APHEXDA- motixafortide injection, powder, lyophilized, for solution
Aphexda by
Drug Labeling and Warnings
Aphexda by is a Prescription medication manufactured, distributed, or labeled by BioLineRx USA Inc, PolyPeptide Laboratories San Diego LLC, Element Materials Technology Pharma US LLC, PPD Development, L.P., ALS Group USA, Corp., BioConnection B.V., Eurofins PROXY Laboratories B.V, BioChem Labor für biologische und chemische Analytik GmbH, Almac Pharma Services Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use APHEXDA safely and effectively. See full prescribing information for APHEXDA.
APHEXDA TM (motixafortide) for injection, for subcutaneous use.
Initial U.S. Approval: 2023INDICATIONS AND USAGE
APHEXDA, a hematopoietic stem cell mobilizer, is indicated in combination with filgrastim (G-CSF) to mobilize hematopoietic stem cells to the peripheral blood for collection and subsequent autologous transplantation in patients with multiple myeloma. ( 1)
DOSAGE AND ADMINISTRATION
- Initiate APHEXDA treatment after filgrastim has been administered daily for 4 days. ( 2.1)
- Recommended dosage is 1.25 mg/kg actual body weight by subcutaneous injection 10 to 14 hours prior to initiation of apheresis. ( 2.2)
- A second dose of APHEXDA can be administered 10 to 14 hours prior to a third apheresis. ( 2.2)
- See Full Prescribing Information for instructions on preparation and administration.
DOSAGE FORMS AND STRENGTHS
For Injection: 62 mg as a lyophilized powder in a single-dose vial for reconstitution. ( 3)
CONTRAINDICATIONS
History of serious hypersensitivity to APHEXDA. ( 4)
WARNINGS AND PRECAUTIONS
- Anaphylactic Shock and Hypersensitivity Reactions: Premedicate all patients with a combination of an H1-antihistamine, an H2 blocker, and a leukotriene inhibitor prior to each APHEXDA dose. Administer APHEXDA in a setting where personnel and therapies are available for immediate treatment. Observe for signs and symptoms and manage promptly. ( 2.1, 2.3, 5.1)
- Injection Site Reactions: The addition of analgesic premedication (e.g., acetaminophen) is recommended. ( 5.2)
- Tumor Cell Mobilization in Patients with Leukemia: APHEXDA may mobilize leukemic cells and should not be used in leukemia patients. ( 5.3)
- Leukocytosis: Increased circulating leukocytes have been observed. Monitor white blood cell counts during APHEXDA use. ( 5.4)
- Potential for Tumor Cell Mobilization: Tumor cells may be released from marrow during HSC mobilization with APHEXDA and filgrastim. Effect of reinfusion of tumor cells is unknown. ( 5.5)
- Embryo-fetal Toxicity: Can cause fetal harm. Advise women of reproductive potential of the potential risk to a fetus and to use effective contraception. ( 5.6, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence >20%) are injection site reactions, injection site pain, injection site erythema, injection site pruritus, pruritus, flushing, and back pain. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact BioLineRx at 1-800-574-9978 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. ( 8.2)
See 17 for PATIENT COUNSELING INFORMATION
Revised: 9/2023
Revised: 9/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSING AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Recommended Dosage
2.3 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactic Shock and Hypersensitivity Reactions
5.2 Injection Site Reactions
5.3 Tumor Cell Mobilization in Patients with Leukemia
5.4 Leukocytosis
5.5 Potential for Tumor Cell Mobilization
5.6 Embryo-fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSING AND ADMINISTRATION
2.1 Important Dosing Information
- Premedicate all patients before each dose of APHEXDA to reduce the risk of hypersensitivity and injection site reactions:
- Administer diphenhydramine (12.5 mg intravenously or 25 mg to 50 mg orally, or another H1-antihistamine), an H2 blocker (e.g., famotidine), and a leukotriene inhibitor (e.g., montelukast) approximately 30 to 60 minutes before injection of APHEXDA.
- The addition of an analgesic medication (e.g., acetaminophen) to the premedication regimen is recommended.
- Administer filgrastim 10 mcg/kg subcutaneously once daily for 4 days prior to the first dose of APHEXDA and on each day prior to each apheresis.
2.2 Recommended Dosage
The recommended dosage of APHEXDA is 1.25 mg/kg administered via slow (approximately 2 minutes) subcutaneous injection 10 to 14 hours prior to the initiation of the first apheresis. Dosing is based on actual body weight.
A second dose of APHEXDA can be administered 10 to 14 hours before a third apheresis, if necessary.
2.3 Preparation and Administration
Use aseptic technique to prepare and administer APHEXDA:
- More than one vial may be needed for a full dose. Calculate the dose, the total volume of reconstituted APHEXDA solution required, and number of APHEXDA vials required based on patient’s actual body weight.
- Remove the APHEXDA vial(s) from the refrigerator and allow to reach room temperature at 20°C to 25°C (68°F to 77°F) for at least 30 minutes.
- Reconstitute each vial with 2 mL of 0.45% Sodium Chloride Injection, USP at room temperature (20°C to 25°C (68°F to 77°F)) resulting in a concentration of 36.5 mg/mL of APHEXDA and permitting withdrawal of up to 1.7 mL (62 mg). Alternatively, you can reconstitute each vial with 1 mL of Sterile Water for Injection, USP and 1 mL of 0.9% Sodium Chloride Injection, USP.
- Gently swirl and invert for up to 3 minutes slowly until completely dissolved.
- Inspect the reconstituted solution for discoloration and particulate matter. The reconstituted solution should appear clear and colorless. Do not use if the reconstituted solution is discolored, is cloudy, or contains visible particulates.
- If needed, store the reconstituted APHEXDA solution refrigerated at 2°C to 8°C (36°F to 46°F) or at room temperature at 20°C to 25°C (68°F to 77°F) for up to 24 hours protected from light.
- Withdraw the required injection volume of APHEXDA from the vial(s) into an appropriately sized syringe.
- Each injection volume should not exceed 2 mL. Divide doses requiring greater than 2 mL into multiple syringes to allow different injection sites.
Administration
- Administer injection into the abdomen, the back or side of the upper arms, or the thighs. Rotate injection sites. An injection should never be given into scar tissue or areas that are reddened, inflamed, or swollen. If injecting into the abdomen, avoid a 5 cm diameter circle around the navel. If more than one injection is needed for a single dose of APHEXDA, the injection sites should be at least 2 cm apart from previous injection locations. Discard unused portion of the drug.
- Monitor patients for one hour after administration [see Warnings and Precautions ( 5.1) ].
- Premedicate all patients before each dose of APHEXDA to reduce the risk of hypersensitivity and injection site reactions:
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
APHEXDA is contraindicated in patients with a history of serious hypersensitivity reactions to motixafortide [see Warnings and Precautions (5.1)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactic Shock and Hypersensitivity Reactions
Anaphylactic shock occurred in 0.7% of APHEXDA-treated patients in clinical studies (n=407). The time to anaphylactic shock was between 5 minutes and 30 minutes after drug administration. Hypersensitivity reactions occurred in 7.6% of APHEXDA-treated patients in the GENESIS study. In addition, pruritus, flushing, urticaria, rash, erythema, vomiting, nausea and chills have been reported.
Premedicate all patients prior to each dose of APHEXDA 30-60 minutes prior to administration with a triple-drug premedication regimen that includes an H1-antihistamine, an H2 blocker, and a leukotriene inhibitor [see Dosage and Administration (2.1)]. Patients receiving concomitant negative chronotropic drugs (e.g., beta blockers) may be more at risk for hypotension in case of hypersensitivity reaction. When appropriate, beta blockers should be replaced with non-chronotropic drugs.
Administer APHEXDA only in a setting where personnel and therapies are immediately available for the treatment of anaphylaxis and other systemic reactions. Monitor patients for signs or symptoms of hypersensitivity reactions for one hour following administration of APHEXDA and manage reactions promptly.
5.2 Injection Site Reactions
Injection site reactions were reported in 73% of patients receiving APHEXDA in the GENESIS trial. Symptoms of injection site reactions included pain, erythema, pruritus, bruising, discomfort, induration, mass, nodule, rash, swelling, and urticaria. Among 92 patients treated with APHEXDA, the highest severity of the reactions was severe in 9%.
Premedicate with an analgesic medication (e.g., acetaminophen) prior to each APHEXDA dose. Use analgesic medication and local treatments postdose, as needed.
5.3 Tumor Cell Mobilization in Patients with Leukemia
For the purpose of hematopoietic stem cell (HSC) mobilization, APHEXDA may cause mobilization of leukemic cells and subsequent contamination of the apheresis product. Therefore, APHEXDA is not intended for HSC mobilization and harvest in patients with leukemia.
5.4 Leukocytosis
Administration of APHEXDA in conjunction with filgrastim increases circulating leukocytes as well as HSC populations. Monitor white blood cell counts during APHEXDA use.
5.5 Potential for Tumor Cell Mobilization
When APHEXDA is used in combination with filgrastim for HSC mobilization, tumor cells may be released from the marrow and subsequently collected in the leukapheresis product. The effect of potential reinfusion of tumor cells has not been well-studied.
5.6 Embryo-fetal Toxicity
Based on its mechanism of action, APHEXDA can cause fetal harm when administered to a pregnant woman. Animal models link dysfunction in CXCR4/SDF-1 signaling to adverse outcomes in mammalian embryo-fetal development and suggest risks to normal placental development.
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with APHEXDA and for 8 days after the final dose [ see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in other sections of the labeling:
- Anaphylactic Shock and Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Injection Site Reactions [see Warnings and Precautions (5.2)]
- Potential for Tumor Cell Mobilization in Patients in Leukemia [see Warnings and Precautions (5.3)]
- Leukocytosis [see Warnings and Precautions (5.4)]
- Potential for Tumor Cell Mobilization [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of APHEXDA was evaluated in the GENESIS study based on data from 92 patients with multiple myeloma who received at least one dose of APHEXDA 1.25 mg/kg subcutaneously and filgrastim and 42 patients who received placebo and filgrastim for mobilization of hematopoietic stem cells for collection and apheresis [see Clinical Studies (14)]. The premedication regimen changed during the conduct of the trial as evidence of hypersensitivity reactions was noted. Of the 92 patients who received at least one dose of APHEXDA, 14 patients received the triple-drug premedication regimen and 78 did not receive the triple-drug premedication regimen (either received no premedication or another premedication regimen).
Serious adverse reactions occurred in 5.4% of patients receiving APHEXDA in combination with filgrastim. Serious adverse reactions included vomiting, injection site reaction, hypersensitivity reaction, injection site cellulitis, hypokalemia and hypoxia.
One patient did not receive the 5th dose of filgrastim due to an elevated white blood cell count following administration of APHEXDA. The most common adverse reactions occurring in GENESIS (>20% and at least 2% higher than the filgrastim + placebo arm) were injection site reactions (pain, erythema and pruritus), pruritus, flushing, and back pain. Table 1 summarizes the common adverse reactions in GENESIS.
Table 1: Common Adverse Reactions in Patients with Multiple Myeloma During Hematopoietic Stem Cell Mobilization and Apheresis in GENESIS a APHEXDA and Filgrastim
n=92
%
Placebo and Filgrastim
n=42
%
Injection site reactions c All Grades a Grade >3 b All Grades a Grade >3 b Injection site reactions 73 8 5 0 Injection site pain 53 7 5 0 Injection site erythema 27 0 0 0 Injection site pruritus 24 0 0 0 Pruritus 38 11 0 0 Flushing d 33 7 0 0 Rash e 16 0 5 0 Urticaria 14 1.1 0 0 Erythema 12 0 0 0 Back pain f 21 0 17 0 Paresthesia g 19 0 17 0 Hypokalemia 15 4.3 12 0 Nausea 14 0 12 0 (a) Adverse reactions that occurred in ≥10% in APHEXDA-treated patients and ≥2% more than placebo-treated patients.
(b) All reactions were grade 3.
(c) Injection site reactions includes: injection site bruising, injection site discomfort, injection site erythema, injection site induration, injection site mass, injection site nodule, injection site pain, injection site pruritus, injection site rash, injection site swelling, injection site urticaria, injection site cellulitis and injection related reaction.
(d) Flushing includes hot flush.
(e) Rash includes: rash erythematous, rash maculo-papular, rash papular and rash pruritic.
(f) Back pain includes spinal pain and sacral pain.
(g) Paresthesia includes: paresthesia oral, hypoesthesia, hypoesthesia oral and burning sensation.
Clinically relevant adverse reactions that occurred in the APHEXDA arm only in <10% of patients include dermatitis exfoliative generalized, ear swelling, pyrexia, chills, dizziness, tremor and hypertension.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, APHEXDA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data with APHEXDA use in pregnant women informing the risk of embryo-fetal toxicity. Animal models link dysfunction in CXCR4/SDF-1 signaling to adverse outcomes in mammalian embryo-fetal development and suggest risk to normal placental development (see Data). No animal studies have been conducted to evaluate the effect of motixafortide on reproduction and fetal development. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriages for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Animal reproduction studies have not been conducted with motixafortide to evaluate its effect on reproduction and embryo-fetal development. Disruption of CXCR4/SDF-1 signaling in mice and other models caused increased embryo-fetal lethality, and impairment of vascularization, cardiac anomalies, reduced hematopoiesis, impaired bone marrow myelopoiesis, disorganized neural layers in cerebellum, and reduced neural innervation of limbs. CXCR4/SDF-1 levels have been shown to play a key role in stimulating trophoblast proliferation and differentiation necessary for appropriate placental growth and function in humans.
8.2 Lactation
Risk Summary
There are no data on the presence of motixafortide in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential serious adverse reactions in the breastfed child, advise females that breastfeeding is not recommended during the APHEXDA treatment and for 8 days after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating APHEXDA.
Contraception
Females
APHEXDA can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with APHEXDA and for 8 days after the final dose [see Use in Specific Populations (8.1)].
-
11 DESCRIPTION
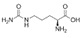
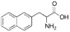
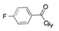
APHEXDA for injection contains motixafortide, which is a hematopoietic stem cell mobilizer. The chemical name of the synthetic motixafortide acetate, the active pharmaceutical ingredient is N-(4-Fluoro-benzoyl)-L-arginyl-L-arginyl-[L-3-naphthyl)alanyl]-L-cysteinyl-L-tyrosyl 5-L-citrullinyl-L-lysyl-D-lysyl-L-prolyl-L-tyrosyl 10-L‑arginyl-L-citrullinyl-L-cysteinyl-L-arginineamide, cyclic (4-13)-disulfide, acetate salt. Motixafortide acetate is a synthetic, cyclic peptide composed of a sequence of amino acids with the following structure:

Cit: Citrulline
Nal: Naphthylalanine
Fba: 4-Florobenzoic acid
Counter Ion: Acetate
Molecular weight: 2159.6 g/mol (as the free base)
APHEXDA is supplied as a sterile white to off-white, preservative-free lyophilized powder in single-dose vials. Each vial contains 73 mg of motixafortide (provided as motixafortide acetate) for reconstitution with 2 mL 0.45% saline, delivering 62 mg motixfortide per 1.7 mL. Motixafortide is present as a salt with 4 to 8.5 molar equivalents of acetate. The inactive ingredients include 26 mg of mannitol and hydrochloric acid for pH adjustment.
Reconstitution with 2 mL of 0.45% sodium chloride for injection (or 1 mL water for injection + 1 mL 0.9% sodium chloride for injection) yields a clear solution of 36.5 mg/mL motixafortide, pH 5.8 to 7.5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Motixafortide is an inhibitor of the C-X-C Motif Chemokine Receptor 4 (CXCR4) and blocks the binding of its cognate ligand, stromal-derived factor-1α (SDF-1α)/C-X-C Motif Chemokine Ligand 12 (CXCL12).
SDF-1α and CXCR4 play a role in the trafficking and homing on human hematopoietic stem cells to the marrow compartment. Once in the marrow, stem cell CXCR4 can help anchor these cells to the marrow matrix, either directly via SDF-1α or through the induction of other adhesion molecules.
Treatment with motixafortide resulted in leukocytosis, and elevations in circulating hematopoietic stem and progenitor cells into the peripheral circulation in mice, rats, dogs, and humans.
Stem cells mobilized by motixafortide were capable of engraftment with long-term repopulating capacity in a rodent transplantation model.
12.2 Pharmacodynamics
In vitro studies showed that the concentration for half-maximal inhibition (IC 50) of motixafortide towards CXCR4 ranges from 0.42 – 4.5 nM, the binding affinity (K D) is 7.9 pM and the dissociation rate (Kd) from the CXCR4 receptor is 3.38e-5 s-1 with receptor occupancy maintained over >72 hours.
The pharmacodynamics of motixafortide was evaluated in clinical studies conducted in healthy volunteers, healthy donors paired with recipients with advanced hematological malignancies, and multiple myeloma patients. Across all clinical studies, mobilization of CD34+ to peripheral blood was observed.
In healthy volunteers after administration of motixafortide as monotherapy, CD34+ cell counts increased with time, reaching maximal levels at 16 hours postdose. Thereafter, CD34+ cell counts declined slightly but remained elevated well above baseline at the last observation timepoint 24 hours postdose.
Data on the fold increase in peripheral blood CD34+ cell count (cells/mcL) by apheresis day were evaluated in the placebo-controlled part of the GENESIS study. The fold increase in CD34+ cell count over the 24-hour period, from the day prior to the first apheresis (before filgrastim administration) to the next morning before filgrastim administration (12 hours after APHEXDA administration) is summarized in Table 2. During this 24-hour period, a single dose of motixafortide or placebo was administered 10 to 14 hours prior to apheresis.
Table 2: Fold Increase in Peripheral Blood CD34+ Cell Count following Pre-treatment with Filgrastim and Administration of APHEXDA in GENESIS
APHEXDA and Filgrastim Placebo and Filgrastim Median Mean (SD) Median Mean (SD) 6.7 7.7 (7.5) 1.5 1.6 (0.7) Cardiac Electrophysiology
A thorough QT study in 38 healthy volunteers showed APHEXDA prolongs the QTcI interval in a concentration-dependent fashion. At 1.7-fold the C max of the maximum approved recommended dose (1.25 mg/kg), the mean placebo-corrected QTcI change from baseline was 11 (90% CI: 8 to 14) msec.
12.3 Pharmacokinetics
The pharmacokinetics of motixafortide were evaluated using both noncompartmental and population PK approaches in various populations, including healthy volunteers, patients with acute myeloid leukemia, and patients with multiple myeloma. Subjects received single or multiple doses ranging between 0.24 mg/kg to 2 mg/kg of APHEXDA administered by subcutaneous injections using a weight-based (mg/kg) dosing strategy as monotherapy or in combination with filgrastim with or without chemotherapy.
The PK characteristics of motixafortide following subcutaneous administration were similar across all study populations.
A population pharmacokinetic analysis was conducted using 2240 plasma motixafortide concentrations from 223 individuals, including 81 healthy subjects, 37 patients with acute myeloid leukemia, and 105 patients with multiple myeloma.
Motixafortide pharmacokinetics following subcutaneous administration were adequately described by a 3-compartment model with first-order absorption, dose-dependent relative bioavailability, and linear elimination kinetics. Minimal to no drug accumulation was observed following once daily dose administration and, as such, the single dose PK profile is considered representative of a steady state profile.
Absorption
After subcutaneous injection as either a single or repeated dose at 0.24 to 2 mg/kg, motixafortide appeared in plasma with time to maximum concentration occurring at approximately 0.25 to 1.17 hours.
Distribution
Motixafortide is highly bound to human plasma proteins (>99%). The estimated volume of distribution of the central compartment in a typical subject is 27 L.
Metabolism
Motixafortide undergoes non-specific degradation into small peptides and individual amino acids and their derivatives by catabolic pathways. Catabolism can occur in both blood and liver microsomes. In in vitro studies using human and animal biomaterials, no prominent unique human metabolites were identified.
Elimination
Motixafortide has an effective half-life in human plasma of approximately 2 hours. The elimination kinetics were similar in healthy subjects and patients with multiple myeloma.
Apparent total clearance of motixafortide for a typical subject is 46.5 L/h.
No mass balance studies were conducted in humans.
In studies conducted in rats and dogs, the total amount of 14C-labeled motixafortide-related material excreted in urine was approximately 80% and 82%, respectively, of the dose administered and no parent drug was detected in urine. In both species, no metabolite exceeded 30% of total clearance.
Specific Populations
Renal Impairment
Based on the population PK analysis, in patients with mildly to moderately decreased renal function, the pharmacokinetic profile of motixafortide was not significantly affected. While the effect of severe renal impairment on the clearance of motixafortide has not been evaluated, a significant effect is not anticipated.
Hepatic Impairment
Motixafortide is catabolized in both the liver and blood, and animal data suggest that the main excretion pathway of its metabolites is via the kidneys and biliary excretion is minimal. Based on the population PK analysis, mildly impaired liver function did not significantly affect the pharmacokinetic profile of motixafortide. The effect of moderate to severe hepatic impairment has not been evaluated, but the risk for adverse reactions due to increased exposure is low because motixafortide is administered as a single dose.
Race/Ethnicity
In the population PK analysis, ethnicity as a covariate was not found to be a statistically significant predictor of motixafortide PK.
Gender
In the population analysis, minimal effect of gender was seen on the overall pharmacokinetic profile of motixafortide. Despite higher relative bioavailability in females compared to males, the mg/kg dosing strategy and generally lower body weights in females result in only 10% higher exposures (AUC 0-24h and C max) in females versus males for a weight-based dose and therefore is not considered clinically meaningful.
Age
In the population PK analysis, age as a covariate was not found to be a statistically significant predictor of motixafortide PK.
Drug Interaction StudiesIn vitro studies showed a lack of significant (>25%) cytochrome P450 (CYP) inhibition in human hepatocytes, a lack of CYP induction potential in human hepatocytes, and a low potential for transporter-mediated interactions. As such, motixafortide has a low potential for both metabolism- and transporter-mediated drug interactions.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies between studies, including those of motixafortide.
No pre-existing or treatment-emergent (up to 8 weeks post-administration) anti-drug antibodies were detected in 84 patients with multiple myeloma who received one or two administrations of motixafortide in combination with filgrastim for hematopoietic stem cell mobilization in the GENESIS study [ see Clinical Studies (14)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with motixafortide have not been conducted. Motixafortide was not genotoxic in an in vitro bacterial mutation assay (Ames test), in an in vitro chromosomal aberration test using V79 Chinese hamster cells, or in an in vivo bone marrow micronucleus test in mice after intravenous injected at doses up to 4 mg/kg (12 mg/m 2).
Fertility studies have not been conducted with motixafortide. No histopathological evidence of toxicity to male or female reproductive organs was observed in 28-day repeat-dose toxicity studies in rats and dogs.
-
14 CLINICAL STUDIES
The efficacy of APHEXDA in combination with filgrastim was evaluated in the GENESIS study (NCT 03246529). In this randomized, double-blind, placebo-controlled study, 122 patients with multiple myeloma were randomized in a 2:1 ratio to receive APHEXDA 1.25 mg/kg subcutaneously (N=80) or placebo (N=42). Prior to receiving APHEXDA or placebo, patients received daily morning doses of filgrastim 10-15 mcg/kg for 4 days. On the evening of Day 4, patients received APHEXDA or placebo. On Day 5, patients received a fifth morning dose of filgrastim within 1 hour prior to their first apheresis (12 hours ± 2 hours from the APHEXDA/placebo administration). The apheresis cell collection goal for the study was ≥ 6 × 10 6 CD34+ cells/kg. The assessment of CD34+ cells was performed by central and local laboratories. Central laboratory assessments were used for the efficacy results. Local laboratory results were used for clinical treatment decisions.
In the event that the cell collection goal was not achieved with the first apheresis on Day 5, patients received another morning dose of filgrastim on Day 6 within 1 hour prior to their second apheresis. In the event that the cell collection goal was still not achieved, patients received a second administration of APHEXDA or placebo on the evening of Day 6 and a seventh dose of filgrastim in the morning of Day 7 within 1 hour prior to a third apheresis. If the collection goal was not achieved, patients received an eighth dose of filgrastim in the morning of Day 8 within 1 hour prior to a fourth apheresis.
The median age of the study population was 63 years (range 34-75); 65% were males, 86% Caucasian, 8% African American, 2% Asian and 10% were of Hispanic or Latino ethnicity. Seventy percent of patients were previously treated with lenalidomide.
The efficacy of APHEXDA was based upon the proportion of patients who achieved a cell collection goal of ≥ 6 × 10 6 CD34+ cells/kg in up to 2 aphereses after administration of filgrastim and a single administration of APHEXDA or placebo.
Efficacy results showed that 67.5% of patients in the APHEXDA treatment arm versus 9.5% in the placebo arm achieved the cell collection goal of ≥ 6 × 10 6 CD34+ cells/kg in up to 2 aphereses after a single administration of APHEXDA or placebo, resulting in an adjusted difference between treatment arms of 56.8% (p < 0.0001) (Table 3).
Table 3: Proportion of Patients Who Achieved CD34+ Cell Collection Goal Following Single Administration of APHEXDA or Placebo (GENESIS)
Cell Collection Goal APHEXDA and Filgrastim
(N = 80)
Placebo and Filgrastim
(N = 42)
p-value* Proportion of patients with ≥ 6 × 10 6 CD34+ cells/kg in up to 2 aphereses – Central laboratory 67.5% 9.5% <0.0001 Proportion of patients with ≥ 6 × 10 6 CD34+ cells/kg in 1 apheresis – Central laboratory 63.8% 2.4% <0.0001 Proportion of patients with ≥ 2 × 10 6 CD34+ cells/kg in 1 apheresis – Central laboratory 87.5% 38.1% <0.0001 *The reported p values are two sided based on the Cochran-Mantel-Haenszel common proportion difference method stratifying for response status (CR or PR) at baseline and to baseline platelet count (<200 × 10 9/L or ≥ 200 × 10 9/L).
Multiple factors can influence time to engraftment and graft durability following stem cell transplantation. In the GENESIS study, time to neutrophil and platelet engraftment and graft durability following transplantation were similar across treatment groups.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
APHEXDA (motixafortide) for injection is supplied as a white to off-white lyophilized powder in a single-dose vial for reconstitution. Each vial delivers 62 mg motixafortide free base.
NDC: 82737-073-01 (Carton containing one vial).
Store at 2°C to 8°C (36°F to 46°F) in original carton to protect from light.
Discard prepared reconstituted solution after 24 hours storage under refrigeration 2°C to 8°C (36°F to 46°F) or at room temperature 20°C to 25°C (68°F to 77°F) protected from light.
-
17 PATIENT COUNSELING INFORMATION
- Advise patients of the risk of anaphylactic and hypersensitivity reactions (such as pruritus, flushing, urticaria, rash, vomiting, nausea and chills) during and after APHEXDA injection and to immediately report such signs and symptoms to healthcare professionals [see Warnings and Precautions (5.1)] .
- Advise patients that APHEXDA may cause injection site reactions, such as pain, redness and swelling [see Warnings and Precautions (5.2), Adverse Reactions (6.1)] .
- Advise females of reproductive potential to use effective contraceptive methods during APHEXDA treatment and for 8 days after the administration of APHEXDA [see Warnings and Precautions (5.2), Use in Specific Populations (8.1, 8.3)] .
- Advise females of reproductive potential of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant or if pregnancy is suspected during treatment with APHEXDA [see Warnings and Precautions (5.6), Use in Specific Populations (8.1, 8.3)].
- Advise women that breastfeeding is not recommended during treatment with APHEXDA and for 8 days following the last dose [see Use in Specific Populations (8.2)] .
Manufactured for:
BioLineRx Ltd TM
Modi’in, Israel
Distributed by:
BioLineRx USA Inc TM
77 Fourth Avenue
Waltham, MA 02451
USA
Revised: 9/2023
-
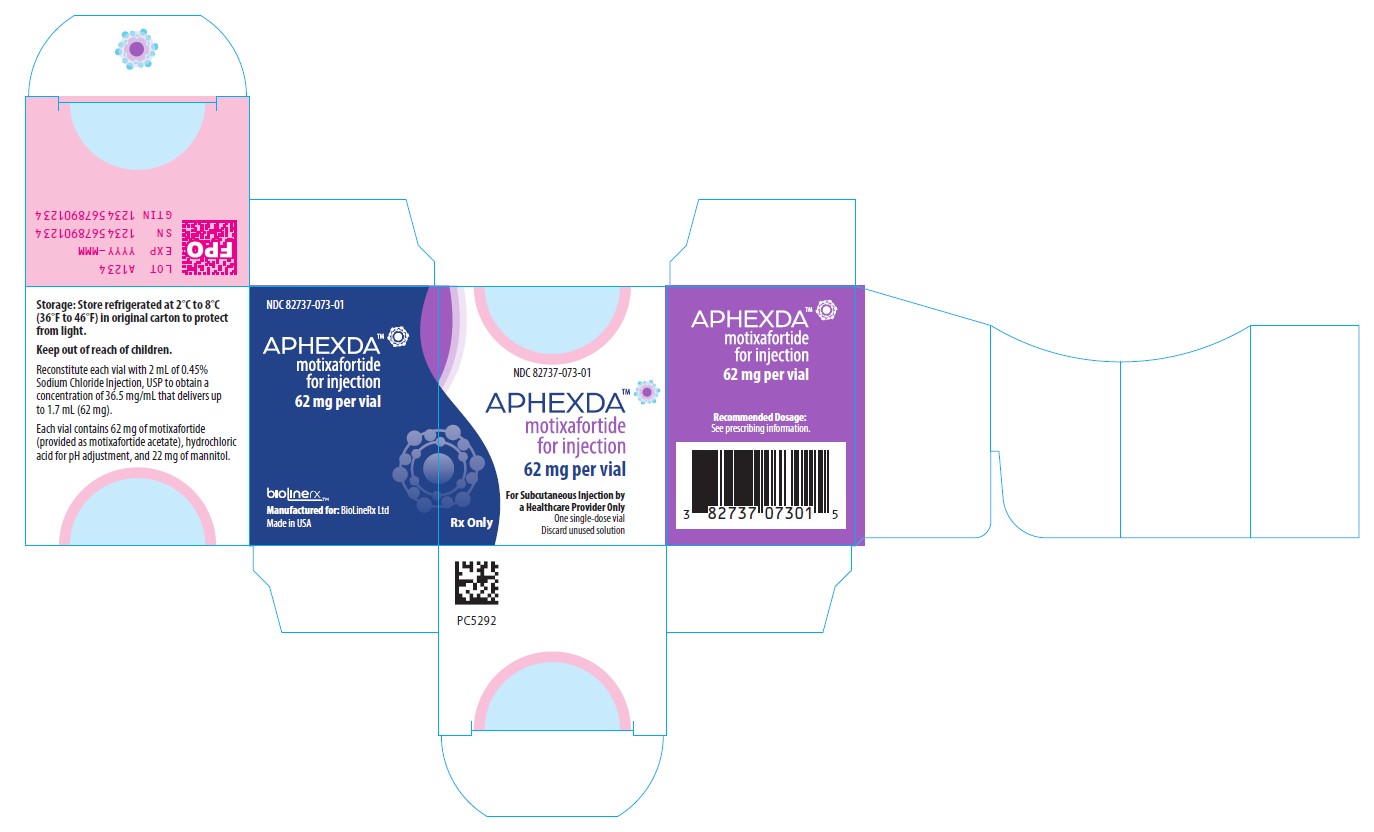
PRINCIPAL DISPLAY PANEL
NDC: 82737-073-01
APHEXDA TM
Motixafortide for injection
62 mg per vial
For Subcutaneous Injection by a Healthcare Provider Only
One single-dose vial
Discard unused solution
Recommended Dosage: See prescribing information.
Storage: Store refrigerated at 2°C to 8°C (36°F to 46°F) in original carton to protect from light.
Keep out of reach of children.
Reconstitute each vial with 2 mL of 0.45% Sodium Chloride Injection, USP to obtain a concentration of 36.5 mg/mL that delivers up to 1.7 mL (62 mg).
Each vial contains 62 mg of motixafortide (provided as motixafortide acetate), hydrochloric acid for pH adjustment, and 22 mg of mannitol.
Manufactured for: BioLineRx Ltd.
Made in USA
-
INGREDIENTS AND APPEARANCE
APHEXDA
motixafortide injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82737-073 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MOTIXAFORTIDE ACETATE (UNII: 3ZPX60DV8A) (MOTIXAFORTIDE - UNII:DA9G065962) MOTIXAFORTIDE 62 mg in 1.7 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82737-073-01 1 in 1 CARTON 09/08/2023 1 1.7 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA217159 09/08/2023 Labeler - BioLineRx USA Inc (118674652) Establishment Name Address ID/FEI Business Operations Element Materials Technology Pharma US LLC 081315325 analysis(82737-073) Establishment Name Address ID/FEI Business Operations ALS Group USA, Corp. 087409727 analysis(82737-073) Establishment Name Address ID/FEI Business Operations Almac Pharma Services Limited 233170864 label(82737-073) , pack(82737-073) , analysis(82737-073) Establishment Name Address ID/FEI Business Operations BioChem Labor für biologische und chemische Analytik GmbH 318354230 analysis(82737-073) Establishment Name Address ID/FEI Business Operations Eurofins PROXY Laboratories B.V 405855359 analysis(82737-073) Establishment Name Address ID/FEI Business Operations BioConnection B.V. 414325873 manufacture(82737-073) Establishment Name Address ID/FEI Business Operations PolyPeptide Laboratories San Diego LLC 621516145 api manufacture(82737-073) , analysis(82737-073) Establishment Name Address ID/FEI Business Operations PPD Development, L.P. 838082055 analysis(82737-073) Establishment Name Address ID/FEI Business Operations Polypeptide Laboratories Inc 962386124 analysis(82737-073) Establishment Name Address ID/FEI Business Operations PHARBIL Pharma GmbH 340705425 analysis(82737-073)
Trademark Results [Aphexda]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 APHEXDA 79358785 not registered Live/Pending |
BioLineRx Ltd. 2022-08-17 |
 APHEXDA 79358780 not registered Live/Pending |
BioLineRx Ltd. 2022-08-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.