and/or progression to cirrhosis [see Warnings and Precautions (5.1)].
Interrupt treatment with OCALIVA in patients with laboratory or clinical evidence of worsening liver function indicating risk of decompensation, and monitor the patient's liver function.
If the patient's condition returns to baseline, weigh the potential risks and benefits of restarting OCALIVA treatment. If OCALIVA is re-initiated, use the recommended starting dosage with adjustment for Child-Pugh classification [see Dosage and Administration (2.2)].
Consider discontinuing OCALIVA in patients who have experienced clinically significant liver-related adverse reactions.
2.4 Management of Patients with Intolerable Pruritus on OCALIVA
For patients with intolerable pruritus on OCALIVA, consider one or more of the following management strategies:
For Non-Cirrhotic or Compensated Cirrhotic Child-Pugh Class A Patients:
- Add an antihistamine or bile acid binding resin [see Dosage and Administration (2.5), Clinical Studies (14)].
- Reduce the dosage of OCALIVA to:
- 5 mg every other day, for patients intolerant to 5 mg once daily.
- 5 mg once daily, for patients intolerant to 10 mg once daily.
- Temporarily interrupt OCALIVA dosing for up to 2 weeks. Restart at a reduced dosage.
For patients whose dosage is reduced or interrupted, titrate the dosage based on biochemical response, tolerability and adjust according to Child-Pugh classification [see Dosage and Administration (2.2)].
For Child-Pugh Class B or C or Patients with a Prior Decompensation Event:
- Add an antihistamine or bile acid binding resin [see Dosage and Administration (2.5), Clinical Studies (14)].
- Temporarily interrupt OCALIVA dosing for up to 2 weeks. Restart at a reduced dosage if applicable. Titrate the dosage based on biochemical response, tolerability and adjust according to Child-Pugh classification [see Dosage and Administration (2.2)].
Treatment Discontinuation
Consider discontinuing OCALIVA treatment in patients who continue to experience persistent, intolerable pruritus despite management strategies [see Warnings and Precautions (5.3)].
2.5 Administration Instructions
- Take OCALIVA with or without food.
- For patients taking a bile acid binding resin, take OCALIVA at least 4 hours before or 4 hours after taking the bile acid binding resin, or at as great an interval as possible [see Drug Interactions (7.1), Clinical Studies (14)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Decompensation and Failure in Incorrectly Dosed PBC Patients with Child-Pugh Class B or C or Decompensated Cirrhosis
In postmarketing reports, hepatic decompensation and failure, in some cases fatal, have been reported in PBC patients with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than the recommended starting dosage of 5 mg once weekly. Reported cases typically occurred within 2 to 5 weeks after starting OCALIVA and were characterized by an acute increase in total bilirubin and/or ALP concentrations in association with clinical signs and symptoms of hepatic decompensation (e.g., ascites, jaundice, gastrointestinal bleeding, worsening of hepatic encephalopathy). A few cases reported improvement after OCALIVA discontinuation; however, some cases reported ongoing symptoms. Because postmarketing cases often contain limited clinical information, there was insufficient information to rule out confounding factors (e.g., concomitant medications) or the role of the patient's underlying advanced disease in the events [see Adverse Reactions (6.2)].
Patients who died due to liver-related complications generally had decompensated cirrhosis prior to treatment and were started on OCALIVA 5 mg once daily, which is 7-fold greater than the once weekly starting regimen in this population [see Dosage and Administration (2.2)].
Patient Management
Routinely monitor patients for progression of PBC disease, including liver-related complications, with laboratory and clinical assessments. Dosage adjustment, interruption or discontinuation may be required [see Dosage and Administration (2.1, 2.2, 2.3)].
Close monitoring is recommended for patients at an increased risk of hepatic decompensation. Severe intercurrent illnesses that may worsen renal function or cause dehydration (e.g., gastroenteritis), may exacerbate the risk of hepatic decompensation.
Interrupt treatment with OCALIVA in patients with laboratory or clinical evidence of worsening liver function indicating risk of decompensation, and monitor the patient's liver function. Consider discontinuing OCALIVA in patients who have experienced clinically significant liver-related adverse reactions [see Dosage and Administration (2.3)].
Discontinue OCALIVA in patients who develop complete biliary obstruction [see Contraindications (4)].
5.2 Liver-Related Adverse Reactions
In two 3-month, placebo-controlled clinical trials in patients with primarily early stage PBC disease, a dose-response relationship was observed for the occurrence of liver-related adverse reactions including jaundice, worsening ascites and primary biliary cholangitis flare with dosages of OCALIVA of 10 mg once daily to 50 mg once daily (up to 5-times the highest recommended dosage), as early as one month after starting treatment with OCALIVA [see Overdosage (10)].
In a pooled analysis of three placebo-controlled trials in patients with primarily early stage PBC disease, the exposure-adjusted incidence rates for all serious and otherwise clinically significant liver-related adverse reactions, and isolated elevations in liver biochemical tests, per 100 patient exposure years (PEY) were: 5.2 in the OCALIVA 10 mg group (highest recommended dosage), 19.8 in the OCALIVA 25 mg group (2.5 times the highest recommended dosage) and 54.5 in the OCALIVA 50 mg group (5 times the highest recommended dosage) compared to 2.4 in the placebo group.
Monitor patients during treatment with OCALIVA for elevations in liver biochemical tests and for the development of liver-related adverse reactions. Close monitoring is recommended for patients at an increased risk of hepatic decompensation. Interrupt treatment with OCALIVA in patients with laboratory or clinical evidence of worsening liver function indicating risk of decompensation, and monitor the patient's liver function [see Dosage and Administration (2.2, 2.3), Warnings and Precautions (5.1)].
5.3 Severe Pruritus
Severe pruritus was reported in 23% of patients in the OCALIVA 10 mg arm, 19% of patients in the OCALIVA titration arm, and 7% of patients in the placebo arm in Trial 1, a 12-month double-blind randomized controlled trial of 216 patients [see Adverse Reactions (6.1)]. Severe pruritus was defined as intense or widespread itching, interfering with activities of daily living, or causing severe sleep disturbance, or intolerable discomfort, and typically requiring medical interventions. In the subgroup of patients in the OCALIVA titration arm who increased their dosage from 5 mg once daily to 10 mg once daily after 6 months of treatment (n=33), the incidence of severe pruritus was 0% from Months 0 to 6 and 15% from Months 6 to 12. The median time to onset of severe pruritus was 11, 158, and 75 days for patients in the OCALIVA 10 mg, OCALIVA titration, and placebo arms, respectively.
Consider clinical evaluation of patients with new onset or worsening severe pruritus. Management strategies include the addition of bile acid resins or antihistamines, OCALIVA dosage reduction, and/or temporary interruption of OCALIVA dosing [see Dosage and Administration (2.4)].
5.4 Reduction in HDL-C
Patients with PBC generally exhibit hyperlipidemia characterized by a significant elevation in total cholesterol primarily due to increased levels of high density lipoprotein-cholesterol (HDL-C). In Trial 1, dose-dependent reductions from baseline in mean HDL-C levels were observed at 2 weeks in OCALIVA-treated patients, 20% and 9% in the 10 mg and titration arms, respectively, compared to 2% in the placebo arm. At month 12, the reduction from baseline in mean HDL-C level was 19% in the OCALIVA 10 mg arm, 12% in the OCALIVA titration arm, and 2% in the placebo arm. Nine patients in the OCALIVA 10 mg arm, 6 patients in the OCALIVA titration arm, versus 3 patients in the placebo arm had reductions in HDL-C to less than 40 mg/dL.
Monitor patients for changes in serum lipid levels during treatment. For patients who do not respond to OCALIVA after 1 year at the highest recommended dosage that can be tolerated (maximum of 10 mg once daily), and who experience a reduction in HDL-C, weigh the potential risks against the benefits of continuing treatment.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hepatic Decompensation and Failure in Incorrectly Dosed PBC Patients with Child-Pugh Class B or C or Decompensated Cirrhosis [see Warnings and Precautions (5.1)]
- Liver-Related Adverse Reactions [see Warnings and Precautions (5.2)]
- Severe Pruritus [see Warnings and Precautions (5.3)]
- Reduction in HDL-C [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 432 patients with PBC were studied in three double-blind placebo-controlled trials. Of these patients, 290 were treated with OCALIVA for at least 6 months, 232 were treated for at least 12 months, and 70 were treated for at least 2 years. There were 131 patients who received OCALIVA 10 mg once daily and 70 who received OCALIVA 5 mg once daily.
In Trial 1, 216 patients were randomized (1:1:1) to receive either:
- OCALIVA 10 mg once daily for the entire 12 months of the trial (n=73);
- OCALIVA titration (5 mg once daily for the initial 6 months, with the option to increase to 10 mg once daily for the last 6 months, in patients who were tolerating OCALIVA, but had ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction) (n=70); or
- placebo (n=73).
During the trial, OCALIVA or placebo was administered in combination with UDCA in 93% of patients and as monotherapy in 7% of patients who were unable to tolerate UDCA. The overall discontinuation rate was 12% in the OCALIVA 10 mg arm, 10% in the OCALIVA titration arm, and 4% in the placebo arm.
The recommended starting dosage of OCALIVA is 5 mg orally once daily for 3 months with titration to 10 mg once daily based upon tolerability and response [see Dosage and Administration (2.2)]. Initiation of therapy with OCALIVA 10 mg once daily is not recommended due to an increased risk of pruritus.
The most common adverse reactions in Trial 1 occurring in at least 5% of patients in either OCALIVA treatment arm and at an incidence at least 1% higher than the placebo treatment arm are shown in Table 3.
Table 3: Most Common Adverse Reactions in Adult Patients with PBC in Trial 1 by Treatment Arm with or without UDCA* Placebo
N = 73
%Adverse Reaction† OCALIVA
10 mg
N = 73
%OCALIVA
Titration‡
N = 70
%- * In the trial there were 16 patients (7%) who were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA titration arm, and 5 patients (7%) in the placebo arm.
- † Occurring in greater than or equal to 5% of patients in either OCALIVA treatment arm and at an incidence greater than or equal to1% higher than in the placebo treatment arm.
- ‡ Patients randomized to OCALIVA titration received OCALIVA 5 mg once daily for the initial 6 month period. At Month 6, patients who were tolerating OCALIVA, but had an ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction were eligible for titration from 5 mg once daily to 10 mg once daily for the final 6 months of the trial.
- § Includes skin eruptions, prurigo, pruritus, pruritus generalized, eye pruritus, ear pruritus, anal pruritus, vulvovaginal pruritus, and rash pruritic.
- ¶ Includes fatigue, tiredness and asthenia.
- # Includes abdominal pain upper, abdominal pain, abdominal discomfort, abdominal pain lower, abdominal tenderness, and gastrointestinal pain.
- Þ Includes urticaria, rash, rash macular, rash papular, rash maculo-papular, heat rash, urticaria cholinergic.
- ß Includes dizziness, syncope, presyncope.
- à Includes thyroxine free decreased, blood thyroid stimulating hormone increased, hypothyroidism.
Pruritus§ 70 56 38 Fatigue¶ 25 19 15 Abdominal pain and discomfort# 10 19 14 RashÞ 10 7 8 Arthralgia 10 6 4 Oropharyngeal pain 8 7 1 Dizzinessß 7 7 5 Constipation 7 7 5 Peripheral Edema 7 3 3 Palpitations 7 3 1 Pyrexia 7 0 1 Thyroid function abnormalityà 4 6 3 Eczema 3 6 0 Liver-Related Adverse Reactions
In Trial 1, the following serious or otherwise clinically significant liver-related adverse reactions were reported at the recommended dosage of OCALIVA: one patient in the OCALIVA 10 mg treatment arm experienced ascites; one patient in the OCALIVA titration treatment arm experienced two episodes of ascites and four episodes of hepatic encephalopathy; one patient in the placebo treatment arm experienced variceal bleeding.
Pruritus
Approximately 60% of patients had a history of pruritus upon enrollment in Trial 1. Treatment-emergent pruritus, including all the terms described in Table 3, generally started within the first month following the initiation of treatment with OCALIVA.
The incidence of pruritus was higher in patients who started on OCALIVA 10 mg once daily relative to the OCALIVA titration arm, 70% and 56%, respectively. Discontinuation rates due to pruritus were also higher in patients who started on OCALIVA 10 mg once daily relative to the OCALIVA titration arm, 10% and 1%, respectively.
The number of patients with pruritus who required an intervention (e.g., dosage adjustment, treatment interruption, or initiation of bile acid binding resin or antihistamine) was 30 of 51 patients (59%) in the OCALIVA 10 mg arm, 24 of 39 patients (62%) in the OCALIVA titration arm, and 14 of 28 patients (50%) in the placebo arm.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of OCALIVA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure, particularly in PBC patients who have progressive liver disease.
Hepatobiliary Disorders: liver failure, new onset cirrhosis, increased direct and total bilirubin, new or worsening of jaundice, ascites, worsening hepatic encephalopathy [see Warnings and Precautions (5.1)].
-
7 DRUG INTERACTIONS
7.1 Bile Acid Binding Resins
Bile acid binding resins such as cholestyramine, colestipol, or colesevelam adsorb and reduce bile acid absorption and may reduce the absorption, systemic exposure, and efficacy of OCALIVA. If taking a bile acid binding resin, take OCALIVA at least 4 hours before or 4 hours after taking the bile acid binding resin, or at as great an interval as possible [see Dosage and Administration (2.5)].
7.2 Warfarin
The International Normalized Ratio (INR) decreased following coadministration of warfarin and OCALIVA [see Clinical Pharmacology (12.3)]. Monitor INR and adjust the dosage of warfarin, as needed, to maintain the target INR range when co-administering OCALIVA and warfarin.
7.3 CYP1A2 Substrates with Narrow Therapeutic Index
Obeticholic acid may increase the exposure to concomitant drugs that are CYP1A2 substrates [see Clinical Pharmacology (12.3)]. Therapeutic monitoring of CYP1A2 substrates with a narrow therapeutic index (e.g. theophylline and tizanidine) is recommended when co-administered with OCALIVA.
7.4 Inhibitors of Bile Salt Efflux Pump
Avoid concomitant use of inhibitors of the bile salt efflux pump (BSEP) such as cyclosporine [see Clinical Pharmacology (12.3)]. Concomitant medications that inhibit canalicular membrane bile acid transporters such as the BSEP may exacerbate accumulation of conjugated bile salts including taurine conjugate of obeticholic acid in the liver and result in clinical symptoms. If concomitant use is deemed necessary, monitor serum transaminases and bilirubin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited available human data on the use of obeticholic acid during pregnancy are not sufficient to inform a drug-associated risk. In animal reproduction studies, no developmental abnormalities or fetal harm was observed when pregnant rats or rabbits were administered obeticholic acid during the period of organogenesis at exposures approximately 13 times and 6 times human exposures, respectively, at the maximum recommended human dose (MRHD) of 10 mg [see Data below].
The estimated background risks of major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, obeticholic acid was administered orally during the period of organogenesis at doses of 5, 25, and 75 mg/kg/day. At 25 mg/kg/day (a dose that produced systemic exposures approximately 13 times those in humans at the MRHD of 10 mg), there was no maternal or developmental toxicity. At 75 mg/kg/day (approximately 40 times the human exposure at the MRHD), decreased fetal body weights and increased numbers of early or late resorptions and nonviable fetuses were observed. In maternal animals, mortality, fetal loss, decreased body weight and food consumption as well as decreased body weight gain were observed at 75 mg/kg/day. Thus, the developmental toxicity observed at this dose may be secondary to maternal toxicity. In rabbits, obeticholic acid was administered orally during the period of organogenesis at doses of 3, 9, and 20 mg/kg/day. Obeticholic acid administered at doses up to 20 mg/kg/day (approximately 6 times the human exposure at the MRHD) was not teratogenic and did not produce any evidence of fetal harm.
In a pre- and post-natal development study, administration of obeticholic acid in rats during organogenesis through lactation at doses of 5, 25, and 40 mg/kg/day did not produce effects on pregnancy, parturition or postnatal development at any dose (the 40 mg/kg/day dose is approximately 21 times the human exposure at the MRHD).
Obeticholic acid exposure margins were calculated using systemic exposure (AUC) values of obeticholic acid plus obeticholic acid's active metabolite conjugates (tauro-obeticholic acid and glycol-obeticholic acid) in animals (at the indicated doses) and in humans at the MRHD of 10 mg.
8.2 Lactation
Risk Summary
There is no information on the presence of obeticholic acid in human milk, the effects on the breast-fed infant or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OCALIVA and any potential adverse effects on the breastfed infant from OCALIVA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of OCALIVA in pediatric patients have not been established.
8.5 Geriatric Use
Of the 201 patients in clinical trials of OCALIVA who received the recommended dosage (5 mg or 10 mg once daily), 41 (20%) were 65 years of age and older, while 9 (4%) were 75 years of age and older. No overall differences in safety or effectiveness were observed between these subjects and subjects less than 65 years of age, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
Hepatic decompensation and failure, in some cases fatal, have been reported postmarketing in PBC patients with decompensated cirrhosis or Child-Pugh B or C hepatic impairment when OCALIVA was dosed more frequently than recommended. In PBC clinical trials, a dose-response relationship was observed for the occurrence of liver-related adverse reactions with OCALIVA [see Warnings and Precautions (5.2)].
Plasma exposure to obeticholic acid and its active conjugates, increases significantly in patients with moderate to severe hepatic impairment (Child-Pugh Classes B and C) [see Clinical Pharmacology (12.3)].
Prior to the initiation of OCALIVA determine the patient's Child-Pugh classification to determine the starting dosage. Re-evaluate the dosing regimen periodically during treatment.
Dosage adjustment is required in patients with Child-Pugh Class B and C. Routinely monitor patients for progression of PBC disease with laboratory and clinical assessments. Dosage adjustment, interruption or discontinuation may be required [see Dosage and Administration (2.1, 2.2, 2.3)].
-
10 OVERDOSAGE
In PBC patients who received OCALIVA 25 mg once daily (2.5 times the highest recommended dosage) or 50 mg once daily (5 times the highest recommended dosage), a dose-dependent increase in the incidence of liver-related adverse reactions, including elevations in liver biochemical tests, ascites, jaundice, portal hypertension, and primary biliary cholangitis flare, was reported. Serious liver-related adverse reactions have been reported postmarketing in PBC patients with decompensated cirrhosis or Child-Pugh Class B or C hepatic impairment when OCALIVA was dosed more frequently than the recommended starting dosage of 5 mg once weekly [see Warnings and Precautions (5.1, 5.2)].
In the case of overdosage, patients should be carefully observed and supportive care administered, as appropriate.
-
11 DESCRIPTION
OCALIVA is a farnesoid X receptor (FXR) agonist. Chemically, obeticholic acid is 3α,7α-dihydroxy-6α-ethyl-5β-cholan-24-oic acid. It is a white to off-white powder. It is soluble in methanol, acetone and ethyl acetate. Its solubility in water is pH dependent. It is slightly soluble at low pH and very soluble at high pH. Its chemical formula is C26H44O4, the molecular weight is 420.63 g/mol and the chemical structure is:
OCALIVA tablets are supplied in 5 mg and 10 mg strengths for oral administration. Each tablet contains obeticholic acid as the active ingredient and the following inactive ingredients: microcrystalline cellulose, sodium starch glycolate, and magnesium stearate. The film coating is Opadry II (Yellow) containing polyvinyl alcohol-part hydrolyzed, titanium dioxide, macrogol (polyethylene glycol 3350), talc, and iron oxide yellow.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Obeticholic acid is an agonist for FXR, a nuclear receptor expressed in the liver and intestine. FXR is a key regulator of bile acid, inflammatory, fibrotic, and metabolic pathways. FXR activation decreases the intracellular hepatocyte concentrations of bile acids by suppressing de novo synthesis from cholesterol as well as by increased transport of bile acids out of the hepatocytes. These mechanisms limit the overall size of the circulating bile acid pool while promoting choleresis, thus reducing hepatic exposure to bile acids.
12.2 Pharmacodynamics
Dose Titration
In Trial 1, ALP reduction was observed to plateau at approximately 3 months in most patients treated with OCALIVA 5 mg once daily. Increasing the dosage of OCALIVA to 10 mg once daily based on tolerability and response provided additional reduction in ALP in the majority of patients [see Dosage and Administration (2.2), Clinical Studies (14)].
Pharmacodynamic Markers
In Trial 1, administration of OCALIVA 10 mg once daily was associated with a 173% increase in concentrations of FGF-19, an FXR-inducible enterokine involved in bile acid homeostasis, from baseline to Month 12. Concentrations of cholic acid and chenodeoxycholic acid were reduced 2.7 micromolar and 1.4 micromolar, respectively, from baseline to Month 12. The clinical relevance of these findings is unknown.
12.3 Pharmacokinetics
Absorption
Following multiple oral doses of OCALIVA 10 mg once daily, peak plasma concentrations (Cmax) of obeticholic acid occurred at a median time (Tmax) of approximately 1.5 hours. The median Tmax for both the glyco- and tauro-conjugates of obeticholic acid was 10 hours. Coadministration with food did not alter the extent of absorption of obeticholic acid [see Dosage and Administration (2.5)].
Following multiple-dose administration of OCALIVA 5, 10, and 25 mg once daily (2.5 times the highest recommend dosage) for 14 days, systemic exposures of obeticholic acid increased dose proportionally. Exposures to glyco-obeticholic acid and tauro-obeticholic acid, and total obeticholic acid (the sum of obeticholic acid and its two active conjugates) increased more than proportionally with dose. The steady-state systemic exposure (AUC0-24h ) achieved on Day14 of total obeticholic acid was 4.2-, 6.6-, and 7.8- fold the systemic exposure (AUC0-24h ) achieved on Day 1 after 5, 10, and 25 mg once daily dosing, respectively.
Distribution
Human plasma protein binding of obeticholic acid and its conjugates is greater than 99%. The volume of distribution of obeticholic acid is 618 L. The volumes of distribution of glyco- and tauro-obeticholic acid have not been determined.
Elimination
Metabolism
Obeticholic acid is conjugated with glycine or taurine in the liver and secreted into bile. These glycine and taurine conjugates of obeticholic acid are absorbed in the small intestine leading to enterohepatic recirculation. The conjugates can be deconjugated in the ileum and colon by intestinal microbiota, leading to the conversion to obeticholic acid that can be reabsorbed or excreted in feces, the principal route of elimination.
After daily administration of obeticholic acid, there was accumulation of the glycine and taurine conjugates of obeticholic acid, which have in vitro pharmacological activities similar to the parent drug, obeticholic acid. The metabolite-to-parent ratios of the glycine and taurine conjugates of obeticholic acid were 13.8 and 12.3 respectively, after daily administration. An additional third obeticholic acid metabolite, 3-glucuronide, was formed but was considered to have minimal pharmacologic activity.
Specific Populations
Body weight, Age, Sex Race/Ethnicity: Based on population pharmacokinetic analysis, body weight was a significant predictor of obeticholic acid pharmacokinetics with lower obeticholic acid exposure expected with higher body weight. The body weight effect is not expected to cause a meaningful impact on efficacy. The pharmacokinetics of obeticholic acid would not be expected to be altered based on age, sex, or race/ethnicity.
Renal Impairment: Obeticholic acid has not been studied in patients with moderate and severe renal impairment (estimated glomerular filtration rate [eGFR] less than 60 mL/min/1.73 m2). In the population pharmacokinetic analysis, an eGFR greater than 50 mL/min/1.73 m2 did not have a meaningful effect on the pharmacokinetics of obeticholic acid and its conjugated metabolites.
Hepatic Impairment: Obeticholic acid is metabolized in the liver. In subjects with mild, moderate and severe hepatic impairment (Child-Pugh Class A, B, and C, respectively), the mean AUC of total obeticholic acid increased 1.1-, 4- and 17-fold, respectively, compared to subjects with normal hepatic function following single-dose administration of 10 mg OCALIVA [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Effect of Obeticholic Acid on Other Drugs
Based on in vitro studies, obeticholic acid can inhibit CYP3A4. However, an in vivo study indicated no inhibition of CYP3A4 by obeticholic acid at the recommended dose of OCALIVA. Obeticholic acid is not expected to inhibit CYPs 2B6, 2C8, 2C9, 2C19, and 2D6, or induce CYPs 1A2, 2B6, 2C8, 2C9, 2C19, and 3A4 at the recommended dose of OCALIVA. Down-regulation of mRNA was observed in a concentration-dependent fashion for CYP1A2 and CYP3A4 by obeticholic acid and its glycine and taurine conjugates.
In vitro studies suggest that there is potential for obeticholic acid and its glycine and taurine conjugates to inhibit OATP1B1 and OATP1B3 (the clinical significance of which is unknown), but not P-gp, BCRP, OAT1, OAT3, OCT2, and MATE transporters, at the recommended dose of OCALIVA.
In vitro studies showed that obeticholic acid and its glycine and taurine conjugates inhibit BSEP in a dose dependent manner. However, an in vivo drug interaction due to inhibition of BSEP in patients using the recommended dosage regimen appears unlikely.
Induction of BSEP can occur by FXR activation by obeticholic acid and its conjugates, which are FXR agonists.
Warfarin: Concomitant administration of 25 mg warfarin as a single dose with OCALIVA 10 mg once daily resulted in 13% increase in systemic exposure to S-warfarin and 11% decrease in maximum INR [see Drug Interactions (7.2)].
Caffeine (CYP1A2 substrate): Concomitant administration of 200 mg caffeine as a single dose with OCALIVA 10 mg once daily resulted in a 42% increase in plasma AUC and 6% increase in Cmax of caffeine [see Drug Interactions (7.3)].
Omeprazole (CYP2C19 substrate): Concomitant administration of 20 mg omeprazole as a single dose with OCALIVA 10 mg once daily resulted in a 32% increase in AUC and a 33% increase in Cmax of omeprazole. The clinical significance is unknown.
No clinically relevant interactions were seen when the following drugs were administered as single doses concomitantly with OCALIVA 10 mg once daily:
Midazolam 2 mg (CYP3A4 substrate): 2% increase in AUC and Cmax of midazolam.
Dextromethorphan 30 mg (CYP2D6 substrate): 11% decrease in AUC and 12% decrease in Cmax of dextromethorphan.
Digoxin 0.25 mg (P-gp substrate): 1% increase in AUC and 3% decrease in Cmax of digoxin.
Rosuvastatin 20 mg (BCRP, OATP1B1, OATP1B3 substrate): 22% increase in AUC and a 27% increase in Cmax of rosuvastatin.
Effect of Other Drugs on Obeticholic Acid
In vitro data suggest that obeticholic acid is not metabolized to any significant extent by CYP450 enzymes.
Proton Pump Inhibitors (omeprazole): Concomitant administration of 20 mg omeprazole once daily with OCALIVA 10 mg once daily resulted in a less than 1.2-fold increase in obeticholic acid exposure. This increase is not expected to be clinically relevant. Concomitant administration of 40 mg omeprazole once daily with OCALIVA 10 mg once daily was not studied.
BSEP inhibitors: In vitro data indicate that tauro-obeticholic acid is a substrate of BSEP [see Drug Interactions (7.4)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic potential of obeticholic acid was assessed in carcinogenicity studies of up to 2 years in duration in mice and rats. In mice, there were no drug-related neoplastic findings at doses up to 25 mg/kg/day obeticholic acid, a dose that produced systemic exposures approximately 12 times those in humans at the MRHD of 10 mg. In rats, obeticholic acid was administered at doses of 2, 7, and 20 mg/kg/day. At 20 mg/kg/day (approximately 12 times the human exposure at the MRHD), obeticholic acid caused an increase in the incidence of benign granulosa cell tumors in the ovaries and benign granular cell tumors in the cervix and vagina of female rats. There were no drug-related neoplastic findings in male rats.
Obeticholic acid was not genotoxic in the Ames test, a human peripheral blood lymphocyte chromosomal aberration test, and a mouse micronucleus test. The glycine conjugate of obeticholic acid was also not genotoxic in an Ames test and human peripheral blood lymphocyte chromosome aberration test. The taurine conjugate of obeticholic acid was not genotoxic in an Ames test, and was negative in a human peripheral blood lymphocyte chromosomal aberration test in the presence of metabolic activation; the findings of the chromosomal aberration assay in the absence of metabolic activation were inconclusive.
Obeticholic acid, administered at oral doses of 5, 25 and 50 mg/kg/day to male rats for 28 days before mating and throughout the mating period, and to female rats from 14 days before mating through mating and until gestation day 7, did not alter male or female fertility or early embryonic development at any dose (the 50 mg/kg/day dose is approximately 13 times the human exposure at the MRHD).
-
14 CLINICAL STUDIES
The recommended starting dosage of OCALIVA is 5 mg orally once daily for 3 months with titration to 10 mg once daily based upon tolerability and response [see Dosage and Administration (2.2)]. Initiation of therapy with a starting dosage OCALIVA 10 mg once daily is not recommended due to an increased risk of pruritus [see Adverse Reactions (6.1)].
Trial 1 was a randomized, double-blind, placebo-controlled, 12-month trial which evaluated the safety and efficacy of OCALIVA in 216 patients with PBC who were taking UDCA for at least 12 months (on a stable dosage for at least 3 months), or who were unable to tolerate UDCA and did not receive UDCA for at least 3 months. Patients were included in the trial if the ALP was 1.67-times upper limit of normal (ULN) or greater and/or if total bilirubin was greater than 1-times ULN but less than 2-times ULN. Patients were excluded from the trial if they had other liver disease, presence of clinically significant hepatic decompensation events (i.e., portal hypertension and its complications, cirrhosis with complications, or hepato-renal syndrome), severe pruritus, or Model for End Stage Liver Disease (MELD) score of 15 or greater.
Patients were randomized (1:1:1) to receive either OCALIVA 10 mg once daily for the entire 12 months of the trial, (n=73); OCALIVA titration (5 mg once daily for the initial 6 months, with the option to increase to 10 mg once daily for the last 6 months if the patient was tolerating OCALIVA but had ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction) (n=70); or placebo (n=73). OCALIVA or placebo was administered in combination with UDCA in 93% of patients during the trial and as monotherapy in 7% of patients who were unable to tolerate UDCA.
The primary endpoint was a responder analysis at Month 12, where response was defined as a composite of three criteria: ALP less than 1.67-times the ULN, total bilirubin less than or equal to ULN, and an ALP decrease of at least 15%. The ULN for ALP was defined as 118 U/L for females and 124 U/L for males. The ULN for total bilirubin was defined as 1.1 mg/dL for females and 1.5 mg/dL for males.
The study population was 91% female and 94% white. The mean age was 56 years (range 29 to 86 years). The mean baseline ALP concentration was 323.2 U/L, corresponding to 2.74-times ULN. Approximately 29% of the patients had ALP concentration levels greater than 3-times the ULN. The mean baseline total bilirubin concentration was 0.65 mg/dL, and was less than or equal to the ULN in 92% of the enrolled patients. Distribution of patients by Rotterdam disease stage criteria at baseline is shown in Table 4. Cirrhosis was present at baseline in 4 patients (5%) in the OCALIVA 10 mg arm, 7 patients (10%) in the OCALIVA titration arm, and 9 patients (12%) in the placebo arm.
Table 4: Rotterdam Disease Stage Criteria at Baseline in Trial 1 by Treatment Arm with or without UDCA * Disease Stage† OCALIVA 10 mg
(N=73)OCALIVA Titration
(N=70)Placebo
(N=73)Percentages are based on non-missing values for each time point. - * In the trial there were 16 patients (7%) who were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA titration arm, and 5 patients (7%) in the placebo arm.
- † Early: normal total bilirubin and normal albumin (values less than or equal to ULN and greater than or equal to the lower limit of normal (LLN), respectively), Moderately advanced: abnormal total bilirubin or abnormal albumin, Advanced: abnormal total bilirubin and abnormal albumin. Total bilirubin ULN: 1.1 mg/dL (females) and 1.5 mg/dL (males). Albumin LLN: 35 g/L (females and males).
Early, n (%) 66 (90) 64 (91) 65 (89) Moderately Advanced, n (%) 7 (10) 6 (9) 8 (11) Advanced, n (%) 0 (0) 0 (0) 0 (0) Table 5 shows the percentage of patients by treatment arm in Trial 1 who achieved a response to the primary composite endpoint at Month 12, and to the individual components of the primary endpoint (i.e., ALP less than 1.67-times the ULN, total bilirubin less than or equal to ULN, and an ALP decrease of at least 15%). A total of 33 patients in the OCALIVA titration arm, who did not achieve a response at 6 months and tolerated OCALIVA, had their dosage increased from 5 mg once daily to 10 mg once daily. Of these 33 patients, 13 (39%) achieved the primary composite endpoint at 12 months.
Table 5: Percentage of Adult Patients with PBC Achieving the Primary Composite Endpoint at Month 12 in Trial 1 by Treatment Arm with or without UDCA* OCALIVA
10 mg
(N = 73)OCALIVA
Titration†
(N = 70)Placebo
(N = 73)- * In the trial there were 16 patients (7%) who were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA titration arm, and 5 patients (7%) in the placebo arm.
- † Patients randomized to OCALIVA titration received OCALIVA 5 mg for the initial 6 month period. At Month 6, patients who were tolerating OCALIVA, but had an ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction were eligible for titration from 5 mg once daily to 10 mg once daily for the final 6 months of the trial.
- ‡ Percentage of patients achieving a response, defined as an ALP less than 1.67-times the ULN, total bilirubin less than or equal to the ULN, and an ALP decrease of at least 15%. Missing values were considered a non-response. The exact test was used to calculate the 95% CIs.
- § p<0.0001 for OCALIVA titration and OCALIVA 10 mg arms versus placebo. P-values are obtained using the Cochran–Mantel–Haenszel General Association test stratified by intolerance to UDCA and pretreatment ALP greater than 3-times ULN and/or AST greater than 2-times ULN and/or total bilirubin greater than ULN.
- ¶ Response rates were calculated based on the observed case analysis (i.e., [n=observed responder]/[N=ITT population]); percentage of patients with Month 12 values are 86%, 91% and 96% for the OCALIVA 10 mg, OCALIVA titration and placebo arms, respectively.
- # The mean baseline total bilirubin value was 0.65 mg/dL, and was less than or equal to the ULN in 92% of the enrolled patients.
Primary Composite Endpoint‡ Responder rate, (%)§
[95% CI]48
[36, 60]46
[34, 58]10
[4, 19]Components of Primary Endpoint¶ ALP less than 1.67-times ULN, n (%) 40 (55) 33 (47) 12 (16) Decrease in ALP of at least 15%, n (%) 57 (78) 54 (77) 21 (29) Total bilirubin less than or equal to ULN#, n (%) 60 (82) 62 (89) 57 (78) Mean Reduction in ALP
Figure 1 shows the mean reductions in ALP in OCALIVA-treated patients compared to placebo. Reductions were observed as early as Week 2, plateaued by Month 3 and were maintained through Month 12 for patients who were maintained on the same dosage throughout 12 months. Although Trial 1 studied titration at 6 months, these data are supportive of titration of OCALIVA after 3 months [see Dosage and Administration (2.2)]. For patients in the OCALIVA titration arm whose OCALIVA dosage was increased from 5 mg once daily to 10 mg once daily, additional reductions in ALP were observed at Month 12 in the majority of patients [see Clinical Pharmacology (12.2)].
Figure 1: Mean ALP over 12 Months in Trial 1 by Treatment Arm with or without UDCAa
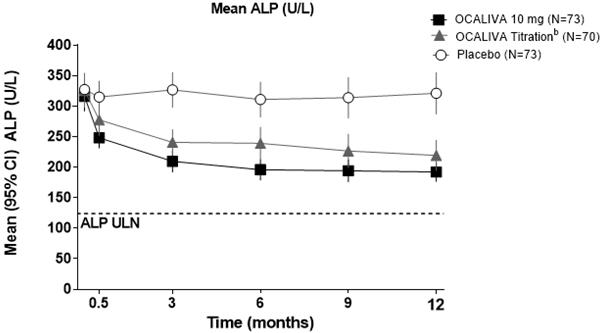
a In the trial there were 16 patients (7%) who were intolerant and did not receive concomitant UDCA: 6 patients (8%) in the OCALIVA 10 mg arm, 5 patients (7%) in the OCALIVA titration arm, and 5 patients (7%) in the placebo arm. b Patients randomized to OCALIVA titration received OCALIVA 5 mg once daily for the initial 6 month period. At Month 6, patients who were tolerating OCALIVA, but had an ALP 1.67-times ULN or greater, and/or total bilirubin greater than ULN, or less than 15% ALP reduction were eligible for titration from 5 mg once daily to 10 mg once daily for the final 6 months of the trial. Mean Reduction in GGT
The mean (95% CI) reduction in gamma-glutamyl transferase (GGT) was 178 (137, 219) U/L in the OCALIVA 10 mg arm, 138 (102, 174) U/L in the OCALIVA titration arm, and 8 (-32, 48) U/L in the placebo arm.
OCALIVA Monotherapy
Fifty-one PBC patients with baseline ALP 1.67-times ULN or greater and/or total bilirubin greater than ULN were evaluated for a biochemical response to OCALIVA as monotherapy (24 patients received OCALIVA 10 mg once daily and 27 patients received placebo) in a pooled analysis of data from Trial 1 and from a randomized, double-blind, placebo-controlled, 3-month trial. At Month 3, 9 (38%) OCALIVA-treated patients achieved a response to the composite endpoint, compared to 1 (4%) placebo-treated patient. The mean (95% CI) reduction in ALP in OCALIVA-treated patients was 246 (165, 327) U/L compared to an increase of 17 (-7, 42) U/L in the placebo-treated patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
OCALIVA tablets are packaged in a 40 mL high density polyethylene bottle closed with a 33 mm polypropylene child resistant cap containing an induction seal. Each bottle contains 30 tablets.
5 mg Tablets
OCALIVA tablets are available as off-white to yellow, round tablets debossed with INT on one side and 5 on the other side. Each tablet contains 5 mg of obeticholic acid.
- NDC: 69516-005-30 5 mg tablets in a bottle (30 count)
10 mg Tablets
OCALIVA tablets are available as off-white to yellow, triangular tablets debossed with INT on one side and 10 on the other side. Each tablet contains 10 mg of obeticholic acid.
- NDC: 69516-010-30 10 mg tablets in a bottle (30 count)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Hepatic Decompensation and Failure in Incorrectly Dosed PBC Patients with Child-Pugh Class B or C or Decompensated Cirrhosis
- Instruct patients and caregivers to immediately contact their healthcare provider if they experience:
- Symptoms of disease progression or worsening liver function, such as ascites, jaundice, gastrointestinal bleeding or worsening of hepatic encephalopathy.
- Symptoms of complete biliary obstruction [see Warnings and Precautions (5.1), Contraindications (4)].
- Instruct patients and caregivers to immediately contact their healthcare provider if they experience severe or persistent non-specific signs and symptoms of impaired health: nausea, vomiting, abdominal pain, diarrhea, weight loss, fever and chills, worsening or new fatigue, weakness, loss of appetite or dehydration.
- Inform patients that they will need to undergo laboratory testing periodically while on OCALIVA treatment to assess liver function.
Severe Pruritus
- Advise patients to contact their healthcare provider if they experience new onset or worsening severe pruritus [see Warnings and Precautions (5.3)].
Reduction in HDL-C
- Advise patients that they may need to undergo laboratory testing to check for changes in lipid levels while on treatment with OCALIVA [see Warnings and Precautions (5.4)].
Administration
Advise patients to take:
- OCALIVA with or without food.
- OCALIVA at least 4 hours before or 4 hours after taking a bile acid binding resin, or at as great an interval as possible [see Drug Interactions (7.1)].
- Instruct patients and caregivers to immediately contact their healthcare provider if they experience:
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: January 2018 MEDICATION GUIDE
OCALIVA® (o-CAL-eh-vah)
(obeticholic acid) tabletsWhat is the most important information I should know about OCALIVA?
If you have primary biliary cholangitis (PBC) with advanced cirrhosis, you may need a lower dose of OCALIVA.- Before you start OCALIVA, and during your treatment with OCALIVA, your healthcare provider will do tests to check your liver. These tests will help your healthcare provider decide how much OCALIVA you should take and how often you should take it.
- If you have worsening liver problems, your dose of OCALIVA may be changed, stopped for a period of time, or stopped completely by your healthcare provider.
Worsening of liver problems, liver failure, in some cases leading to death, have happened in people with PBC with advanced liver cirrhosis when OCALIVA was taken more often than recommended.- Tell your healthcare provider right away if you have any of the following symptoms of worsening liver problems during treatment with OCALIVA:
- swelling of your stomach-area from a build-up of fluid
- yellowing of your skin or the whites of your eyes
- black, tarry, or bloody stools
- coughing up or vomiting blood, or your vomit looks like "coffee grounds"
- mental changes such as confusion, sleepier than usual or harder to wake up, slurred speech, mood swings, or changes in personality
- Tell your healthcare provider right away if you have any of the following symptoms during treatment with OCALIVA and they are severe or do not go away:
- stomach-area pain
- nausea, vomiting, or diarrhea
- loss of appetite or weight loss
- new or worsening fatigue
- weakness
- fever and chills
- light-headedness
- less frequent urination
What is OCALIVA?
OCALIVA is a prescription medicine used to treat primary biliary cholangitis (PBC) in combination with ursodeoxycholic acid (UDCA) in adults who have not responded well enough to UDCA, or alone in adults who cannot tolerate UDCA.
It is not known if taking OCALIVA will improve your chance of survival or improve your symptoms of PBC.
It is not known if OCALIVA is safe and effective in children.Do not take OCALIVA if you: - have a complete blockage of the bile ducts in your liver or gall bladder.
Before taking OCALIVA, tell your healthcare provider about all of your medical conditions, including if you: - are pregnant or plan to become pregnant. It is not known if OCALIVA will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if OCALIVA passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you take OCALIVA.
How should I take OCALIVA? - Take OCALIVA exactly as your healthcare provider tells you to.
- Do not take more OCALIVA than your healthcare provider tells you to.
- Take OCALIVA with or without food.
- If you take a bile acid binding resin, take OCALIVA at least 4 hours before or 4 hours after you take your bile acid binding resin. If this is not possible, space the time between taking OCALIVA and your bile acid binding resin as far apart as possible.
- If you take too much OCALIVA, call your healthcare provider or get emergency medical help right away.
What are the possible side effects of OCALIVA?
OCALIVA can cause serious side effects, including:- See "What is the most important information I should know about OCALIVA?"
- Severe itching (pruritus). Itching is a common side effect of OCALIVA and can sometimes become severe (intense itching or itching over much of your body). Severe itching can cause discomfort, problems sleeping, and problems doing daily activities and usually needs to be treated. Tell your healthcare provider if you get severe itching or if your itching gets worse.
- Lower HDL-C ("good" cholesterol). OCALIVA can lower high levels of HDL-C. Your healthcare provider will check your cholesterol levels during your treatment with OCALIVA.
- tiredness
- stomach pain and discomfort
- rash
- joint pain
- mouth and throat pain
- dizziness
- constipation
- swelling in your hands, ankles, or feet
- fast or irregular heartbeat
- fever
- changes in how your thyroid gland works
- dryness, irritation, redness, crusting or drainage of the skin (eczema)
These are not all the possible side effects of OCALIVA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store OCALIVA? - Store OCALIVA at room temperature between 68°F to 77°F (20°C to 25°C).
General information about the safe and effective use of OCALIVA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use OCALIVA for a condition for which it was not prescribed. Do not give OCALIVA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about OCALIVA that is written for health professionals.What are the ingredients in OCALIVA?
Active ingredient: obeticholic acid
Inactive ingredients: microcrystalline cellulose, sodium starch glycolate, magnesium stearate
Film coating: Opadry II (Yellow) containing polyvinyl alcohol-part hydrolyzed, titanium dioxide, macrogol (polyethylene glycol 3350), talc, iron oxide yellow
Distributed by: Intercept Pharmaceuticals, Inc., New York, NY 10001
OCALIVA is a registered trademark of Intercept Pharmaceuticals, Inc.
For more information, go to www.OCALIVA.com or call 1-844-782-4278. - PRINCIPAL DISPLAY PANEL - 5 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 10 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
OCALIVA
obeticholic acid tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69516-005 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Obeticholic Acid (UNII: 0462Z4S4OZ) (Obeticholic Acid - UNII:0462Z4S4OZ) Obeticholic Acid 5 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) Sodium Starch Glycolate Type A Potato (UNII: 5856J3G2A2) Magnesium stearate (UNII: 70097M6I30) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium dioxide (UNII: 15FIX9V2JP) Polyethylene glycol 3350 (UNII: G2M7P15E5P) Talc (UNII: 7SEV7J4R1U) Ferric Oxide Yellow (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape ROUND Size 8mm Flavor Imprint Code INT;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69516-005-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 05/27/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA207999 05/27/2016 OCALIVA
obeticholic acid tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69516-010 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Obeticholic Acid (UNII: 0462Z4S4OZ) (Obeticholic Acid - UNII:0462Z4S4OZ) Obeticholic Acid 10 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) Sodium Starch Glycolate Type A Potato (UNII: 5856J3G2A2) Magnesium stearate (UNII: 70097M6I30) Polyvinyl Alcohol, Unspecified (UNII: 532B59J990) Titanium dioxide (UNII: 15FIX9V2JP) Polyethylene glycol 3350 (UNII: G2M7P15E5P) Talc (UNII: 7SEV7J4R1U) Ferric Oxide Yellow (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape TRIANGLE Size 8mm Flavor Imprint Code INT;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69516-010-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 05/27/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA207999 05/27/2016 Labeler - Intercept Pharmaceuticals Inc (966658416)


Trademark Results [Ocaliva]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 OCALIVA 86710924 5033038 Live/Registered |
INTERCEPT PHARMACEUTICALS, INC. 2015-07-31 |
 OCALIVA 86710865 5014396 Live/Registered |
INTERCEPT PHARMACEUTICALS, INC. 2015-07-31 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.