RANOLAZINE tablet, extended release
Ranolazine by
Drug Labeling and Warnings
Ranolazine by is a Prescription medication manufactured, distributed, or labeled by Viona Pharmaceuticals Inc, Zydus Lifesciences Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RANOLAZINE EXTENDED-RELEASE TABLETS safely and effectively. See full prescribing information for RANOLAZINE EXTENDED-RELEASE TABLETS.
RANOLAZINE extended-release tablets, for oral use
Initial U.S. Approval: 2006INDICATIONS AND USAGE
Ranolazine extended-release tablet is an antianginal indicated for the treatment of chronic angina. (1)
DOSAGE AND ADMINISTRATION
500 mg twice daily and increase to 1000 mg twice daily, based on clinical symptoms (2.1)
DOSAGE FORMS AND STRENGTHS
Extended-release tablets: 500 mg, 1000 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- QT interval prolongation: Can occur with ranolazine. Little data available on high doses, long exposure, use with QT interval-prolonging drugs, potassium channel variants causing prolonged QT interval, in patients with a family history of (or congenital) long QT syndrome, or in patients with known acquired QT interval prolongation. (5.1)
- Renal failure: Monitor renal function after initiation and periodically in patients with moderate to severe renal impairment (CrCL<60 mL/min). If acute renal failure develops, discontinue ranolazine. (5.2)
ADVERSE REACTIONS
Most common adverse reactions (>4% and more common than with placebo) are dizziness, headache, constipation, nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Viona Pharmaceuticals Inc. at 1-888-304-5011 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Moderate CYP3A inhibitors (e.g., diltiazem, verapamil, erythromycin): Limit ranolazine to 500 mg twice daily. (7.1)
- P-gp inhibitors (e.g., cyclosporine): Ranolazine exposure increased. Titrate ranolazine based on clinical response. (7.1)
- CYP3A substrates: Limit simvastatin to 20 mg when used with ranolazine. Doses of other sensitive CYP3A substrates (e.g., lovastatin) and CYP3A substrates with narrow therapeutic range (e.g., cyclosporine, tacrolimus, sirolimus) may need to be reduced with ranolazine. (7.2)
- OCT2 substrates: Limit the dose of metformin to 1700 mg daily when used with ranolazine 1000 mg twice daily. Doses of other OCT2 substrates may require adjusted doses. (7.2)
- Drugs transported by P-gp (e.g., digoxin), or drugs metabolized by CYP2D6 (e.g., tricyclic antidepressants) may need reduced doses when used with ranolazine. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Dose Adjustments in Specific Populations
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 QT Interval Prolongation
5.2 Renal Failure
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Ranolazine
7.2 Effects of Ranolazine on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Patients with Hepatic Impairment
8.7 Use in Patients with Renal Impairment
8.8 Use in Patients with Heart Failure
8.9 Use in Patients with Diabetes Mellitus
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Chronic Stable Angina
14.2 Lack of Benefit in Acute Coronary Syndrome
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
Initiate ranolazine extended-release tablet dosing at 500 mg twice daily and increase to 1000 mg twice daily, as needed, based on clinical symptoms. Take ranolazine extended-release tablet with or without meals. Swallow ranolazine extended-release tablet whole; do not crush, break, or chew.
The maximum recommended daily dose of ranolazine extended-release tablet is 1000 mg twice daily.
If a dose of ranolazine extended-release tablet is missed, take the prescribed dose at the next scheduled time; do not double the next dose.
2.2 Dose Adjustments in Specific Populations
Dose adjustments may be needed when ranolazine extended-release tablet is taken in combination with certain other drugs [see Drug Interactions (7.1)]. Limit the maximum dose of ranolazine extended-release tablet to 500 mg twice daily in patients on moderate CYP3A inhibitors such as diltiazem, verapamil, and erythromycin. Use of ranolazine extended-release tablet with strong CYP3A inhibitors is contraindicated [see Contraindications (4), Drug Interactions (7.1)]. Use of P-gp inhibitors, such as cyclosporine, may increase exposure to ranolazine extended-release tablet. Titrate ranolazine extended-release tablet based on clinical response [see Drug Interactions (7.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Ranolazine is contraindicated in patients:
- Taking strong inhibitors of CYP3A [seeDrug Interactions (7.1)]
- Taking inducers of CYP3A [see Drug Interactions (7.1)]
- With liver cirrhosis [see Use in Specific Populations (8.6)]
-
5 WARNINGS AND PRECAUTIONS
5.1 QT Interval Prolongation
Ranolazine blocks IKr and prolongs the QTc interval in a dose-related manner.
Clinical experience in an acute coronary syndrome population did not show an increased risk of proarrhythmia or sudden death [see Clinical Studies (14.2)]. However, there is little experience with high doses (>1000 mg twice daily) or exposure, other QT-prolonging drugs, potassium channel variants resulting in a long QT interval, in patients with a family history of (or congenital) long QT syndrome, or in patients with known acquired QT interval prolongation.
5.2 Renal Failure
Acute renal failure has been observed in some patients with severe renal impairment (creatinine clearance [CrCL] <30 mL/min) while taking ranolazine. If acute renal failure develops (e.g., marked increase in serum creatinine associated with an increase in blood urea nitrogen [BUN]), discontinue ranolazine and treat appropriately [see Use in Specific Populations (8.7)].
Monitor renal function after initiation and periodically in patients with moderate to severe renal impairment (CrCL <60 mL/min) for increases in serum creatinine accompanied by an increase in BUN.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 2018 patients with chronic angina were treated with ranolazine in controlled clinical trials. Of the patients treated with ranolazine, 1026 were enrolled in three double-blind, placebo-controlled, randomized studies (CARISA, ERICA, MARISA) of up to 12 weeks' duration. In addition, upon study completion, 1251 patients received treatment with ranolazine in open-label, long-term studies; 1227 patients were exposed to ranolazine for more than 1 year, 613 patients for more than 2 years, 531 patients for more than 3 years, and 326 patients for more than 4 years.
At recommended doses, about 6% of patients discontinued treatment with ranolazine because of an adverse event in controlled studies in angina patients compared to about 3% on placebo. The most common adverse events that led to discontinuation more frequently on ranolazine than placebo were dizziness (1.3% versus 0.1%), nausea (1% versus 0%), asthenia, constipation, and headache (each about 0.5% versus 0%). Doses above 1000 mg twice daily are poorly tolerated.
In controlled clinical trials of angina patients, the most frequently reported treatment-emergent adverse reactions (>4% and more common on ranolazine than on placebo) were dizziness (6.2%), headache (5.5%), constipation (4.5%), and nausea (4.4%). Dizziness may be dose-related. In open-label, long-term treatment studies, a similar adverse reaction profile was observed.
The following additional adverse reactions occurred at an incidence of 0.5 to 4.0% in patients treated with ranolazine and were more frequent than the incidence observed in placebo-treated patients:
Cardiac Disorders – bradycardia, palpitations
Ear and Labyrinth Disorders – tinnitus, vertigo
Eye Disorders – blurred vision
Gastrointestinal Disorders – abdominal pain, dry mouth, vomiting, dyspepsia
General Disorders and Administrative Site Adverse Events – asthenia, peripheral edema
Metabolism and Nutrition Disorders – anorexia
Nervous System Disorders – syncope (vasovagal)
Psychiatric Disorders – confusional state
Renal and Urinary Disorders – hematuria
Respiratory, Thoracic, and Mediastinal Disorders – dyspnea
Skin and Subcutaneous Tissue Disorders – hyperhidrosis
Vascular Disorders – hypotension, orthostatic hypotension
Other (<0.5%) but potentially medically important adverse reactions observed more frequently with ranolazine than placebo treatment in all controlled studies included: angioedema, renal failure, eosinophilia, chromaturia, blood urea increased, hypoesthesia, paresthesia, tremor, pulmonary fibrosis, thrombocytopenia, leukopenia, and pancytopenia.
A large clinical trial in acute coronary syndrome patients was unsuccessful in demonstrating a benefit for ranolazine, but there was no apparent proarrhythmic effect in these high-risk patients [see Clinical Studies (14.2)].
Laboratory Abnormalities:
Ranolazine produces elevations of serum creatinine by 0.1 mg/dL, regardless of previous renal function, likely because of inhibition of creatinine's tubular secretion. In general, the elevation has a rapid onset, shows no signs of progression during long-term therapy, is reversible after discontinuation of ranolazine, and is not accompanied by changes in BUN. In healthy volunteers, ranolazine 1000 mg twice daily had no effect upon the glomerular filtration rate. More marked and progressive increases in serum creatinine, associated with increases in BUN or potassium, indicating acute renal failure, have been reported after initiation of ranolazine in patients with severe renal impairment [see Warnings and Precautions (5.2), Use in Specific Populations (8.7)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ranolazine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Nervous System Disorders – Abnormal coordination, myoclonus, paresthesia, tremor, and other serious neurologic adverse events have been reported to occur, sometimes concurrently, in patients taking ranolazine. The onset of events was often associated with an increase in ranolazine dose or exposure. Many patients reported symptom resolution following drug discontinuation or dose decrease.
Metabolism and Nutrition Disorders – Cases of hypoglycemia have been reported in diabetic patients on antidiabetic medication.
Psychiatric Disorders – hallucination
Renal and Urinary Disorders – dysuria, urinary retention
Skin and Subcutaneous Tissue Disorders – angioedema, pruritus, rash
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on Ranolazine
Do not use ranolazine with strong CYP3A inhibitors, including ketoconazole, itraconazole, clarithromycin, nefazodone, nelfinavir, ritonavir, indinavir, and saquinavir [see Contraindications (4), Clinical Pharmacology (12.3)].
Moderate CYP3A Inhibitors
Limit the dose of ranolazine to 500 mg twice daily in patients on moderate CYP3A inhibitors, including diltiazem, verapamil, erythromycin, fluconazole, and grapefruit juice or grapefruit-containing products [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
P-gp Inhibitors
Concomitant use of ranolazine and P-gp inhibitors, such as cyclosporine, may result in increases in ranolazine concentrations. Titrate ranolazine based on clinical response in patients concomitantly treated with predominant P-gp inhibitors such as cyclosporine [see Dosage and Administration (2.2)].
CYP3A Inducers
Do not use ranolazine with CYP3A inducers such as rifampin, rifabutin, rifapentine, phenobarbital, phenytoin, carbamazepine, and St. John's wort [see Contraindications (4), Clinical Pharmacology (12.3)].
7.2 Effects of Ranolazine on Other Drugs
Limit the dose of simvastatin in patients on any dose of ranolazine to 20 mg once daily, when ranolazine is co-administered. Dose adjustment of other sensitive CYP3A substrates (e.g., lovastatin) and CYP3A substrates with a narrow therapeutic range (e.g., cyclosporine, tacrolimus, sirolimus) may be required as ranolazine may increase plasma concentrations of these drugs [see Clinical Pharmacology (12.3)].
Drugs Transported by P-gp
Concomitant use of ranolazine and digoxin results in increased exposure to digoxin. The dose of digoxin may have to be adjusted [see Clinical Pharmacology (12.3)].
Drugs Metabolized by CYP2D6
The exposure to CYP2D6 substrates, such as tricyclic antidepressants and antipsychotics, may be increased during co-administration with ranolazine, and lower doses of these drugs may be required.
Drugs Transported by OCT2
In subjects with type 2 diabetes mellitus, concomitant use of ranolazine 1000 mg twice daily and metformin results in increased plasma levels of metformin. When ranolazine 1000 mg twice daily is co-administered with metformin, metformin dose should not exceed 1700 mg/day. Monitor blood glucose levels and risks associated with high exposures of metformin.
Metformin exposure was not significantly increased when given with ranolazine 500 mg twice daily [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no available data on ranolazine use in pregnant women to inform any drug-associated risks. Studies in rats and rabbits showed no evidence of fetal harm at exposures 4 times the maximum recommended human dose (MRHD) (see Data).
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage of clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Embryofetal toxicity studies were conducted in rats and rabbits orally administered ranolazine during organogenesis. In rats, decreased fetal weight and reduced ossification were observed at doses (corresponding to 4-fold the AUC for the MRHD) that caused maternal weight loss. No adverse fetal effects were observed in either species exposed (AUC) to ranolazine at exposures (AUC) equal to the MRHD.
8.2 Lactation
There are no data on the presence of ranolazine in human milk, the effects on the breastfed infant, or the effects on milk production. However, ranolazine is present in rat milk [see Use in Specific Populations (8.1)]. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ranolazine and any potential adverse effects on the breastfed infant from ranolazine or from the underlying maternal condition.
Adult female rats were administered ranolazine orally from gestation day 6 through postnatal day 20. No adverse effects on pup development, behavior, or reproduction parameters were observed at a maternal dosage level of 60 mg/kg/day (equal to the MHRD based on AUC). At maternally toxic doses, male and female pups exhibited increased mortality and decreased body weight, and female pups showed increased motor activity. The pups were potentially exposed to low amounts of ranolazine via the maternal milk.
8.5 Geriatric Use
Of the chronic angina patients treated with ranolazine in controlled studies, 496 (48%) were ≥65 years of age, and 114 (11%) were ≥75 years of age. No overall differences in efficacy were observed between older and younger patients. There were no differences in safety for patients ≥65 years compared to younger patients, but patients ≥75 years of age on ranolazine, compared to placebo, had a higher incidence of adverse events, serious adverse events, and drug discontinuations due to adverse events. In general, dose selection for an elderly patient should usually start at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease, or other drug therapy.
8.6 Use in Patients with Hepatic Impairment
Ranolazine is contraindicated in patients with liver cirrhosis. In a study of cirrhotic patients, the Cmax of ranolazine was increased 30% in cirrhotic patients with mild (Child-Pugh Class A) hepatic impairment, but increased 80% in cirrhotic patients with moderate (Child-Pugh Class B) hepatic impairment compared to patients without hepatic impairment. This increase was not enough to account for the 3-fold increase in QT prolongation seen in cirrhotic patients with mild to moderate hepatic impairment [see Clinical Pharmacology (12.2)].
8.7 Use in Patients with Renal Impairment
A pharmacokinetic study of ranolazine in subjects with severe renal impairment (CrCL <30 mL/min) was stopped when 2 of 4 subjects developed acute renal failure after receiving ranolazine 500 mg twice daily for 5 days (lead-in phase) followed by 1000 mg twice a day (1 dose in one subject and 11 doses in the other). Increases in creatinine, BUN, and potassium were observed in 3 subjects during the 500 mg lead-in phase. One subject required hemodialysis, while the other 2 subjects improved upon drug discontinuation [see Warnings and Precautions (5.2)]. Monitor renal function periodically in patients with moderate to severe renal impairment. Discontinue ranolazine if acute renal failure develops.
In a separate study, Cmax was increased between 40% and 50% in patients with mild, moderate, or severe renal impairment compared to patients with no renal impairment, suggesting a similar increase in exposure in patients with renal failure independent of the degree of impairment. The pharmacokinetics of ranolazine has not been assessed in patients on dialysis.
8.8 Use in Patients with Heart Failure
Heart failure (NYHA Class I to IV) had no significant effect on ranolazine pharmacokinetics. Ranolazine had minimal effects on heart rate and blood pressure in patients with angina and heart failure NYHA Class I to IV. No dose adjustment of ranolazine is required in patients with heart failure.
8.9 Use in Patients with Diabetes Mellitus
A population pharmacokinetic evaluation of data from angina patients and healthy subjects showed no effect of diabetes on ranolazine pharmacokinetics. No dose adjustment is required in patients with diabetes.
Ranolazine produces small reductions in HbA1c in patients with diabetes, the clinical significance of which is unknown. Ranolazine should not be considered a treatment for diabetes.
-
10 OVERDOSAGE
Hypotension, QT prolongation, bradycardia, myoclonic activity, severe tremor, unsteady gait/incoordination, dizziness, nausea, vomiting, dysphasia, and hallucinations have been seen in cases of oral overdose of ranolazine. In cases of extreme overdose of ranolazine fatal outcomes have been reported. In clinical studies, high intravenous exposure resulted in diplopia, paresthesia, confusion, and syncope.
In addition to general supportive measures, continuous ECG monitoring may be warranted in the event of overdose.
Since ranolazine is about 62% bound to plasma proteins, hemodialysis is unlikely to be effective in clearing ranolazine.
-
11 DESCRIPTION
Ranolazine extended-release tablets are available as a film-coated, non-scored, extended-release tablet for oral administration.
Ranolazine is a racemic mixture, chemically described as 1-piperazineacetamide, N(2,6-dimethylphenyl)-4-[2-hydroxy-3-(2-methoxyphenoxy)propyl]-, (±)-. It has an empirical formula of C24H33N3O4, a molecular weight of 427.54 g/mole, and the following structural formula:
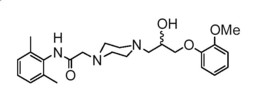
Ranolazine is a white to off-white solid. Ranolazine is soluble in dichloromethane and methanol.
Ranolazine extended-release tablets contain 500 mg or 1000 mg of ranolazine and the following inactive ingredients: ferrosoferric oxide, hypromellose, iron oxide yellow, magnesium stearate, methacrylic acid copolymer type C (contains polysorbate 80 and sodium lauryl sulfate), microcrystalline cellulose, polyethylene glycol, sodium hydroxide, titanium dioxide and talc. Additional inactive ingredient for the 500 mg tablets include iron oxide red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of ranolazine's antianginal effects has not been determined. Ranolazine has anti-ischemic and antianginal effects that do not depend upon reductions in heart rate or blood pressure. It does not affect the rate-pressure product, a measure of myocardial work, at maximal exercise. Ranolazine at therapeutic levels can inhibit the cardiac late sodium current (INa). However, the relationship of this inhibition to angina symptoms is uncertain.
The QT prolongation effect of ranolazine on the surface electrocardiogram is the result of inhibition of IKr, which prolongs the ventricular action potential.
12.2 Pharmacodynamics
Patients with chronic angina treated with ranolazine in controlled clinical studies had minimal changes in mean heart rate (<2 bpm) and systolic blood pressure (<3 mm Hg). Similar results were observed in subgroups of patients with CHF NYHA Class I or II, diabetes, or reactive airway disease, and in elderly patients.
Electrocardiographic Effects
Dose and plasma concentration-related increases in the QTc interval [see Warnings and Precautions (5.1)], reductions in T wave amplitude, and, in some cases, notched T waves, have been observed in patients treated with ranolazine. These effects are believed to be caused by ranolazine and not by its metabolites. The relationship between the change in QTc and ranolazine plasma concentrations is linear, with a slope of about 2.6 msec/1000 ng/mL, through exposures corresponding to doses several-fold higher than the maximum recommended dose of 1000 mg twice daily. The variable blood levels attained after a given dose of ranolazine give a wide range of effects on QTc. At Tmax following repeat dosing at 1000 mg twice daily, the mean change in QTc is about 6 msec, but in the 5% of the population with the highest plasma concentrations, the prolongation of QTc is at least 15 msec. In cirrhotic subjects with mild or moderate hepatic impairment, the relationship between plasma level of ranolazine and QTc is much steeper [see Contraindications (4)].
Age, weight, gender, race, heart rate, congestive heart failure, diabetes, and renal impairment did not alter the slope of the QTc-concentration relationship of ranolazine.
No proarrhythmic effects were observed on 7-day Holter recordings in 3162 acute coronary syndrome patients treated with ranolazine. There was a significantly lower incidence of arrhythmias (ventricular tachycardia, bradycardia, supraventricular tachycardia, and new atrial fibrillation) in patients treated with ranolazine (80%) versus placebo (87%), including ventricular tachycardia ≥3 beats (52% versus 61%). However, this difference in arrhythmias did not lead to a reduction in mortality, a reduction in arrhythmia hospitalization, or a reduction in arrhythmia symptoms.
12.3 Pharmacokinetics
Ranolazine is extensively metabolized in the gut and liver and its absorption is highly variable. For example, at a dose of 1000 mg twice daily, the mean steady-state Cmax was 2600 ng/mL with 95% confidence limits of 400 and 6100 ng/mL. The pharmacokinetics of the (+) R-and (-) S-enantiomers of ranolazine are similar in healthy volunteers. The apparent terminal half-life of ranolazine is 7 hours. Steady state is generally achieved within 3 days of twice-daily dosing with ranolazine. At steady state over the dose range of 500 to 1000 mg twice daily, Cmax and AUC0-T increase slightly more than proportionally to dose, 2.2-and 2.4-fold, respectively. With twice-daily dosing, the trough:peak ratio of the ranolazine plasma concentration is 0.3 to 0.6. The pharmacokinetics of ranolazine is unaffected by age, gender, or food.
Absorption and Distribution
After oral administration of ranolazine, peak plasma concentrations of ranolazine are reached between 2 and 5 hours. After oral administration of 14C-ranolazine as a solution, 73% of the dose is systemically available as ranolazine or metabolites. The bioavailability of ranolazine from ranolazine extended-release tablet relative to that from a solution of ranolazine is 76%. Because ranolazine is a substrate of P-gp, inhibitors of P-gp may increase the absorption of ranolazine.
Food (high-fat breakfast) has no important effect on the Cmax and AUC of ranolazine. Therefore, ranolazine may be taken without regard to meals. Over the concentration range of 0.25 to 10 μg/mL, ranolazine is approximately 62% bound to human plasma proteins.
Metabolism and Excretion
Ranolazine is metabolized mainly by CYP3A and, to a lesser extent, by CYP2D6. Following a single oral dose of ranolazine solution, approximately 75% of the dose is excreted in urine and 25% in feces. Ranolazine is metabolized rapidly and extensively in the liver and intestine; less than 5% is excreted unchanged in urine and feces. The pharmacologic activity of the metabolites has not been well characterized. After dosing to steady state with 500 mg to 1500 mg twice daily, the four most abundant metabolites in plasma have AUC values ranging from about 5 to 33% that of ranolazine, and display apparent half-lives ranging from 6 to 22 hours.
Drug Interactions
Effect of Other Drugs on Ranolazine
In vitro data indicate that ranolazine is a substrate of CYP3A and, to a lesser degree, of CYP2D6. Ranolazine is also a substrate of P-glycoprotein.
Strong CYP3A Inhibitors
Plasma levels of ranolazine with ranolazine 1000 mg twice daily are increased by 220% when co-administered with ketoconazole 200 mg twice daily [see Contraindications (4)].
Moderate CYP3A Inhibitors
Plasma levels of ranolazine with ranolazine 1000 mg twice daily are increased by 50 to 130% by diltiazem 180 to 360 mg, respectively. Plasma levels of ranolazine with ranolazine 750 mg twice daily are increased by 100% by verapamil 120 mg three times daily [see Drug Interactions (7.1)].
Weak CYP3A Inhibitors
The weak CYP3A inhibitors simvastatin (20 mg once daily) and cimetidine (400 mg three times daily) do not increase the exposure to ranolazine in healthy volunteers.
CYP3A Inducers
Rifampin 600 mg once daily decreases the plasma concentrations of ranolazine (1000 mg twice daily) by approximately 95% [see Contraindications (4)].
CYP2D6 Inhibitors
Paroxetine 20 mg once daily increased ranolazine concentrations by 20% in healthy volunteers receiving ranolazine 1000 mg twice daily. No dose adjustment of ranolazine is required in patients treated with CYP2D6 inhibitors.
Digoxin
Plasma concentrations of ranolazine are not significantly altered by concomitant digoxin at 0.125 mg once daily.
Effect of Ranolazine on Other Drugs
In vitro ranolazine and its O-demethylated metabolite are weak inhibitors of CYP3A and moderate inhibitors of CYP2D6 and P-gp. In vitro ranolazine is an inhibitor of OCT2.
CYP3A Substrates
The plasma levels of simvastatin, a CYP3A substrate, and its active metabolite are increased by 100% in healthy volunteers receiving 80 mg once daily and ranolazine 1000 mg twice daily [see Drug Interactions (7.2)]. Mean exposure to atorvastatin (80 mg daily) is increased by 40% following co-administration with ranolazine (1000 mg twice daily) in healthy volunteers. However, in one subject the exposure to atorvastatin and metabolites was increased by ~400% in the presence of ranolazine.
Diltiazem
The pharmacokinetics of diltiazem is not affected by ranolazine in healthy volunteers receiving diltiazem 60 mg three times daily and ranolazine 1000 mg twice daily.
P-gp Substrates
Ranolazine increases digoxin concentrations by 50% in healthy volunteers receiving ranolazine 1000 mg twice daily and digoxin 0.125 mg once daily [see Drug Interactions (7.2)].
CYP2D6 Substrates
Ranolazine 750 mg twice daily increases the plasma concentrations of a single dose of immediate release metoprolol (100 mg), a CYP2D6 substrate, by 80% in extensive CYP2D6 metabolizers with no need for dose adjustment of metoprolol. In extensive metabolizers of dextromethorphan, a substrate of CYP2D6, ranolazine inhibits partially the formation of the main metabolite dextrorphan.
OCT2 Substrates
In subjects with type 2 diabetes mellitus, the exposure to metformin is increased by 40% and 80% following administration of ranolazine 500 mg twice daily and 1000 mg twice daily, respectively. If co-administered with ranolazine 1000 mg twice daily, do not exceed metformin doses of 1700 mg/day [see Drug Interactions (7.2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Ranolazine tested negative for genotoxic potential in the following assays: Ames bacterial mutation assay, Saccharomyces assay for mitotic gene conversion, chromosomal aberrations assay in Chinese hamster ovary (CHO) cells, mammalian CHO/HGPRT gene mutation assay, and mouse and rat bone marrow micronucleus assays.
There was no evidence of carcinogenic potential in mice or rats. The highest oral doses used in the carcinogenicity studies were 150 mg/kg/day for 21 months in rats (900 mg/m2/day) and 50 mg/kg/day for 24 months in mice (150 mg/m2/day). These maximally tolerated doses are 0.8 and 0.1 times, respectively, the daily maximum recommended human dose (MRHD) of 2000 mg on a surface area basis. A published study reported that ranolazine promoted tumor formation and progression to malignancy when given to transgenic APC (min/+) mice at a dose of 30 mg/kg twice daily [see References (15)]. The clinical significance of this finding is unclear.
In male and female rats, oral administration of ranolazine that produced exposures (AUC) approximatelty 3-fold or 5-fold higher, respectively, than the MRHD had no effect on fertility.
-
14 CLINICAL STUDIES
14.1 Chronic Stable Angina
CARISA (Combination Assessment of Ranolazine In Stable Angina) was a study in 823 chronic angina patients randomized to receive 12 weeks of treatment with twice-daily ranolazine 750 mg, 1000 mg, or placebo, who also continued on daily doses of atenolol 50 mg, amlodipine 5 mg, or diltiazem CD 180 mg. Sublingual nitrates were used in this study as needed.
In this trial, statistically significant (p <0.05) increases in modified Bruce treadmill exercise duration and time to angina were observed for each ranolazine dose versus placebo, at both trough (12 hours after dosing) and peak (4 hours after dosing) plasma levels, with minimal effects on blood pressure and heart rate. The changes versus placebo in exercise parameters are presented in Table 1. Exercise treadmill results showed no increase in effect on exercise at the 1000 mg dose compared to the 750 mg dose.
Table 1 Exercise Treadmill Results (CARISA)
a p-value ≤0.05
b p-value ≤0.005
Mean Difference from Placebo (sec)
Study
CARISA (N=791)
Ranolazine Twice-daily Dose
750 mg
1000 mg
Exercise Duration
Trough
Peak
24a
34b
24a
26a
Time to Angina
Trough
Peak
30a
38b
26a
38b
Time to 1 mm ST-Segment
Depression
Trough
Peak
20
41b
21
35b
The effects of ranolazine on angina frequency and nitroglycerin use are shown in Table 2.
Table 2 Angina Frequency and Nitroglycerin Use (CARISA)
a Twice daily
Placebo
Ranolazine
750 mga
Ranolazine
1000 mga
Angina Frequency
N
258
272
261
(attacks/week)
Mean
3.3
2.5
2.1
P-value vs placebo
-
0.006
<0.001
Nitroglycerin
N
252
262
244
Use(doses/week)
Mean
3.1
2.1
1.8
P-value vs placebo
-
0.016
<0.001
Tolerance to ranolazine did not develop after 12 weeks of therapy. Rebound increases in angina, as measured by exercise duration, have not been observed following abrupt discontinuation of ranolazine.
Ranolazine has been evaluated in patients with chronic angina who remained symptomatic despite treatment with the maximum dose of an antianginal agent. In the ERICA (Efficacy of Ranolazine In Chronic Angina) trial, 565 patients were randomized to receive an initial dose of Ranolazine extended-release tablets 500 mg twice daily or placebo for 1 week, followed by 6 weeks of treatment with Ranolazine extended-release tablets 1000 mg twice daily or placebo, in addition to concomitant treatment with amlodipine 10 mg once daily. In addition, 45% of the study population also received long-acting nitrates. Sublingual nitrates were used as needed to treat angina episodes. Results are shown in Table 3. Statistically significant decreases in angina attack frequency (p=0.028) and nitroglycerin use (p=0.014) were observed with ranolazine compared to placebo. These treatment effects appeared consistent across age and use of long-acting nitrates.
Table 3 Angina Frequency and Nitroglycerin Use (ERICA)
a 1000 mg twice daily
Placebo
Ranolazine a
Angina Frequency
N
281
277
(attacks/week)
Mean
4.3
3.3
Median
2.4
2.2
Nitroglycerin Use
N
281
277
(doses/week)
Mean
3.6
2.7
Median
1.7
1.3
Effects on angina frequency and exercise tolerance were considerably smaller in women than in men. In CARISA, the improvement in Exercise Tolerance Test (ETT) in females was about 33% of that in males at the 1000 mg twice-daily dose level. In ERICA, where the primary endpoint was angina attack frequency, the mean reduction in weekly angina attacks was 0.3 for females and 1.3 for males.
Race
There were insufficient numbers of non-Caucasian patients to allow for analyses of efficacy or safety by racial subgroup.
14.2 Lack of Benefit in Acute Coronary Syndrome
In a large (n=6560) placebo-controlled trial (MERLIN-TIMI 36) in patients with acute coronary syndrome, there was no benefit shown on outcome measures. However, the study is somewhat reassuring regarding proarrhythmic risks, as ventricular arrhythmias were less common on ranolazine [see Clinical Pharmacology (12.2)], and there was no difference between ranolazine and placebo in the risk of all-cause mortality (relative risk ranolazine:placebo 0.99 with an upper 95% confidence limit of 1.22).
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Ranolazine extended-release tablets, 500 mg are light orange colored, oval shaped, beveled edge, biconvex, film coated tablets debossed with "588" on one side and plain on other side and are supplied as follows:
NDC: 72578-064-14 in bottles of 60 tablets
NDC: 72578-064-01 in bottles of 100 tablets
NDC: 72578-064-05 in bottles of 500 tablets
NDC: 72578-064-77 in unit-dose blister cartons of 100 Tablets (10 x 10 Unit-dose)
Ranolazine extended-release tablets, 1000 mg are pale yellow colored, oval shaped, beveled edge, biconvex, film coated tablets debossed with "589" on one side and plain on other side and are supplied as follows:
NDC: 72578-065-14 in bottles of 60 tablets
NDC: 72578-065-01 in bottles of 100 tablets
NDC: 72578-065-05 in bottles of 500 tablets
NDC: 72578-065-77 in unit-dose blister cartons of 100 Tablets (10 x 10 Unit-dose)
Store at 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients that ranolazine will not abate an acute angina episode.
Strong CY3PA Inhibitors, CYP3A Inducers, Liver Cirrhosis
- Inform patients that ranolazine should not be used with drugs that are strong CYP3A inhibitors (e.g., ketoconazole, clarithromycin, nefazodone, ritonavir) [(see Contraindications (4), Drug Interactions (7.1)].
- Inform patients that ranolazine should not be used with drugs that are inducers of CYP3A (e.g., rifampin, rifabutin, rifapentine, barbiturates, carbamazepine, phenytoin, St. John's wort) [(see Contraindications (4), Drug Interactions (7.1)].
- Inform patients that ranolazine should not be used in patients with liver cirrhosis [(see Contraindications (4), Use in Specific Populations (8.6)].
Moderate CYP3A Inhibitors, P-gp Inhibitors, Grapefruit Products
- Advise patients to inform their physician if they are receiving drugs that are moderate CYP3A inhibitors (e.g., diltiazem, verapamil, erythromycin) [see Drug Interactions (7)].
- Advise patients to inform their physician if they are receiving drugs that are P-gp inhibitors (e.g., cyclosporine) [seeDrug Interactions (7)].
- Advise patients to limit grapefruit juice or grapefruit products when taking ranolazine [see Drug Interactions (7)].
- Inform patients that ranolazine may produce changes in the electrocardiogram (QTc interval prolongation) [seeWarnings and Precautions (5.1)].
- Advise patients to inform their physician of any personal or family history of QTc prolongation, congenital long QT syndrome, or if they are receiving drugs that prolong the QTc interval such as Class Ia (e.g., quinidine) or Class III (e.g., dofetilide, sotalol, amiodarone) antiarrhythmic agents, erythromycin, and certain antipsychotics (e.g., thioridazine, ziprasidone) [see Warnings and Precautions (5.1)].
Use in Patients with Renal Impairment
Patients with severe renal impairment may be at risk of renal failure while on ranolazine. Advise patients to inform their physician if they have impaired renal function before or while taking ranolazine [see Warnings and Precautions (5.2)].
Dizziness, Fainting
- Inform patients that ranolazine may cause dizziness and lightheadedness. Patients should know how they react to ranolazine before they operate an automobile or machinery, or engage in activities requiring mental alertness or coordination [see Adverse Reactions (6.1)].
- Advise patients to contact their physician if they experience fainting spells while taking ranolazine.
- Instruct patients to swallow ranolazine extended-release tablet whole, with or without meals, and not to crush, break, or chew tablets. Inform patients that if a dose is missed, to take the usual dose at the next scheduled time. The next dose should not be doubled. Inform patients that doses of ranolazine higher than 1000 mg twice daily should not be used [see Dosage and Administration (2)].
- Advise patients to inform their physician of any other medications taken concurrently with ranolazine, including over-the-counter medications.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
Ranolazine (ra NOE la zeen)
extended-release tablets
Dosing Strengths:
500 mg tablets
1000 mg tablets
Read this Patient Information before you start taking ranolazine extended-release tablet and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or treatment.
What are ranolazine extended-release tablet?
Ranolazine extended-release tablet is a prescription medicine used to treat angina that keeps coming back (chronic angina).
Ranolazine extended-release tablet may be used with other medicines that are used for heart problems and blood pressure control.
It is not known if ranolazine extended-release tablet is safe and effective in children.
Who should not take ranolazine extended-release tablet?
Do not take ranolazine extended-release tablet if:
- you take any of the following medicines:
- for fungus infection: ketoconazole (Nizoral®), itraconazole (Sporanox®, OnmelTM)
- for infection: clarithromycin (Biaxin®)
- for depression: nefazodone
- for HIV: nelfinavir (Viracept®), ritonavir (Norvir®), lopinavir and ritonavir (Kaletra®), indinavir (Crixivan®), saquinavir (Invirase®)
- for tuberculosis (TB): rifampin (Rifadin®), rifabutin (Mycobutin®), rifapentine (Priftin®)
- for seizures: phenobarbital, phenytoin (Phenytek®, Dilantin®, Dilantin125®), carbamazepine (Tegretol®)
- St. John's wort (Hypericum perforatum)
- you have scarring (cirrhosis) of your liver
What should I tell my doctor before taking ranolazine extended-release tablet?
Before you take ranolazine extended-release tablet, tell your doctor if you:
- have or have a family history of a heart problem, called 'QT prolongation' or 'long QT syndrome'.
- have liver problems.
- have kidney problems.
- are pregnant or plan to become pregnant. It is not known if ranolazine extended-release tablet will harm your unborn baby.
- are breast-feeding or plan to breast-feed. It is not known if ranolazine extended-release tablet passes into your breast milk. You and your doctor should decide if you will breast-feed.
Tell your doctor about all the medicines you take, including all prescription and nonprescription medicines, vitamins, and herbal supplements. Ranolazine extended-release tablet may affect the way other medicines work and other medicines may affect how ranolazine extended-release tablet works.
Tell your doctor if you take medicines:
- for your heart
- for cholesterol
- for diabetes
- for infection
- for fungus
- for transplant
- for nausea and vomiting because of cancer treatments
- for mental problems
Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine.
How should I take ranolazine extended-release tablet?
- Take ranolazine extended-release tablet exactly as your doctor tells you.
- Your doctor will tell you how much ranolazine extended-release tablet to take and when to take it.
- Do not change your dose unless your doctor tells you to.
- Tell your doctor if you still have symptoms of angina after starting ranolazine extended-release tablet.
- Take ranolazine extended-release tablet by mouth, with or without food.
- Swallow the ranolazine extended-release tablet whole. Do not crush, break, or chew ranolazine extended-release tablet before swallowing.
- If you miss a dose of ranolazine extended-release tablet, wait to take the next dose of ranolazine extended-release tablet at your regular time. Do not make up for the missed dose. Do not take more than 1 dose at a time.
- If you take too much ranolazine extended-release tablet, call your doctor, or go to the nearest emergency room right away.
What should I avoid while taking ranolazine extended-release tablet?
- Grapefruit and grapefruit juice. Limit products that have grapefruit in them. They can cause your blood levels of ranolazine extended-release tablet to increase.
- Ranolazine extended-release tablet can cause dizziness, lightheadedness, or fainting. If you have these symptoms, do not drive a car, use machinery, or do anything that needs you to be alert.
What are the possible side effects of ranolazine extended-release tablet?
Ranolazine extended-release tablet may cause serious side effects, including:
- changes in the electrical activity of your heart called QT prolongation. Your doctor may check the electrical activity of your heart with an ECG. Tell your doctor right away if you feel faint, lightheaded, or feel your heart beating irregularly or fast while taking ranolazine extended-release tablet. These may be symptoms related to QT prolongation.
- kidney failure in people who already have severe kidney problems. Your doctor may need to do tests to check how your kidneys are working.
The most common side effects of ranolazine extended-release tablet include:
- dizziness
- headache
- constipation
- nausea
Tell your doctor if you have any side effect that bothers you or does not go away.
These are not all the possible side effects of ranolazine extended-release tablet. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
How should I store ranolazine extended-release tablet?
Store ranolazine extended-release tablets between 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Keep ranolazine extended-release tablet and all medicines out of the reach of children.
General information about ranolazine extended-release tablet.
Medicines are sometimes prescribed for purposes other than those listed in the Patient Information. Do not use ranolazine extended-release tablet for a condition for which it was not prescribed. Do not give ranolazine extended-release tablet to other people, even if they have the same condition you have. It may harm them.
The Patient Information summarizes the most important information about ranolazine extended-release tablet. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about ranolazine extended-release tablet that is written for health professionals.
For more information, call Viona Pharmaceuticals Inc. at 1-888-304-5011.
What is chronic angina?
Chronic angina means pain or discomfort in the chest, jaw, shoulder, back, or arm that keeps coming back. There are other possible signs and symptoms of angina including shortness of breath. Angina usually comes on when you are active or under stress. Chronic angina is a symptom of a heart problem called coronary heart disease (CHD), also known as coronary artery disease (CAD). When you have CHD, the blood vessels in your heart become stiff and narrow. Oxygen-rich blood cannot reach your heart muscle easily. Angina comes on when too little oxygen reaches your heart muscle.
What are the ingredients in ranolazine extended-release tablet?
Active ingredient: ranolazine
Inactive ingredients:
500 mg tablet: ferrosoferric oxide, hypromellose, iron oxide red, iron oxide yellow, magnesium stearate, methacrylic acid copolymer type C (contains polysorbate 80 and sodium lauryl sulfate), microcrystalline cellulose, polyethylene glycol, sodium hydroxide, titanium dioxide and talc.
1000 mg tablet: ferrosoferric oxide, hypromellose, iron oxide yellow, magnesium stearate, methacrylic acid copolymer type C (contains polysorbate 80 and sodium lauryl sulfate), microcrystalline cellulose, polyethylene glycol, sodium hydroxide, titanium dioxide and talc.
This Patient Information has been approved by the U.S. Food and Drug Administration.
All other trademarks referenced herein are the property of their respective owners.
- you take any of the following medicines:
- SPL UNCLASSIFIED SECTION
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Ranolazine extended-release tablets, 500 mg
NDC: 72578-064-14
60 Tablets
Rx only
Viona

Ranolazine extended-release tablets, 1000 mg
NDC: 72578-065-14
60 Tablets
Rx only
Viona

-
INGREDIENTS AND APPEARANCE
RANOLAZINE
ranolazine tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72578-064 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RANOLAZINE (UNII: A6IEZ5M406) (RANOLAZINE - UNII:A6IEZ5M406) RANOLAZINE 500 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) SODIUM HYDROXIDE (UNII: 55X04QC32I) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color ORANGE (Light Orange) Score no score Shape OVAL Size 17mm Flavor Imprint Code 588 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72578-064-14 60 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 2 NDC: 72578-064-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 3 NDC: 72578-064-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 4 NDC: 72578-064-77 10 in 1 CARTON 09/10/2019 4 NDC: 72578-064-30 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA210188 09/10/2019 RANOLAZINE
ranolazine tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72578-065 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RANOLAZINE (UNII: A6IEZ5M406) (RANOLAZINE - UNII:A6IEZ5M406) RANOLAZINE 1000 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) SODIUM HYDROXIDE (UNII: 55X04QC32I) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color YELLOW (Pale Yellow) Score no score Shape OVAL Size 22mm Flavor Imprint Code 589 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72578-065-14 60 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 2 NDC: 72578-065-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 3 NDC: 72578-065-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 09/10/2019 4 NDC: 72578-065-77 10 in 1 CARTON 09/10/2019 4 NDC: 72578-065-30 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA210188 09/10/2019 Labeler - Viona Pharmaceuticals Inc (081468959) Registrant - Cadila Healthcare Limited (650199482) Establishment Name Address ID/FEI Business Operations Cadila Healthcare Limited 677605858 ANALYSIS(72578-064, 72578-065) , MANUFACTURE(72578-064, 72578-065)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.