ELAHERE- mirvetuximab soravtansine injection, solution
ELAHERE by
Drug Labeling and Warnings
ELAHERE by is a Prescription medication manufactured, distributed, or labeled by ImmunoGen, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ELAHERE safely and effectively. See full prescribing information for ELAHERE.
ELAHERE® (mirvetuximab soravtansine-gynx) injection, for intravenous use.
Initial U.S. Approval: 2022
WARNING: OCULAR TOXICITY
See full prescribing information for complete boxed warning.
-
ELAHERE can cause severe ocular toxicities, including visual impairment, keratopathy, dry eye, photophobia, eye pain, and uveitis. (5.1, 6.1)
-
Conduct an ophthalmic exam including visual acuity and slit lamp exam prior to initiation of ELAHERE, every other cycle for the first 8 cycles, and as clinically indicated. (2.3)
-
Administer prophylactic artificial tears and ophthalmic topical steroids. (2.3, 5.1)
-
Withhold ELAHERE for ocular toxicities until improvement and resume at the same or reduced dose. (2.4, 5.1)
- Discontinue ELAHERE for Grade 4 ocular toxicities. (2.4, 5.1)
RECENT MAJOR CHANGES
Dosage and Administration (2.5) 07/2025 INDICATIONS AND USAGE
ELAHERE is a folate receptor alpha (FRα)-directed antibody and microtubule inhibitor conjugate indicated for the treatment of adult patients with FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received one to three prior systemic treatment regimens. Select patients for therapy based on an FDA-approved test. (1, 2.1)
DOSAGE AND ADMINISTRATION
- Administer ELAHERE as an intravenous infusion only after dilution in 5% Dextrose Injection, USP. ELAHERE is incompatible with normal saline. (2.5)
- The recommended dose of ELAHERE is 6 mg/kg adjusted ideal body weight administered as an intravenous infusion every 3 weeks until disease progression or unacceptable toxicity. (2.2)
- Premedicate with a corticosteroid, antihistamine, and antipyretic. (2.3)
- Premedicate with an antiemetic, ophthalmic topical steroids, and lubricating eye drops. (2.3, 5.1)
- See full Prescribing Information for preparation and administration instructions and dose modifications for adverse reactions. (2)
DOSAGE FORMS AND STRENGTHS
- Injection: 100 mg/20 mL (5 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Pneumonitis: Withhold ELAHERE for persistent or recurrent Grade 2 pneumonitis and consider dose reduction. Permanently discontinue ELAHERE for Grade 3 or 4 pneumonitis. (2.4, 5.2)
- Peripheral Neuropathy: Monitor patients for new or worsening peripheral neuropathy. Withhold dosage, dose reduce, or permanently discontinue ELAHERE based on the severity of peripheral neuropathy. (2.4, 5.3)
- Embryo-Fetal Toxicity: ELAHERE can cause fetal harm. Advise of the potential risk to a fetus and to use effective contraception. (5.4, 8.1, 8.3)
ADVERSE REACTIONS
The most common (≥20 %) adverse reactions, including lab abnormalities, were increased aspartate aminotransferase, fatigue, increased alanine aminotransferase, blurred vision, nausea, increased alkaline phosphatase, diarrhea, abdominal pain, keratopathy, peripheral neuropathy, musculoskeletal pain, decreased lymphocytes, decreased platelets, decreased magnesium, decreased hemoglobin, dry eye, constipation, decreased leukocytes, vomiting, decreased albumin, decreased appetite, and decreased neutrophils. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
Strong CYP3A4 Inhibitors: Closely monitor for ELAHERE adverse reactions. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2025
-
ELAHERE can cause severe ocular toxicities, including visual impairment, keratopathy, dry eye, photophobia, eye pain, and uveitis. (5.1, 6.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: OCULAR TOXICITY
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Premedication and Required Eye Care
2.4 Dosage Modifications
2.5 Instructions for Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Ocular Disorders
5.2 Pneumonitis
5.3 Peripheral Neuropathy
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on ELAHERE
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: OCULAR TOXICITY
-
ELAHERE can cause severe ocular toxicities, including visual impairment, keratopathy, dry eye, photophobia, eye pain, and uveitis [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
-
Conduct an ophthalmic exam including visual acuity and slit lamp exam prior to initiation of ELAHERE, every other cycle for the first 8 cycles, and as clinically indicated [see Dosage and Administration (2.3)].
-
Administer prophylactic artificial tears and ophthalmic topical steroids [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
-
Withhold ELAHERE for ocular toxicities until improvement and resume at the same or reduced dose [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
- Discontinue ELAHERE for Grade 4 ocular toxicities [see Dosage and Administration (2.4) and Warnings and Precautions (5.1)].
-
ELAHERE can cause severe ocular toxicities, including visual impairment, keratopathy, dry eye, photophobia, eye pain, and uveitis [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
-
1 INDICATIONS AND USAGE
ELAHERE® is indicated for the treatment of adult patients with folate receptor-alpha (FRα) positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer, who have received one to three prior systemic treatment regimens. Select patients for therapy based on an FDA-approved test [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the treatment of platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer with ELAHERE based on the presence of FRα tumor expression [see Indications & Usage (1) and Clinical Studies (14)] using an FDA-approved test.
Information on FDA-approved tests for the measurement of FRα tumor expression is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of ELAHERE is 6 mg/kg adjusted ideal body weight (AIBW) administered once every 3 weeks (21-day cycle) as an intravenous infusion until disease progression or unacceptable toxicity [see Dosage and Administration (2.5)]. Dosing based on AIBW reduces exposure variability for patients who are either under or overweight.
The total dose of ELAHERE is calculated based on each patient’s AIBW using the following formula:
AIBW = Ideal Body Weight (IBW [kg]) + 0.4*(Actual weight [kg] – IBW)
Female IBW [kg] = 0.9*height[cm] – 92
2.3 Premedication and Required Eye Care
Premedication
Administer the premedications in Table 1 prior to each infusion of ELAHERE to reduce the incidence and severity of infusion related reactions (IRRs), nausea, and vomiting.
Table 1: Premedication Prior to Each ELAHERE Infusion Premedication Route of Administration Examples (or equivalent) Administration Time Prior to ELAHERE Infusion Corticosteroid intravenous dexamethasone 10 mg Antihistamine oral or intravenous diphenhydramine 25 mg to 50 mg At least 30 minutes prior Antipyretic oral or intravenous acetaminophen 325 mg to 650 mg Antiemetic oral or intravenous 5-HT3 serotonin receptor antagonist or appropriate alternatives Before each dose and thereafter as needed Consider additional premedications including corticosteroids the day prior to ELAHERE administration for patients who experienced IRRs.
Ophthalmic Exams and Premedication
Ophthalmic Exam: Conduct an ophthalmic exam including visual acuity and slit lamp exam prior to initiation of ELAHERE, every other cycle for the first 8 cycles, and as clinically indicated.
Ophthalmic Topical Steroids: The use of ophthalmic topical steroids is recommended. The initial prescription and renewals of any corticosteroid medication should be made only after examination with a slit lamp. Administer one drop of ophthalmic topical steroids in each eye 6 times daily starting the day prior to each infusion until day 4; then administer one drop in each eye 4 times daily for days 5-8 of each cycle of ELAHERE [see Warnings and Precautions (5.1)].
Lubricating Eye Drops: The use of lubricating eye drops at least four times daily and as needed is recommended during treatment with ELAHERE. Instruct patients to use lubricating eye drops and advise to wait at least 10 minutes after ophthalmic topical steroid administration before instilling lubricating eye drops [see Warnings and Precautions (5.1)].
2.4 Dosage Modifications
Table 2 provides dose reduction levels and Table 3 provides dosage modifications for ELAHERE due to adverse reactions.
Table 2: Dosage Reduction Schedule ELAHERE Dose Levels First Dose Reduction 5 mg/kg AIBW once every 3 weeks (21-day cycle) Second Dose Reduction 4 mg/kg AIBW once every 3 weeks (21-day cycle) * * Permanently discontinue in patients who cannot tolerate 4 mg/kg AIBW.
Table 3: Dosage Modifications for Adverse Reactions Adverse Reaction Severity of Adverse Reaction* Dosage Modification Nonconfluent superficial keratitis Monitor. Keratitis/Keratopathy
[see Warnings and Precautions (5.1) and Adverse Reactions (6.1)]Confluent superficial keratitis, a cornea epithelial defect, or 3-line or more loss in best corrected visual acuity Withhold until improved or resolved, then maintain at same dose level or consider dose reduction. Corneal ulcer or stromal opacity or best corrected distance visual acuity 20/200 or worse Withhold until improved or resolved, then reduce by one dose level. Corneal perforation Permanently discontinue. Grade 1/ Rare cell in anterior chamber Monitor. Uveitis
[see Warnings and Precautions (5.1) and Adverse Reactions (6.1)]Grade 2/ 1-2+ Cell or Flare in anterior chamber Withhold until Grade 1 or less, then maintain dose at same dose level. Grade 3/ 3+ Cell or Flare in anterior chamber Withhold until Grade 1 or less, then reduce dose by one dose level. Grade 4/ Hypopyon Permanently discontinue. Grade 1 Monitor. Pneumonitis
[see Warnings and Precautions (5.2) and Adverse Reactions (6.1)]Grade 2 Withhold until Grade 1 or less, then maintain at same dose level or consider dose reduction. Grade 3 or 4 Permanently discontinue. Peripheral Neuropathy
[see Warnings and Precautions (5.3) and Adverse Reactions (6.1)]Grade 2 Withhold until Grade 1 or less, then reduce by one dose level. Grade 3 or 4 Permanently discontinue. Grade 1 Maintain infusion rate. Infusion-Related Reactions/Hypersensitivity
[see Adverse Reactions (6.1)]Grade 2 - Interrupt infusion and administer supportive treatment.
- After recovery from symptoms, resume the infusion at 50% of the previous rate, and if no further symptoms appear, increase rate as appropriate until infusion is completed [see Dosage and Administration (2.5)].
- Administer additional premedication for future cycles [see Dosage and Administration (2.3)].
Grade 3 or 4 - Immediately stop infusion and administer supportive treatment.
- Advise patient to seek emergency treatment and immediately notify their healthcare provider if the infusion-related symptoms recur.
- Permanently discontinue.
Hematological
[see Adverse Reactions (6.1)]Grade 3 or 4 Withhold until Grade 1 or less, then resume at one lower dose level. Other Adverse Reactions Grade 3 Withhold until Grade 1 or less, then resume at one lower dose level. [see Adverse Reactions (6.1)] Grade 4 Permanently discontinue. * Unless otherwise specified, National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0.
2.5 Instructions for Preparation and Administration
Preparation
- ELAHERE is a hazardous drug. Follow applicable special handling and disposal procedures1.
- Calculate the dose (mg) (based on the patient’s AIBW), total volume (mL) of solution required, and the number of vials of ELAHERE needed [see Recommended Dosage (2.2) and Dose Modifications (2.4)]. More than one vial will be needed for a full dose.
- Remove the vials of ELAHERE from the refrigerator and allow to warm to room temperature.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. ELAHERE is a clear to slightly opalescent, colorless solution.
- Gently swirl and inspect each vial prior to withdrawing the calculated dose volume of ELAHERE. Do not shake the vial.
- Using aseptic technique, withdraw the calculated dose volume of ELAHERE for subsequent dilution.
- ELAHERE contains no preservatives and is intended for single-dose only. Discard any unused drug remaining in the vial.
Dilution
- ELAHERE must be diluted prior to administration with 5% Dextrose Injection, USP to a final concentration of 1 mg/mL to 2 mg/mL.
- ELAHERE is incompatible with 0.9% Sodium Chloride Injection. ELAHERE must not be mixed with any other drugs or intravenous fluids.
- Determine the volume of 5% Dextrose Injection, USP required to achieve the final diluted drug concentration. Either remove excess 5% Dextrose Injection, USP from a prefilled intravenous bag or add the calculated volume of 5% Dextrose Injection, USP to a sterile empty intravenous bag. Then add the calculated dose volume of ELAHERE to the intravenous bag.
- Gently mix the diluted drug solution by slowly inverting the bag several times to assure uniform mixing. Do not shake or agitate.
If the diluted infusion solution is not used immediately, store solution either at ambient temperature [(18°C to 25°C (64.4°F to 77°F)] for no more than 8 hours (including infusion time), or under refrigeration at 2°C to 8°C (36°F to 46°F) for no more than 24 hours. If refrigerated, allow the infusion bag to reach room temperature prior to administration. After refrigeration, administer diluted infusion solutions within 8 hours (including infusion time).
- Do not freeze prepared infusion solution.
Administration
- Inspect the ELAHERE intravenous infusion bag visually for particulate matter and discoloration prior to administration.
- Administer pre-medications prior to ELAHERE administration [see Premedication and Prophylactic Regimen (2.3)].
- Administer ELAHERE as an intravenous infusion only, using a 0.2 or 0.22 µm polyethersulfone (PES) in-line filter. Do not substitute other membrane materials.
- Administer the initial dose as an intravenous infusion at the rate of 1 mg/min. If well tolerated after 30 minutes at 1 mg/min, the infusion rate can be increased to 3 mg/min. If well tolerated after 30 minutes at 3 mg/min, the infusion rate can be increased to 5 mg/min.
- If no infusion-related reactions occur with the previous dose, subsequent infusions should be started at the maximally tolerated rate and may be increased up to a maximum infusion rate of 5 mg/min, as tolerated.
- Following the infusion, flush the intravenous line with 5% Dextrose Injection, USP to ensure delivery of the full dose. Do not use any other intravenous fluids for flushing.
- Interrupt infusion and administer supportive treatment.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Ocular Disorders
ELAHERE can cause severe ocular adverse reactions, including visual impairment, keratopathy (corneal disorders), dry eye, photophobia, eye pain, and uveitis.
Ocular adverse reactions occurred in 59% of patients with ovarian cancer treated with ELAHERE. Eleven percent (11%) of patients experienced Grade 3 ocular adverse reactions, including blurred vision, keratopathy (corneal disorders), dry eye, cataract, photophobia, and eye pain; two patients (0.3%) experienced Grade 4 events (keratopathy and cataract). The most common (≥5%) ocular adverse reactions were blurred vision (48%), keratopathy (36%), dry eye (27%), cataract (16%), photophobia (14%), and eye pain (10%). [see Adverse Reactions (6.1)].
The median time to onset for first ocular adverse reaction was 5.1 weeks (range: 0.1 to 68.6). Of the patients who experienced ocular events, 53% had complete resolution; 38% had partial improvement (defined as a decrease in severity by one or more grades from the worst grade) at last follow up. Ocular adverse reactions led to permanent discontinuation of ELAHERE in 1% of patients.
Premedication and use of lubricating and ophthalmic topical steroid eye drops during treatment with ELAHERE are recommended [see Dosage and Administration (2.3)]. Advise patients to avoid use of contact lenses during treatment with ELAHERE unless directed by a healthcare provider.
Refer patients to an eye care professional for an ophthalmic exam including visual acuity and slit lamp exam prior to treatment initiation, every other cycle for the first 8 cycles, and as clinically indicated. Promptly refer patients to an eye care professional for any new or worsening ocular signs and symptoms.
Monitor for ocular toxicity and withhold, reduce, or permanently discontinue ELAHERE based on severity and persistence of ocular adverse reactions. [see Dosage and Administration (2.4)].
5.2 Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD), including pneumonitis, can occur in patients treated with ELAHERE.
Pneumonitis occurred in 10% of patients treated with ELAHERE, including 1% with Grade 3 events and 1 patient (0.1%) with a Grade 4 event. One patient (0.1%) died due to respiratory failure in the setting of pneumonitis and lung metastases. One patient (0.1%) died due to respiratory failure of unknown etiology.
Pneumonitis led to permanent discontinuation of ELAHERE in 3% of patients.
Monitor patients for pulmonary signs and symptoms of pneumonitis, which may include hypoxia, cough, dyspnea, or interstitial infiltrates on radiologic exams. Infectious, neoplastic, and other causes for such symptoms should be excluded through appropriate investigations. Withhold ELAHERE for patients who develop persistent or recurrent Grade 2 pneumonitis until symptoms resolve to ≤ Grade 1 and consider dose reduction. Permanently discontinue ELAHERE in all patients with Grade 3 or 4 pneumonitis [see Dosage and Administration (2.4)]. Patients who are asymptomatic may continue dosing of ELAHERE with close monitoring.
5.3 Peripheral Neuropathy
Peripheral neuropathy occurred in 36% of patients with ovarian cancer treated with ELAHERE across clinical trials; 3% of patients experienced Grade 3 peripheral neuropathy. Peripheral neuropathy adverse reactions included peripheral neuropathy (20%), peripheral sensory neuropathy (9%), paraesthesia (6%), neurotoxicity (3%), hypoesthesia (1%), peripheral motor neuropathy (0.9%), polyneuropathy (0.3%), and peripheral sensorimotor neuropathy (0.1%).
The median time to onset of peripheral neuropathy was 5.9 weeks (range 0.1 to 126.7). Of the patients who experienced peripheral neuropathy, 23% had complete resolution and 12% had partial improvement (defined as a decrease in severity by one or more grades from the worst grade) at last follow up. Peripheral neuropathy led to discontinuation of ELAHERE in 0.7% of patients.
Monitor patients for signs and symptoms of neuropathy, such as paresthesia, tingling or a burning sensation, neuropathic pain, muscle weakness, or dysesthesia. For patients experiencing new or worsening peripheral neuropathy, withhold dosage, dose reduce, or permanently discontinue ELAHERE based on the severity of peripheral neuropathy [see Dosage and Administration (2.4)].
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, ELAHERE can cause embryo-fetal harm when administered to a pregnant woman because it contains a genotoxic compound (DM4) and affects actively dividing cells.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ELAHERE and for 7 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed elsewhere in the labeling:
- Ocular Disorders [see Warnings and Precautions (5.1)].
- Pneumonitis [see Warnings and Precautions (5.2)].
- Peripheral Neuropathy [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in Warnings and Precautions reflect exposure to ELAHERE in 682 patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer at 6 mg/kg AIBW administered intravenously once every 3 weeks until disease progression or unacceptable toxicity in Study 0416, Study 0417, Study 0403 (NCT02631876), and Study 0401 (NCT01609556). The median duration of treatment was 4.4 months (range: 1.0 to 30.0). In the pooled safety population, the most common (≥20%) adverse reactions, including laboratory abnormalities, were increased aspartate aminotransferase, fatigue, increased alanine aminotransferase, blurred vision, nausea, increased alkaline phosphatase, diarrhea, abdominal pain, keratopathy, peripheral neuropathy, musculoskeletal pain, decreased lymphocytes, decreased platelets, decreased magnesium, decreased hemoglobin, dry eye, constipation, decreased leukocytes, vomiting, decreased albumin, decreased appetite, and decreased neutrophils.
Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer
Study 0416
The safety of ELAHERE was evaluated in Study 0416, a multicenter, open-label, active-controlled, randomized, two-arm, study in patients (n=453) with platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer [see Clinical Studies (14)]. Patients received ELAHERE 6 mg/kg AIBW once every 3 weeks until disease progression or unacceptable toxicity. The median duration of treatment was 5 months (range: 0.69 to 27.4).
Serious adverse reactions occurred in 24% of patients treated with ELAHERE. The most common (≥2%) serious adverse reactions were intestinal obstruction (5%), abdominal pain (3%), and pleural effusion (3%). Fatal adverse reactions occurred in 3% of patients, including intestinal obstruction, dyspnea in the setting of subileus, neutropenic sepsis, cardiopulmonary failure, respiratory failure, ischemic stroke, and pulmonary embolus.
Permanent discontinuation of ELAHERE due to adverse reactions occurred in 9% of patients. The most common (≥1%) adverse reactions leading to permanent discontinuation were pneumonitis (2%), blurred vision (1%), and peripheral neuropathy (1%).
Dosage delays of ELAHERE due to an adverse reaction occurred in 54% of patients treated with ELAHERE. Adverse reactions which required dosage delays in ≥3% of patients included blurred vision (22%), keratopathy (19%), dry eye (7%), neutropenia (6%), pneumonitis (6%), photophobia (5%), cataract (4%), and peripheral neuropathy (4%).
Dose reductions of ELAHERE due to an adverse reaction occurred in 34% of patients. Adverse reactions which required dose reductions in ≥3% of patients included blurred vision (14%), keratopathy (10%), peripheral neuropathy (6%), and dry eye (5%).
Tables 4 and 5 summarize adverse reactions and laboratory abnormalities, respectively, occurring in ≥10% of patients who received ELAHERE in Study 0416.
Table 4: Adverse Reactions Occurring in ≥10% of Patients with Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer Who Received ELAHERE in Study 0416 Adverse Reaction ELAHERE
(n=218)Chemotherapy¥
(n=207)All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)Gastrointestinal disorders Abdominal pain* 34 3 23 2 Diarrhea 29 1 17 0.5 Constipation 27 0 19 1 Nausea 27 2 29 2 Vomiting 18 3 18 1 Eye disorders Blurred vision※ 45 9 3 0 Keratopathy† 37 11 0 0 Dry eye‡ 29 3 5 0 Photophobia 18 0.5 0.5 0 Cataract˄ 16 3 0.5 0 General disorders and administration site conditions Fatigue⸙ 47 3 41 7 Nervous system disorders Peripheral neuropathy¶ 37 4 23 4 Headache 14 0 10 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain♦ 31 1 21 2 Metabolism and nutrition disorders Decreased appetite 18 1 14 1 Respiratory, thoracic, and mediastinal disorders Pneumonitis± 10 0.5 0.5 0 ¥ Chemotherapy: paclitaxel, pegylated liposomal doxorubicin (PLD), topotecan.
※ Blurred vision includes vision blurred, vitreous floaters, visual acuity reduced, diplopia, accommodation disorder, and visual impairment.
† Keratopathy includes corneal disorder, corneal epithelial microcysts, keratitis, keratopathy, corneal deposits, punctate keratitis, and corneal opacity.
‡ Dry eye includes dry eye and lacrimation increased.
˄ Cataract includes cataract and cataract nuclear.
⸙ Fatigue includes fatigue and asthenia.
* Abdominal pain includes abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort.
¶ Peripheral neuropathy includes neuropathy peripheral, peripheral sensory neuropathy, peripheral motor neuropathy, paresthesia, hypoesthesia, polyneuropathy, neurotoxicity, and peripheral sensorimotor neuropathy.
♦ Musculoskeletal pain includes back pain, myalgia, neck pain, arthralgia, musculoskeletal pain, non-cardiac chest pain, bone pain, pain in extremity, musculoskeletal stiffness, musculoskeletal chest pain, and musculoskeletal discomfort.
± Pneumonitis includes pneumonitis, interstitial lung disease, respiratory failure, and organizing pneumonia.
Clinically relevant adverse reactions occurring in <10% of patients who received ELAHERE in Study 0416 included infusion related reactions/hypersensitivity (8%).
Table 5: Select Laboratory Abnormalities ≥10% for All Grades, in Patients Who Received ELAHERE in Study 0416 Laboratory Abnormality ELAHERE
(n=218)Chemotherapy
(n=207)All Grades
%Grades 3-4
%All Grades
%Grades 3-4
%Liver Function Tests Increased aspartate aminotransferase 57 0 14 0 Increased alanine aminotransferase 38 1 15 1 Increased alkaline phosphatase 30 1 13 1 Chemistry Decreased albumin 21 1 27 2 Decreased magnesium 21 1 29 2 Decreased sodium 16 0 18 0 Decreased potassium 15 1 11 1 Increased calcium 12 0 5 0 Decreased bicarbonate 11 0 11 0 Increased creatinine 10 0 11 0 Hematology* Decreased lymphocytes 27 3 42 11 Decreased leukocytes 23 1 53 10 Decreased neutrophils 22 1 45 17 Decreased hemoglobin 18 1 63 8 Decreased platelets 17 1 20 5 * The denominator used to calculate the rate varied from 63 to 214 (ELAHERE); 63 to 194 (IC Chemo) based on the number of patients with a baseline value and at least one post-treatment value.
Study 0417
The safety of ELAHERE was evaluated in Study 0417, a single-arm, open-label study in patients (n=106) with platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer [see Clinical Studies (14)]. Patients received ELAHERE 6 mg/kg AIBW once every 3 weeks until disease progression or unacceptable toxicity. The median duration of treatment was 4.2 months (range: 0.7 to 13.3).
Serious adverse reactions occurred in 31% of patients treated with ELAHERE. The most common (≥2%) serious adverse reactions were intestinal obstruction (8%), ascites (4%), infection (3%), and pleural effusion (3%). Fatal adverse reactions occurred in 2% of patients, including small intestinal obstruction (1%) and pneumonitis (1%).
Permanent discontinuation of ELAHERE due to adverse reactions occurred in 11% of patients. The most common (≥2%) adverse reactions leading to permanent discontinuation were intestinal obstruction (2%) and thrombocytopenia (2%). One patient (0.9%) permanently discontinued ELAHERE due to visual impairment (unilateral decrease to BCVA ≤ 20/200 that resolved to baseline after discontinuation).
Dosage delays of ELAHERE due to an adverse reaction occurred in 39% of patients treated with ELAHERE. Adverse reactions which required dosage delays in ≥3% of patients included blurred vision (15%), keratopathy (11%), neutropenia (6%), dry eye (5%), cataracts (3%), and increased gamma-glutamyltransferase (3%).
Dose reductions of ELAHERE due to an adverse reaction occurred in 20% of patients. Adverse reactions which required dose reductions in ≥3% of patients included blurred vision (9%) and keratopathy (7%).
Table 6 summarizes the adverse reactions (≥10%) in patients treated with ELAHERE in Study 0417.
Table 6: Adverse Reactions (≥10%) in Patients with Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer Who Received ELAHERE in Study 0417 Adverse Reaction All Grades
N=106
(%)Grade 3-4
N=106
(%)Eye disorders Blurred Vision※ 50 7 Keratopathy† 37 9 Dry eye‡ 27 2 Cataract 18 3 Photophobia 17 0 Eye pain§ 10 0 General disorders Fatigue 49 3 Gastrointestinal disorders Nausea 40 0 Abdominal pain* 36 7 Diarrhea 31 3 Constipation 30 1 Vomiting 19 0 Abdominal distension 11 0 Nervous system disorders Peripheral neuropathy¶ 33 2 Metabolism and nutrition disorders Decreased appetite 18 1 Musculoskeletal and connective tissue disorders Arthralgia 17 0 Myalgia 10 0 Respiratory, thoracic, and mediastinal disorders Dyspnea^ 12 0 ※ Blurred vision includes vision blurred, vitreous floaters, visual acuity reduced, diplopia, presbyopia, accommodation disorder, visual impairment, and refraction disorder.
† Keratopathy includes corneal disorder, corneal epithelial microcysts, corneal epithelial defect, keratitis, keratopathy, corneal deposits, and punctate keratitis.
‡ Dry eye includes dry eye and lacrimation increased.
§ Eye pain includes eye pain and ocular discomfort.
⸙ Fatigue includes fatigue and asthenia.
* Abdominal pain includes abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort.
¶ Peripheral neuropathy includes neuropathy peripheral, peripheral sensory neuropathy, peripheral motor neuropathy, paresthesia, hypoesthesia, polyneuropathy, and neurotoxicity.
^ Dyspnea includes dyspnea and exertional dyspnea.
Clinically relevant adverse reactions occurring in <10% of patients who received ELAHERE in Study 0417 included infusion related reactions/hypersensitivity (9%), pneumonitis (8%), and uveitis (1%).
Table 7 summarizes the laboratory abnormalities in Study 0417.
Table 7: Select Laboratory Abnormalities ≥10% for All Grades, or ≥2% for Grades 3-4 in Patients Who Received ELAHERE Laboratory Abnormality ELAHERE* All Grades
(%)Grade 3-4
(%)Liver Function Tests Increased aspartate aminotransferase 50 2 Increased alanine aminotransferase 39 2 Increased alkaline phosphatase 30 1 Hematology* Decreased lymphocytes 35 7 Decreased leukocytes 26 1 Decreased neutrophils 26 3 Decreased hemoglobin 25 3 Decreased platelets 18 2 Chemistry Decreased albumin 31 1 Decreased magnesium 27 2 Increased creatinine 16 0 Decreased potassium 15 4 * The denominator used to calculate the rate varied from 98 to 101 based on the number of patients with a baseline value and at least one post-treatment value.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions In studies 0416, 0417, 0401, and 0403 in patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer who received ELAHERE at 6 mg/kg AIBW administered intravenously once every 3 weeks, 9% (57/626) developed anti-drug antibodies. Infusion reactions (including bronchospasm, erythema, eyelid erythema, flushing, hypersensitivity, periorbital edema, rash, allergic rhinitis, face edema) occurred in 26% (15/57) of patients with anti-drug antibodies and in 7% (41/569) who did not develop anti-drug antibodies [see Clinical Pharmacology (12.6)].
- Ocular Disorders [see Warnings and Precautions (5.1)].
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on ELAHERE
Strong CYP3A4 Inhibitors
DM4 is a CYP3A4 substrate. Concomitant use of ELAHERE with strong CYP3A4 inhibitors may increase unconjugated DM4 exposure [see Clinical Pharmacology (12.3)], which may increase the risk of ELAHERE adverse reactions [see Adverse Reactions (6)]. Closely monitor patients for adverse reactions with ELAHERE when used concomitantly with strong CYP3A4 inhibitors [see Warnings and Precautions (5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, ELAHERE can cause embryo-fetal harm when administered to a pregnant woman because it contains a genotoxic compound (DM4) and affects actively dividing cells [see Clinical Pharmacology (12.1), Nonclinical Toxicology (13.1)]. Human immunoglobulin G (IgG) is known to cross the placental barrier; therefore, ELAHERE has the potential to be transmitted from the mother to the developing fetus. There are no available human data on ELAHERE use in pregnant women to inform a drug-associated risk. No reproductive or developmental animal toxicity studies were conducted with mirvetuximab soravtansine-gynx. Advise patients of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data: No reproductive or developmental animal toxicity studies have been conducted with mirvetuximab soravtansine-gynx. The cytotoxic component of ELAHERE, DM4, disrupts microtubule function, is genotoxic, and can be toxic to actively dividing cells, suggesting it has the potential to cause embryotoxicity and teratogenicity.
8.2 Lactation
Risk Summary
There are no data on the presence of mirvetuximab soravtansine-gynx in human milk or the effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with ELAHERE and for 1 month after the last dose.
8.3 Females and Males of Reproductive Potential
ELAHERE can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating ELAHERE.
Contraception
Females: Advise females of reproductive potential to use effective contraception during treatment with ELAHERE and for 7 months after the last dose.
8.4 Pediatric Use
Safety and effectiveness of ELAHERE have not been established in pediatric patients.
8.5 Geriatric Use
Of the 682 patients with epithelial ovarian cancer who were treated with ELAHERE across studies, 44% of patients were ≥65 years old. Grade ≥3 adverse reactions occurred in 51% of patients ≥65 years and in 45% <65 years. No clinically meaningful differences in efficacy or safety were observed between patients ≥65 years of age compared to younger patients.
Age does not have a clinically meaningful effect on the pharmacokinetics of ELAHERE [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of ELAHERE is recommended for patients with mild to moderate renal impairment (CLcr 30 to 89 mL/min). The effect of severe renal impairment (CLcr 15 to < 30 mL/min) or end-stage renal disease on ELAHERE is unknown [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Avoid use of ELAHERE in patients with moderate or severe hepatic impairment (total bilirubin >1.5 ULN).
No dosage adjustment of ELAHERE is recommended for patients with mild hepatic impairment (total bilirubin ≤ULN and AST >ULN or total bilirubin >1 to 1.5 times ULN and any AST) [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Mirvetuximab soravtansine-gynx is a folate receptor alpha (FRα)-directed antibody-drug conjugate (ADC) consisting of three components: 1) an anti-FRα monoclonal antibody of IgG1 subtype 2) the small molecule anti-tubulin agent DM4 (a maytansine derivative) and 3) a linker, sulfo-SPDB (1-(2,5-dioxopyrrolidin-1-yl)oxy-1-oxo-4-(pyridin-2-yldisulfanyl)butane-2-sulfonic acid) that covalently attaches DM4 to the mirvetuximab antibody. Mirvetuximab soravtansine-gynx has an approximate molecular weight of 150 kDa. An average of 3.4 molecules of DM4 are attached to each antibody molecule. Mirvetuximab soravtansine-gynx is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
Mirvetuximab soravtansine-gynx has the following structure:
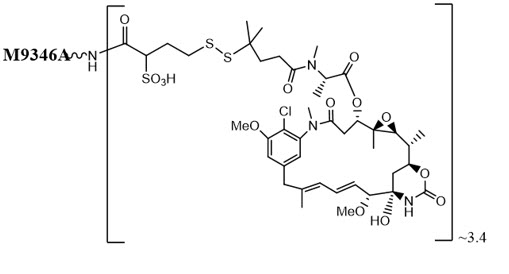
ELAHERE (mirvetuximab soravtansine-gynx) injection is supplied as a sterile, preservative-free, clear to slightly opalescent, colorless solution containing 100 mg/20 mL of mirvetuximab soravtansine-gynx in single-dose vials. Each mL of solution contains 5 mg of mirvetuximab soravtansine-gynx, and glacial acetic acid (0.22 mg), polysorbate 20 (0.1 mg), sodium acetate (0.53 mg), sucrose (90 mg), and Water for Injection. The pH is approximately 5.0.
The ELAHERE vial stoppers are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Mirvetuximab soravtansine-gynx is an antibody-drug conjugate (ADC). The antibody is a chimeric IgG1 directed against folate receptor alpha (FRα). The small molecule, DM4, is a microtubule inhibitor attached to the antibody via a cleavable linker. Upon binding to FRα, mirvetuximab soravtansine-gynx is internalized followed by intracellular release of DM4 via proteolytic cleavage. DM4 disrupts the microtubule network within the cell, resulting in cell cycle arrest and apoptotic cell death.
12.2 Pharmacodynamics
Exposure-Response Relationships
Higher exposure to mirvetuximab soravtansine-gynx was associated with higher overall response rates and longer median PFS and OS; higher exposure to mirvetuximab soravtansine-gynx was also associated with higher incidence of ocular adverse reactions as well as marginally increased peripheral neuropathy.
Cardiac Electrophysiology
At the approved recommended dose, ELAHERE did not cause large mean increases (>10 msec) in the QTc interval.
12.3 Pharmacokinetics
The pharmacokinetics were characterized in patients who received mirvetuximab soravtansine-gynx 0.16 mg/kg to 8.7 mg/kg adjusted ideal body weight (AIBW) (0.03 times to 1.4 times the approved recommended dose of 6 mg/kg AIBW), unless otherwise noted.
Table 8 summarizes the exposure parameters of mirvetuximab soravtansine-gynx, unconjugated DM4, and its metabolite S-methyl-DM4 following administration after the first cycle (3-weeks). Peak mirvetuximab soravtansine-gynx concentrations were observed near the end of intravenous infusion, while peak unconjugated DM4 concentrations were observed on the second day after administration and the peak S-methyl-DM4 concentrations were observed approximately 3 days after administration. Steady state concentrations of mirvetuximab soravtansine-gynx, DM4, and S-methyl-DM4 were reached after one 3-week cycle. Accumulation of the mirvetuximab soravtansine-gynx, DM4, and S-methyl-DM4 was minimal following multiple cycles.
Table 8: Exposure Parameters of Mirvetuximab Soravtansine-gynx, Unconjugated DM4, and S-methyl DM4 After First Cycle at a Dosage of 6 mg/kg Mirvetuximab Soravtansine-gynx
Mean (±SD)Unconjugated DM4
Mean (±SD)S-methyl-DM4
Mean (±SD)Cmax 137.3 (±62.3) µg/mL 4.1 (±2.3) ng/mL 7.0 (±6.8) ng/mL AUCtau 20.6 (±6.8) h*mg/mL 530 (±245) h*ng/mL 1848 (±1585) h*ng/mL Cmax = maximum concentration, AUCtau = area under the concentration vs. time curve over the dosing interval (21 days).
Distribution
The mean (±SD) steady state volume of distribution of mirvetuximab soravtansine-gynx was 2.6 (±2.9) L.
Human plasma protein binding of DM4 and S-methyl DM4 was >99%, in vitro.
Elimination
For mirvetuximab soravtansine-gynx, total plasma clearance (mean [CV%]) of was 19 mL/hour (52%) and the mean terminal phase half-life after the first dose was 4.8 days leading to a steady state at approximately 24 days.
For the unconjugated DM4, the total plasma clearance (mean [CV%]) was 14 L/hour (31%) and the mean terminal phase half-life was 2.8 days.
For S-methyl-DM4, the total plasma clearance (mean [CV%]) was 4.3 L/hour (64%) and the mean terminal phase half-life was 5 days.
Metabolism
The monoclonal antibody portion of mirvetuximab soravtansine-gynx is expected to be metabolized into small peptides by catabolic pathways. Unconjugated DM4 and S-methyl-DM4 undergo metabolism by CYP3A4. In human plasma, DM4 and S-methyl DM4 were identified as the main circulating metabolites, accounting for approximately 0.4% and 1.4% of mirvetuximab soravtansine-gynx AUC, respectively.
Excretion
S-methyl DM4 and DM4-sulfo-SPDB-lysine were detected in urine within 24 hours of infusion as the main metabolites.
Specific Populations
No clinically significant differences in the pharmacokinetics of mirvetuximab soravtansine-gynx were observed based on age (32 to 89 years), race (White, Black, or Asian), body weight (36 to 136 kg), mild hepatic impairment (total bilirubin ≤ULN and any AST >ULN or total bilirubin >1 to 1.5 times ULN and any AST), or mild to moderate renal impairment (CLcr 30 to 89 mL/min).
The pharmacokinetics of mirvetuximab soravtansine-gynx in patients with moderate to severe hepatic impairment (total bilirubin >1.5 ULN with any AST) or severe renal impairment (CLcr 15 to 30 mL/min) is unknown.
Drug Interaction Studies
Clinical Studies and Model Informed Approaches
No clinical studies to evaluate the drug-drug interaction potential of mirvetuximab soravtansine-gynx were conducted.
There were no differences in exposure between patients who received concomitant weak or moderate CYP3A4 inhibitors or P-glycoprotein (P-gp) inhibitors and those who did not.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Unconjugated DM4 is a time-dependent inhibitor of CYP3A4. Unconjugated DM4 and S-methyl DM4 are not inhibitors of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A. DM4 and S-methyl DM4 are not inducers of CYP1A2, CYP2B6, or CYP3A4.
Transporter Systems: Unconjugated DM4 and S-methyl DM4 are substrates of P-gp but are not inhibitors of P-gp.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA), including neutralizing antibody, is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADAs in the studies described below with the incidence of ADAs to mirvetuximab soravtansine-gynx in other studies.
With a median treatment duration of 4.4 months in Studies 0416, 0417, 0401, and 0403, in patients with epithelial ovarian, fallopian tube, or primary peritoneal cancer who received mirvetuximab soravtansine-gynx at 6 mg/kg AIBW intravenously every 3 weeks, 9% (57/626) of patients developed anti-mirvetuximab soravtansine-gynx antibodies. Neutralizing antibodies were detected in 47% (27/57) of patients who were ADA-positive.
No clinically meaningful difference was observed in the trough concentrations of mirvetuximab soravtansine-gynx between ADA-positive and ADA-negative patients. Anti-mirvetuximab soravtansine-gynx antibody formation was associated with a higher incidence of infusion-related reactions [see Adverse Reactions (6.1)]. The effect of anti-drug antibodies on effectiveness has not been fully characterized. Based on limited data, the presence of anti-mirvetuximab soravtansine-gynx antibodies may be associated with decreased efficacy in ADA-positive patients when compared to ADA-negative patients.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with mirvetuximab soravtansine-gynx or DM4.
DM4 and the metabolite, S-methyl DM4, were clastogenic in the in vivo rat bone marrow micronucleus study. DM4 and S-methyl DM4 were not mutagenic in the bacterial reverse mutation (Ames) assay.
Fertility studies have not been conducted with mirvetuximab soravtansine-gynx or DM4.
-
14 CLINICAL STUDIES
Study 0416
The efficacy of ELAHERE was evaluated in Study 0416 (MIRASOL, NCT04209855), a multicenter, open-label, active-controlled, randomized, two-arm, trial in patients (n=453) with FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer. Patients were permitted to receive up to three prior lines of systemic therapy. The trial enrolled patients whose tumors were positive for FRα expression as determined by the VENTANA FOLR1 (FOLR1-2.1) RxDx Assay. Patients were excluded if they had corneal disorders, ocular conditions requiring ongoing treatment, Grade >1 peripheral neuropathy, or noninfectious interstitial lung disease.
Patients were randomized (1:1) to receive ELAHERE 6 mg/kg (based on adjusted ideal body weight) as an intravenous infusion every 3 weeks or investigator’s choice of chemotherapy (paclitaxel, pegylated liposomal doxorubicin [PLD], or topotecan) until disease progression or unacceptable toxicity. Tumor response assessments occurred every 6 weeks for the first 36 weeks and every 12 weeks thereafter. Randomization was stratified by the following factors: number of prior lines of therapy (1 vs. 2 vs. 3) and chemotherapy (paclitaxel vs. PLD vs. topotecan) chosen prior to randomization.
The major efficacy outcome measures were investigator-assessed progression-free survival (PFS), confirmed overall response rate (ORR), and overall survival (OS). PFS and ORR were evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.
The median age was 63 years (range: 29 to 88); 66% were White, 12% were Asian, 3% were Black or African American, and 18% did not have race reported. Six percent of patients were Hispanic or Latino. Nearly all patients had an ECOG PS of 0 (55%) or 1 (44%). Fourteen percent of patients had received 1 prior line of systemic therapy, 39% of patients had received 2 prior lines of systemic therapy, and 47% of patients had received 3 prior lines of systemic therapy. Thirty-seven percent of patients received prior systemic therapy for platinum-resistant disease. Sixty-two percent of patients received prior bevacizumab and 55% had received a prior PARP inhibitor.
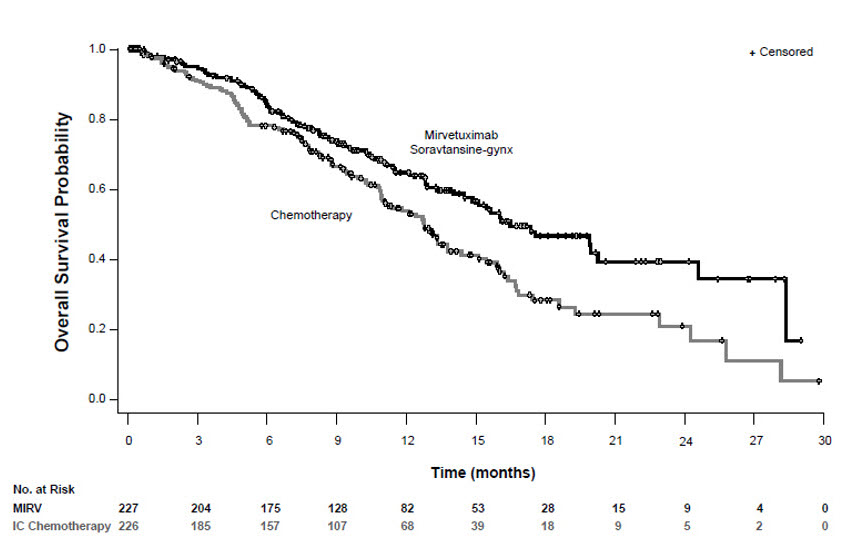
ELAHERE demonstrated a statistically significant improvement in PFS, ORR, and OS for patients randomized to ELAHERE as compared with chemotherapy.
Efficacy results for Study 0416 are summarized in Table 9 and Figures 1 and 2.
Table 9: Efficacy Results in Study 0416 ELAHERE
n=227Chemotherapy*
n=226Progression-free survival (PFS) Number (%) of patients with events 176 (78) 166 (73) Median, months (95% CI) 5.6 (4.3, 5.9) 4.0 (2.9, 4.5) Hazard ratio (95% CI) 0.65 (0.52, 0.81) p-valuea <0.0001 Overall Survival (OS) Number (%) of patients with events 90 (40) 114 (50) Median, months (95% CI) 16.5 (14.5, 24.6) 12.7 (10.9, 14.4) Hazard Ratio (95% CI) 0.67 (0.50, 0.88) p-valuea 0.0046 Confirmed overall response rate (ORR) Number of patients with measurable disease at baseline 225 224 ORR (95% CI) 42% (36, 49) 16% (12, 22) Complete response 5% 0% Partial response 37% 16% p-valueb <0.0001 * Chemotherapy: paclitaxel, PLD, or topotecan.
a Two-sided p-value based on stratified log-rank test.
b Two-sided p-value based upon Cochran-Mantel-Haenszel (CMH) test.
Figure 1: Kaplan-Meier Curve for Progression-free Survival in Study 0416
Figure 2: Kaplan-Meier Curve for Overall Survival in Study 0416
Study 0417
The efficacy of ELAHERE was evaluated in Study 0417 (SORAYA, NCT04296890), a single-arm trial of patients with FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer (n=106). Patients were permitted to receive up to three prior lines of systemic therapy. All patients were required to have received prior bevacizumab. The trial enrolled patients whose tumors were positive for FRα expression as determined by the VENTANA FOLR1 (FOLR1-2.1) RxDx Assay. Patients were excluded if they had corneal disorders, ocular conditions requiring ongoing treatment, Grade >1 peripheral neuropathy, or noninfectious interstitial lung disease.
Patients received ELAHERE 6 mg/kg (based on adjusted ideal body weight) as an intravenous infusion every 3 weeks until disease progression or unacceptable toxicity. Tumor response assessments occurred every 6 weeks for the first 36 weeks and every 12 weeks thereafter.
The major efficacy outcome measures were investigator-assessed overall response rate (ORR) and duration of response (DOR) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.
The efficacy evaluable population included 104 patients with platinum-resistant disease, who had measurable disease, and received at least one dose of ELAHERE. In these 104 patients, the median age was 62 years (range: 35 to 85); 96% were White, 2% were Asian, and 2% did not have race reported. Two percent of patients were Hispanic or Latino. All patients had an ECOG PS of 0 (57%) or 1 (43%). Ten percent of patients had received 1 prior line of systemic therapy, 39% of patients had received 2 prior lines of systemic therapy, and 50% of patients had received 3 prior lines of systemic therapy. All patients had received prior bevacizumab and 47% had received a prior PARP inhibitor.
Efficacy results for Study 0417 are summarized in Table 10.
Table 10: Efficacy Results in Study 0417 ELAHERE
(N=104)Confirmed Overall Response Ratea
(95% CI)32%
(23, 42)Complete response rate 5% Partial response rate 27% Duration of Response N=33 Median duration of response, months
(95% CI)6.9
(5.6, 9.7)a Investigator assessment.
Response assessment results using independent radiology review were consistent with investigator assessment.
-
15 REFERENCES
1“OSHA Hazardous Drugs.” OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Each ELAHERE (mirvetuximab soravtansine-gynx) injection carton (NDC: 72903-853-01) contains:
- One single-dose vial containing 100 mg of mirvetuximab soravtansine-gynx in 20 mL (5 mg/mL) of clear to slightly opalescent, colorless sterile solution.
Storage and Handling
Store ELAHERE vials upright in a refrigerator at 2°C to 8°C (36°F to 46°F) until the time of preparation in the original carton to protect from light.
Do not freeze or shake.
ELAHERE is a hazardous drug. Follow applicable special handling and disposal procedures1.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Ocular Disorders
Inform patients about the need for eye exams before and during treatment with ELAHERE.
Advise patients to contact their healthcare provider promptly if they experience any visual changes. Advise patients to use steroid eye drops and artificial tear substitutes [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
Pneumonitis
Advise patients to immediately report new or worsening respiratory symptoms [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise female patients to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (5.4, 8.1, 8.3)].
Advise females of reproductive potential to use effective contraception during treatment with ELAHERE and for 7 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with ELAHERE and for 1 month after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
AbbVie Inc.
North Chicago, IL 60064, USA
U.S. License 1889©2025 AbbVie Inc. All rights reserved.
ELAHERE and its design are trademarks of ImmunoGen, Inc., an AbbVie company.
752962-02 -
MEDICATION GUIDE
MEDICATION GUIDE
ELAHERE® (el-ah-HERE)
(mirvetuximab soravtansine-gynx)
injection, for intravenous useWhat is the most important information I should know about ELAHERE?
ELAHERE can cause serious side effects, including:
-
Eye problems. Eye problems are common with ELAHERE and can also be severe. Tell your healthcare provider right away if you develop any eye problems during treatment with ELAHERE, including blurred vision, dry eyes, sensitivity to light, eye pain, eye redness, or new or worsening vision changes.
- Your healthcare provider will send you to see an eye care professional to check your eyes before you start treatment with ELAHERE, during treatment with ELAHERE, and as needed for any worsening signs and symptoms of eye problems.
- Your healthcare provider will prescribe steroid eye drops and lubricating eye drops before you start and during your treatment with ELAHERE. You should use eye drops as directed by your healthcare provider.
- Do not wear contact lenses throughout your treatment with ELAHERE unless you are told to use them by your healthcare provider.
- Your healthcare provider will send you to see an eye care professional to check your eyes before you start treatment with ELAHERE, during treatment with ELAHERE, and as needed for any worsening signs and symptoms of eye problems.
What is ELAHERE?
ELAHERE is a prescription medicine used to treat adults with folate receptor-alpha positive ovarian cancer, fallopian tube cancer, or primary peritoneal cancer who:
- have not responded to or are no longer responding to treatment with platinum-based chemotherapy, and
- have received 1 to 3 prior types of chemotherapy.
It is not known if ELAHERE is safe and effective in children.Before receiving ELAHERE, tell your healthcare provider about all of your medical conditions, including if you:
- have vision or eye problems.
- have numbness or tingling in your hands or feet.
- have liver problems.
- are pregnant or plan to become pregnant. ELAHERE can harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with ELAHERE.
Patients who are able to become pregnant:- Your healthcare provider should do a pregnancy test before you start treatment with ELAHERE.
- You should use an effective birth control (contraception) during treatment and for 7 months after your last dose of ELAHERE.
- Your healthcare provider should do a pregnancy test before you start treatment with ELAHERE.
- are breastfeeding or plan to breastfeed. It is not known if ELAHERE passes into your breast milk. Do not breastfeed during treatment and for 1 month after your last dose of ELAHERE.
How will I receive ELAHERE? - ELAHERE will be given to you by infusion into your vein (intravenous or IV).
- Before each dose of ELAHERE you will receive medicines to help prevent infusion related reactions, nausea, and vomiting.
- ELAHERE is usually given every 3 weeks (21-day cycle). Your healthcare provider will decide how many cycles you need.
What are the possible side effects of ELAHERE?
ELAHERE can cause serious side effects, including:
- See “What is the most important information I should know about ELAHERE?”
-
Lung problems (pneumonitis). ELAHERE can cause severe or life-threatening inflammation of the lungs that may lead to death. Tell your healthcare provider right away if you get new or worsening symptoms, including trouble breathing, shortness of breath, cough, or chest pain.
- Peripheral neuropathy. Nerve problems called peripheral neuropathy are common with ELAHERE and can also be severe. Your healthcare provider will monitor you for signs and symptoms of nerve problems. Tell your healthcare provider if you get new or worsening numbness, tingling, burning sensation or pain in your hands or feet or muscle weakness.
- increased liver enzymes in the blood
- decreased platelets
- feeling tired
- decreased magnesium level in the blood
- blurred vision
- dry eye
- nausea
- constipation
- diarrhea
- vomiting
- stomach-area (abdominal) pain
- decreased albumin level in the blood
- changes in the cornea (part of the eye)
- decreased appetite
- peripheral neuropathy
- muscle, bone, or joint pain
- decreased red or white blood cell counts
Your healthcare provider may change your dose of ELAHERE, delay treatment, or completely stop treatment if you have certain side effects.
These are not all of the possible side effects of ELAHERE.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of ELAHERE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you would like more information about ELAHERE, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about ELAHERE that is written for healthcare professionals.What are the ingredients in ELAHERE?
Active ingredient: mirvetuximab soravtansine-gynx
Inactive ingredients: glacial acetic acid, polysorbate 20, sodium acetate, sucrose, Water for Injection.
Manufactured by: AbbVie Inc., North Chicago, IL 60064, USA
U.S. License 1889
©2025 AbbVie Inc. All rights reserved.
ELAHERE and its design are trademarks of ImmunoGen, Inc., an AbbVie company.
752962-02
For more information, go to www.elahere.com or call 1-833-ELAHERE.This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised 07/2025
-
Eye problems. Eye problems are common with ELAHERE and can also be severe. Tell your healthcare provider right away if you develop any eye problems during treatment with ELAHERE, including blurred vision, dry eyes, sensitivity to light, eye pain, eye redness, or new or worsening vision changes.
-
PRINCIPAL DISPLAY PANEL
NDC: 72903-853-01
Rx Only
ELAHERE®
mirvetuximab soravtansine-gynx
Injection
100mg/20ml
(5mg/ml)
For Intravenous Infusion after
Dilution in 5% Dextrose Injection, USP
Single-dose vial.
Discard unused portion
Warning: Hazardous Drug
Dispense the enclosed
Medication Guide to each patient.
abbvie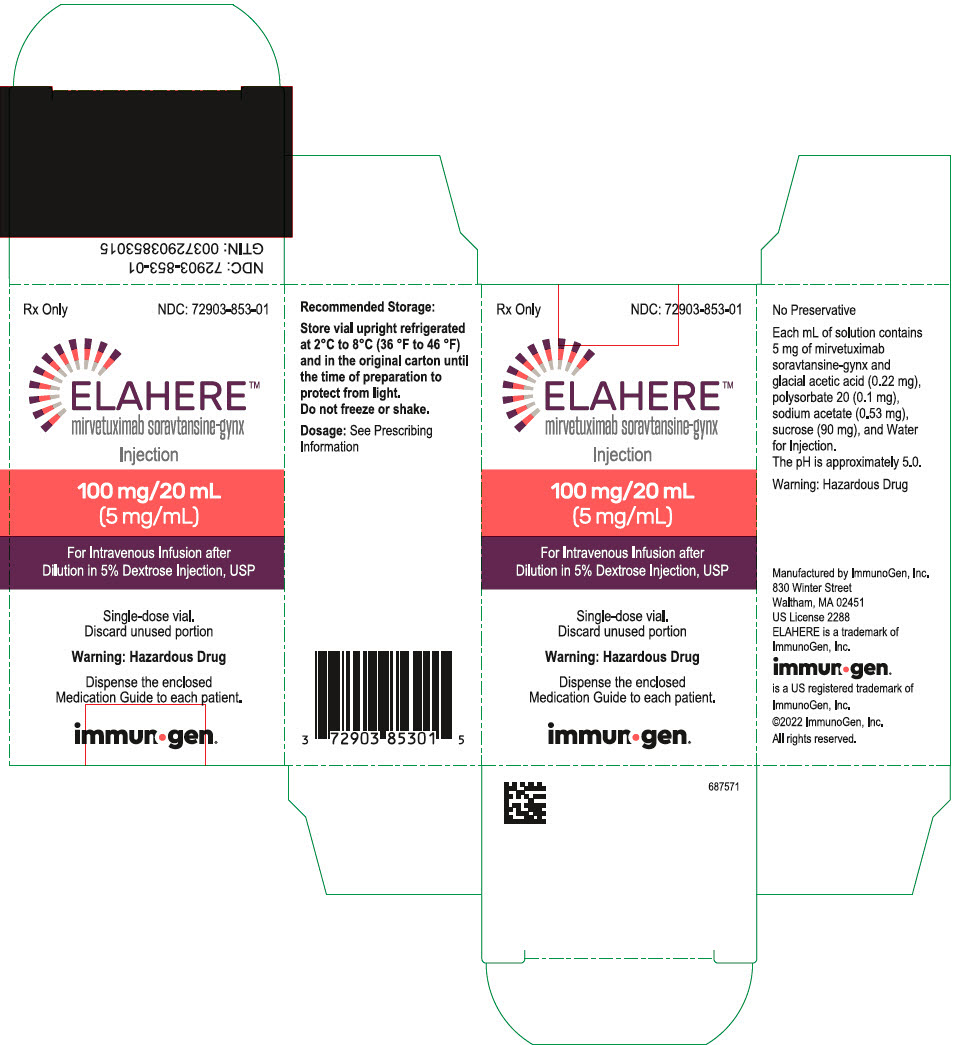
-
INGREDIENTS AND APPEARANCE
ELAHERE
mirvetuximab soravtansine injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72903-853 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MIRVETUXIMAB SORAVTANSINE (UNII: 98DE7VN88D) (MIRVETUXIMAB SORAVTANSINE - UNII:98DE7VN88D) MIRVETUXIMAB SORAVTANSINE 5 mg in 1 mL Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) 0.22 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.1 mg in 1 mL SODIUM ACETATE (UNII: 4550K0SC9B) 0.53 mg in 1 mL SUCROSE (UNII: C151H8M554) 90 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72903-853-01 1 in 1 CARTON 11/14/2022 1 20 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761310 11/14/2022 Labeler - ImmunoGen, Inc. (011991874)
Trademark Results [ELAHERE]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ELAHERE 97693875 not registered Live/Pending |
ImmunoGen, Inc. 2022-11-28 |
 ELAHERE 97676672 not registered Live/Pending |
ImmunoGen, Inc. 2022-11-14 |
 ELAHERE 97676668 not registered Live/Pending |
ImmunoGen, Inc. 2022-11-14 |
 ELAHERE 90488884 not registered Live/Pending |
ImmunoGen, Inc. 2021-01-26 |
 ELAHERE 90009729 not registered Live/Pending |
ImmunoGen, Inc. 2020-06-18 |
 ELAHERE 88268164 not registered Live/Pending |
ImmunoGen, Inc. 2019-01-18 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.