DOCETAXEL- docetaxel anhydrous injection, solution
Docetaxel by
Drug Labeling and Warnings
Docetaxel by is a Prescription medication manufactured, distributed, or labeled by Hospira, Inc., Zydus Hospira Oncology Private Limited, Hospira Australia Pty Ltd. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DOCETAXEL INJECTION safely and effectively. See full prescribing information for DOCETAXEL INJECTION.
DOCETAXEL injection, for intravenous use
Initial U.S. Approval: 1996WARNING: TOXIC DEATHS, HEPATOTOXICITY, NEUTROPENIA, HYPERSENSITIVITY REACTIONS, and FLUID RETENTION
See full prescribing information for complete boxed warning.
- Treatment-related mortality increases with abnormal liver function, at higher doses, and in patients with NSCLC and prior platinum-based therapy receiving docetaxel at 100 mg/m2 (5.1)
- Avoid use of Docetaxel Injection if bilirubin > ULN, or if AST and/or ALT >1.5 × ULN concomitant with alkaline phosphatase >2.5 × ULN. LFT elevations increase risk of severe or life-threatening complications. Obtain LFTs before each treatment cycle (5.2)
- Do not administer Docetaxel Injection to patients with neutrophil counts <1500 cells/mm3. Obtain frequent blood counts to monitor for neutropenia (4, 5.3)
- Severe hypersensitivity, including fatal anaphylaxis, has been reported in patients who received dexamethasone premedication. Severe reactions require immediate discontinuation of Docetaxel Injection and administration of appropriate therapy (5.5)
- Contraindicated if history of severe hypersensitivity reactions to docetaxel or to drugs formulated with polysorbate 80 (4)
- Severe fluid retention may occur despite dexamethasone (5.6)
INDICATIONS AND USAGE
Docetaxel Injection is a microtubule inhibitor indicated for:
- Breast Cancer (BC): single agent for locally advanced or metastatic BC after chemotherapy failure; and with doxorubicin and cyclophosphamide as adjuvant treatment of operable node-positive BC (1.1)
- Non-small Cell Lung Cancer (NSCLC): single agent for locally advanced or metastatic NSCLC after platinum therapy failure; and with cisplatin for unresectable, locally advanced or metastatic untreated NSCLC (1.2)
- Castration-Resistant Prostate Cancer (CRPC): with prednisone in metastatic castration-resistant prostate cancer (1.3)
- Gastric Adenocarcinoma (GC): with cisplatin and fluorouracil for untreated, advanced GC, including the gastroesophageal junction (1.4)
- Squamous Cell Carcinoma of the Head and Neck (SCCHN): with cisplatin and fluorouracil for induction treatment of locally advanced SCCHN (1.5)
DOSAGE AND ADMINISTRATION
Administer in a facility equipped to manage possible complications (e.g., anaphylaxis). Administer intravenously (IV) over 1 hour every 3 weeks. PVC equipment is not recommended. Use only a 21 gauge needle to withdraw Docetaxel Injection from the vial.
- BC locally advanced or metastatic: 60 mg/m2 to 100 mg/m2 single agent (2.1)
- BC adjuvant: 75 mg/m2 administered 1 hour after doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2 every 3 weeks for 6 cycles (2.1)
- NSCLC: after platinum therapy failure: 75 mg/m2 single agent (2.2)
- NSCLC: chemotherapy-naïve: 75 mg/m2 followed by cisplatin 75 mg/m2 (2.2)
- CRPC: 75 mg/m2 with 5 mg prednisone twice a day continuously (2.3)
- GC: 75 mg/m2 followed by cisplatin 75 mg/m2 (both on day 1 only) followed by fluorouracil 750 mg/m2 per day as a 24-hour IV (days 1–5), starting at end of cisplatin infusion (2.4)
- SCCHN: 75 mg/m2 followed by cisplatin 75 mg/m2 IV (day 1), followed by fluorouracil 750 mg/m2 per day as a 24-hour IV (days 1–5), starting at end of cisplatin infusion; for 4 cycles (2.5)
- SCCHN: 75 mg/m2 followed by cisplatin 100 mg/m2 IV (day 1), followed by fluorouracil 1000 mg/m2 per day as a 24-hour IV (days 1–4); for 3 cycles (2.5)
For all patients:
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Second primary malignancies: In patients treated with Docetaxel Injection-containing regimens, monitor for delayed AML, MDS, NHL, and renal cancer. (5.7)
- Cutaneous reactions: Reactions including erythema of the extremities with edema followed by desquamation may occur. Severe cutaneous adverse reactions have been reported. Severe skin toxicity may require dose adjustment or permanent treatment discontinuation. (5.8)
- Neurologic reactions: Reactions including paresthesia, dysesthesia, and pain may occur. Severe neurosensory symptoms require dose adjustment or discontinuation if persistent. (5.9)
- Eye disorders: Cystoid macular edema (CME) has been reported and requires treatment discontinuation. (5.10)
- Asthenia: Severe asthenia may occur and may require treatment discontinuation. (5.11)
- Embryo-fetal toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.12, 8.1, 8.3)
- Alcohol content: The alcohol content in a dose of Docetaxel Injection may affect the central nervous system. This may include impairment of a patient's ability to drive or use machines immediately after infusion. (5.13)
- Tumor lysis syndrome: Tumor lysis syndrome has been reported. Patients at risk should be well hydrated and closely monitored during treatment. (5.14)
ADVERSE REACTIONS
Most common adverse reactions across all docetaxel indications are infections, neutropenia, anemia, febrile neutropenia, hypersensitivity, thrombocytopenia, neuropathy, dysgeusia, dyspnea, constipation, anorexia, nail disorders, fluid retention, asthenia, pain, nausea, diarrhea, vomiting, mucositis, alopecia, skin reactions, and myalgia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Hospira, Inc. at 1-800-441-4100 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Cytochrome P450 3A4 inducers, inhibitors, or substrates: May alter docetaxel metabolism. (7)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: TOXIC DEATHS, HEPATOTOXICITY, NEUTROPENIA, HYPERSENSITIVITY REACTIONS, and FLUID RETENTION
1 INDICATIONS AND USAGE
1.1 Breast Cancer
1.2 Non-small Cell Lung Cancer
1.3 Prostate Cancer
1.4 Gastric Adenocarcinoma
1.5 Head and Neck Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Breast Cancer
2.2 Non-small Cell Lung Cancer
2.3 Prostate Cancer
2.4 Gastric Adenocarcinoma
2.5 Head and Neck Cancer
2.6 Premedication Regimen
2.7 Dosage Adjustments during Treatment
2.8 Administration Precautions
2.9 Preparation and Administration
2.10 Stability
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Toxic Deaths
5.2 Hepatic Impairment
5.3 Hematologic Effects
5.4 Enterocolitis and Neutropenic Colitis
5.5 Hypersensitivity Reactions
5.6 Fluid Retention
5.7 Second Primary Malignancies
5.8 Cutaneous Reactions
5.9 Neurologic Reactions
5.10 Eye Disorders
5.11 Asthenia
5.12 Embryo-Fetal Toxicity
5.13 Alcohol Content
5.14 Tumor Lysis Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic Breast Cancer
14.2 Adjuvant Treatment of Breast Cancer
14.3 Non-small Cell Lung Cancer (NSCLC)
14.4 Castration-Resistant Prostate Cancer
14.5 Gastric Adenocarcinoma
14.6 Head and Neck Cancer
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
16.3 Handling and Disposal
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: TOXIC DEATHS, HEPATOTOXICITY, NEUTROPENIA, HYPERSENSITIVITY REACTIONS, and FLUID RETENTION
Treatment-related mortality associated with docetaxel is increased in patients with abnormal liver function, in patients receiving higher doses, and in patients with non-small cell lung carcinoma and a history of prior treatment with platinum-based chemotherapy who receive docetaxel as a single agent at a dose of 100 mg/m2 [see Warnings and Precautions (5.1)].
Avoid the use of Docetaxel Injection in patients with bilirubin > upper limit of normal (ULN), or to patients with AST and/or ALT >1.5 × ULN concomitant with alkaline phosphatase >2.5 × ULN. Patients with elevations of bilirubin or abnormalities of transaminase concurrent with alkaline phosphatase are at increased risk for the development of severe neutropenia, febrile neutropenia, infections, severe thrombocytopenia, severe stomatitis, severe skin toxicity, and toxic death. Patients with isolated elevations of transaminase >1.5 × ULN also had a higher rate of febrile neutropenia. Measure bilirubin, AST or ALT, and alkaline phosphatase prior to each cycle of Docetaxel Injection [see Warnings and Precautions (5.2)].
Do not administer Docetaxel Injection to patients with neutrophil counts of <1500 cells/mm3. Monitor blood counts frequently as neutropenia may be severe and result in infection [see Warnings and Precautions (5.3)].
Do not administer Docetaxel Injection to patients who have a history of severe hypersensitivity reactions to docetaxel or to other drugs formulated with polysorbate 80 [see Contraindications (4)]. Severe hypersensitivity reactions have been reported in patients despite dexamethasone premedication. Hypersensitivity reactions require immediate discontinuation of the Docetaxel Injection infusion and administration of appropriate therapy [see Warnings and Precautions (5.5)].
Severe fluid retention occurred in 6.5% (6/92) of patients despite use of dexamethasone premedication. It was characterized by one or more of the following events: poorly tolerated peripheral edema, generalized edema, pleural effusion requiring urgent drainage, dyspnea at rest, cardiac tamponade, or pronounced abdominal distention (due to ascites) [see Warnings and Precautions (5.6)].
-
1 INDICATIONS AND USAGE
1.1 Breast Cancer
Docetaxel Injection is indicated for the treatment of patients with locally advanced or metastatic breast cancer after failure of prior chemotherapy.
Docetaxel Injection in combination with doxorubicin and cyclophosphamide is indicated for the adjuvant treatment of patients with operable node-positive breast cancer.
1.2 Non-small Cell Lung Cancer
Docetaxel Injection as a single agent is indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of prior platinum-based chemotherapy.
Docetaxel Injection in combination with cisplatin is indicated for the treatment of patients with unresectable, locally advanced or metastatic non-small cell lung cancer who have not previously received chemotherapy for this condition.
1.3 Prostate Cancer
Docetaxel Injection in combination with prednisone is indicated for the treatment of patients with metastatic castration-resistant prostate cancer.
-
2 DOSAGE AND ADMINISTRATION
For all indications, toxicities may warrant dosage adjustments [see Dosage and Administration (2.7)].
Administer in a facility equipped to manage possible complications (e.g., anaphylaxis).
2.1 Breast Cancer
- For locally advanced or metastatic breast cancer after failure of prior chemotherapy, the recommended dose of Docetaxel Injection is 60 mg/m2 to 100 mg/m2 administered intravenously over 1 hour every 3 weeks.
- For the adjuvant treatment of operable node-positive breast cancer, the recommended Docetaxel Injection dose is 75 mg/m2 administered 1 hour after doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2 every 3 weeks for 6 courses. Prophylactic G-CSF may be used to mitigate the risk of hematological toxicities [see Dosage and Administration (2.7)].
2.2 Non-small Cell Lung Cancer
- For treatment after failure of prior platinum-based chemotherapy, docetaxel was evaluated as monotherapy, and the recommended dose is 75 mg/m2 administered intravenously over 1 hour every 3 weeks. A dose of 100 mg/m2 in patients previously treated with chemotherapy was associated with increased hematologic toxicity, infection, and treatment-related mortality in randomized controlled trials [see Boxed Warning, Dosage and Administration (2.7), Warnings and Precautions (5), Clinical Studies (14)].
- For chemotherapy-naïve patients, docetaxel was evaluated in combination with cisplatin. The recommended dose of Docetaxel Injection is 75 mg/m2 administered intravenously over 1 hour immediately followed by cisplatin 75 mg/m2 over 30–60 minutes every 3 weeks [see Dosage and Administration (2.7)].
2.3 Prostate Cancer
- For metastatic castration-resistant prostate cancer, the recommended dose of Docetaxel Injection is 75 mg/m2 every 3 weeks as a 1 hour intravenous infusion. Prednisone 5 mg orally twice daily is administered continuously [see Dosage and Administration (2.7)].
2.4 Gastric Adenocarcinoma
- For gastric adenocarcinoma, the recommended dose of Docetaxel Injection is 75 mg/m2 as a 1 hour intravenous infusion, followed by cisplatin 75 mg/m2, as a 1 to 3 hour intravenous infusion (both on day 1 only), followed by fluorouracil 750 mg/m2 per day given as a 24-hour continuous intravenous infusion for 5 days, starting at the end of the cisplatin infusion. Treatment is repeated every three weeks. Patients must receive premedication with antiemetics and appropriate hydration for cisplatin administration [see Dosage and Administration (2.7)].
2.5 Head and Neck Cancer
Patients must receive premedication with antiemetics, and appropriate hydration (prior to and after cisplatin administration). Prophylaxis for neutropenic infections should be administered. All patients treated on the Docetaxel Injection containing arms of the TAX323 and TAX324 studies received prophylactic antibiotics.
Induction Chemotherapy Followed by Radiotherapy (TAX323)
For the induction treatment of locally advanced inoperable SCCHN, the recommended dose of Docetaxel Injection is 75 mg/m2 as a 1 hour intravenous infusion followed by cisplatin 75 mg/m2 intravenously over 1 hour, on day one, followed by fluorouracil as a continuous intravenous infusion at 750 mg/m2 per day for five days. This regimen is administered every 3 weeks for 4 cycles. Following chemotherapy, patients should receive radiotherapy [see Dosage and Administration (2.7)].
Induction Chemotherapy Followed by Chemoradiotherapy (TAX324)
For the induction treatment of patients with locally advanced (unresectable, low surgical cure, or organ preservation) SCCHN, the recommended dose of Docetaxel Injection is 75 mg/m2 as a 1 hour intravenous infusion on day 1, followed by cisplatin 100 mg/m2 administered as a 30-minute to 3 hour infusion, followed by fluorouracil 1000 mg/m2/day as a continuous infusion from day 1 to day 4. This regimen is administered every 3 weeks for 3 cycles. Following chemotherapy, patients should receive chemoradiotherapy [see Dosage and Administration (2.7)].
2.6 Premedication Regimen
All patients should be premedicated with oral corticosteroids (see below for prostate cancer) such as dexamethasone 16 mg per day (e.g., 8 mg twice daily) for 3 days starting 1 day prior to Docetaxel Injection administration in order to reduce the incidence and severity of fluid retention as well as the severity of hypersensitivity reactions [see Boxed Warning, Warnings and Precautions (5.5)].
For metastatic castration-resistant prostate cancer, given the concurrent use of prednisone, the recommended premedication regimen is oral dexamethasone 8 mg at 12 hours, 3 hours, and 1 hour before the Docetaxel Injection infusion [see Warnings and Precautions (5.5)].
2.7 Dosage Adjustments during Treatment
Breast Cancer
Patients who are dosed initially at 100 mg/m2 and who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than 1 week, or severe or cumulative cutaneous reactions during Docetaxel Injection therapy should have the dosage adjusted from 100 mg/m2 to 75 mg/m2. If the patient continues to experience these reactions, the dosage should either be decreased from 75 mg/m2 to 55 mg/m2 or the treatment should be discontinued. Conversely, patients who are dosed initially at 60 mg/m2 and who do not experience febrile neutropenia, neutrophils <500 cells/mm3 for more than 1 week, severe or cumulative cutaneous reactions, or severe peripheral neuropathy during Docetaxel Injection therapy may tolerate higher doses. Patients who develop ≥grade 3 peripheral neuropathy should have Docetaxel Injection treatment discontinued entirely.
Combination Therapy with Docetaxel Injection in the Adjuvant Treatment of Breast Cancer
Docetaxel Injection in combination with doxorubicin and cyclophosphamide should be administered when the neutrophil count is ≥1500 cells/mm3. Patients who experience febrile neutropenia should receive G-CSF in all subsequent cycles. Patients who continue to experience this reaction should remain on G-CSF and have their Docetaxel Injection dose reduced to 60 mg/m2. Patients who experience grade 3 or 4 stomatitis should have their Docetaxel Injection dose decreased to 60 mg/m2. Patients who experience severe or cumulative cutaneous reactions or moderate neurosensory signs and/or symptoms during Docetaxel Injection therapy should have their dosage of Docetaxel Injection reduced from 75 mg/m2 to 60 mg/m2. If the patient continues to experience these reactions at 60 mg/m2, treatment should be discontinued.
Non-small Cell Lung Cancer
Monotherapy with Docetaxel Injection for NSCLC Treatment after Failure of Prior Platinum-Based Chemotherapy
Patients who are dosed initially at 75 mg/m2 and who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than one week, severe or cumulative cutaneous reactions, or other grade 3/4 non-hematological toxicities during Docetaxel Injection treatment should have treatment withheld until resolution of the toxicity and then resumed at 55 mg/m2. Patients who develop ≥grade 3 peripheral neuropathy should have Docetaxel Injection treatment discontinued entirely.
Combination Therapy with Docetaxel Injection for Chemotherapy-Naïve NSCLC
For patients who are dosed initially at Docetaxel Injection 75 mg/m2 in combination with cisplatin, and whose nadir of platelet count during the previous course of therapy is <25,000 cells/mm3, in patients who experience febrile neutropenia, and in patients with serious non-hematologic toxicities, the Docetaxel Injection dosage in subsequent cycles should be reduced to 65 mg/m2. In patients who require a further dose reduction, a dose of 50 mg/m2 is recommended. For cisplatin dosage adjustments, see manufacturers' prescribing information.
Prostate Cancer
Combination Therapy with Docetaxel Injection for Metastatic Castration-Resistant Prostate Cancer
Docetaxel Injection should be administered when the neutrophil count is ≥1500 cells/mm3. Patients who experience either febrile neutropenia, neutrophils <500 cells/mm3 for more than one week, severe or cumulative cutaneous reactions or moderate neurosensory signs and/or symptoms during Docetaxel Injection therapy should have the dosage of Docetaxel Injection reduced from 75 mg/m2 to 60 mg/m2. If the patient continues to experience these reactions at 60 mg/m2, the treatment should be discontinued.
Gastric or Head and Neck Cancer
Docetaxel Injection in Combination with Cisplatin and Fluorouracil in Gastric Cancer or Head and Neck Cancer
Patients treated with Docetaxel Injection in combination with cisplatin and fluorouracil must receive antiemetics and appropriate hydration according to current institutional guidelines. In both studies, G-CSF was recommended during the second and/or subsequent cycles in case of febrile neutropenia, or documented infection with neutropenia, or neutropenia lasting more than 7 days. If an episode of febrile neutropenia, prolonged neutropenia or neutropenic infection occurs despite G-CSF use, the Docetaxel Injection dose should be reduced from 75 mg/m2 to 60 mg/m2. If subsequent episodes of complicated neutropenia occur the Docetaxel Injection dose should be reduced from 60 mg/m2 to 45 mg/m2. In case of grade 4 thrombocytopenia the Docetaxel Injection dose should be reduced from 75 mg/m2 to 60 mg/m2. Do not retreat patients with subsequent cycles of Docetaxel Injection until neutrophils recover to a level >1500 cells/mm3 [see Contraindications (4)]. Avoid retreating patients until platelets recover to a level >100,000 cells/mm3. Discontinue treatment if these toxicities persist [see Warnings and Precautions (5.3)].
Recommended dose modifications for toxicities in patients treated with Docetaxel Injection in combination with cisplatin and fluorouracil are shown in Table 1.
Table 1: Recommended Dose Modifications for Toxicities in Patients Treated with Docetaxel Injection in Combination with Cisplatin and Fluorouracil Toxicity
Dosage Adjustment
Diarrhea grade 3
First episode: reduce fluorouracil dose by 20%.
Second episode: then reduce Docetaxel Injection dose by 20%.Diarrhea grade 4
First episode: reduce Docetaxel Injection and fluorouracil doses by 20%.
Second episode: discontinue treatment.Stomatitis/mucositis grade 3
First episode: reduce fluorouracil dose by 20%.
Second episode: stop fluorouracil only, at all subsequent cycles.
Third episode: reduce Docetaxel Injection dose by 20%.Stomatitis/mucositis grade 4
First episode: stop fluorouracil only, at all subsequent cycles.
Second episode: reduce Docetaxel Injection dose by 20%.Liver dysfunction: In case of AST/ALT >2.5 to ≤5 × ULN and AP ≤2.5 × ULN, or AST/ALT >1.5 to ≤5 × ULN and AP >2.5 to ≤5 × ULN, Docetaxel Injection should be reduced by 20%.
In case of AST/ALT >5 × ULN and/or AP >5 × ULN Docetaxel Injection should be stopped.
The dose modifications for cisplatin and fluorouracil in the gastric cancer study are provided below.
Cisplatin Dose Modifications and Delays
Peripheral Neuropathy: A neurological examination should be performed before entry into the study, and then at least every 2 cycles and at the end of treatment. In the case of neurological signs or symptoms, more frequent examinations should be performed and the following dose modifications can be made according to NCI-CTCAE grade:
- Grade 2: Reduce cisplatin dose by 20%.
- Grade 3: Discontinue treatment.
Nephrotoxicity: In the event of a rise in serum creatinine ≥grade 2 (>1.5 × normal value) despite adequate rehydration, CrCl should be determined before each subsequent cycle and the following dose reductions should be considered (see Table 2).
For other cisplatin dosage adjustments, also refer to the manufacturers' prescribing information.
Table 2: Dose Reductions for Evaluation of Creatinine Clearance CrCl = Creatinine clearance Creatinine Clearance Result Before Next Cycle
Cisplatin Dose Next Cycle
CrCl ≥60 mL/min
Full dose of cisplatin was given. CrCl was to be repeated before each treatment cycle.
CrCl between 40 and 59 mL/min
Dose of cisplatin was reduced by 50% at subsequent cycle. If CrCl was >60 mL/min at end of cycle, full cisplatin dose was reinstituted at the next cycle.
If no recovery was observed, then cisplatin was omitted from the next treatment cycle.CrCl <40 mL/min
Dose of cisplatin was omitted in that treatment cycle only.
If CrCl was still <40 mL/min at the end of cycle, cisplatin was discontinued.
If CrCl was >40 and <60 mL/min at end of cycle, a 50% cisplatin dose was given at the next cycle.
If CrCl was >60 mL/min at end of cycle, full cisplatin dose was given at next cycle.Fluorouracil Dose Modifications and Treatment Delays
For diarrhea and stomatitis, see Table 1.
In the event of grade 2 or greater plantar-palmar toxicity, fluorouracil should be stopped until recovery. The fluorouracil dosage should be reduced by 20%.
For other greater than grade 3 toxicities, except alopecia and anemia, chemotherapy should be delayed (for a maximum of 2 weeks from the planned date of infusion) until resolution to grade ≤1 and then recommenced, if medically appropriate.
For other fluorouracil dosage adjustments, also refer to the manufacturers' prescribing information.
Combination Therapy with Strong CYP3A4 Inhibitors
Avoid using concomitant strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin and voriconazole). There are no clinical data with a dose adjustment in patients receiving strong CYP3A4 inhibitors. Based on extrapolation from a pharmacokinetic study with ketoconazole in 7 patients, consider a 50% docetaxel dose reduction if patients require coadministration of a strong CYP3A4 inhibitor [see Drug Interactions (7), Clinical Pharmacology (12.3)].
2.8 Administration Precautions
Docetaxel Injection is a hazardous anticancer drug and, as with other potentially toxic compounds, caution should be exercised when handling and preparing Docetaxel Injection solutions. The use of gloves is recommended [see How Supplied/Storage and Handling (16.3)].
If Docetaxel Injection or final dilution for infusion should come into contact with the skin, immediately and thoroughly wash with soap and water. If Docetaxel Injection or final dilution for infusion should come into contact with mucosa, immediately and thoroughly wash with water.
Contact of the Docetaxel Injection with plasticized PVC equipment or devices used to prepare solutions for infusion is not recommended. In order to minimize patient exposure to the plasticizer DEHP (di-2-ethylhexyl phthalate), which may be leached from PVC infusion bags or sets, the final Docetaxel Injection dilution for infusion should be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.
Docetaxel Injection requires NO prior dilution with a diluent and is ready to add to the infusion solution.
Please follow the preparation instructions provided below.
2.9 Preparation and Administration
Docetaxel Injection (10 mg/mL) requires NO prior dilution with a diluent and is ready to add to the infusion solution. Use only a 21 gauge needle to withdraw Docetaxel Injection from the vial because larger bore needles (e.g., 18 and 19 gauge) may result in stopper coring and rubber particulates.
Dilution for Infusion
- 1. Docetaxel Injection unopened vials should be stored between 20°C and 25°C (68°F and 77°F). After first use and following multiple needle entries and product withdrawals, Docetaxel Injection multiple-dose vials should be stored between 2°C and 8°C (36°F and 46°F). If the vials are stored under refrigeration, allow the appropriate number of vials of Docetaxel Injection vials to stand at room temperature for approximately 5 minutes before use.
- 2.
Using only a 21 gauge needle, aseptically withdraw the required amount of Docetaxel Injection (10 mg docetaxel/mL) with a calibrated syringe and inject via a single injection (one shot) into a 250 mL infusion bag or bottle of either 0.9% Sodium Chloride solution or 5% Dextrose solution to produce a final concentration of 0.3 mg/mL to 0.74 mg/mL.
If a dose greater than 200 mg of docetaxel is required, use a larger volume of the infusion vehicle so that a concentration of 0.74 mg/mL docetaxel is not exceeded. - 3. Thoroughly mix the infusion by gentle manual rotation.
- 4. As with all parenteral products, Docetaxel Injection should be inspected visually for particulate matter or discoloration prior to administration whenever the solution and container permit. If the Docetaxel Injection dilution for intravenous infusion is not clear or appears to have precipitation, it should be discarded.
- 5. Docetaxel Injection infusion solution is supersaturated, therefore may crystallize over time. If crystals appear, the solution must no longer be used and shall be discarded.
The Docetaxel Injection dilution for infusion should be administered intravenously as a 1-hour infusion under ambient room temperature (below 25°C) and lighting conditions.
2.10 Stability
Docetaxel Injection final dilution for infusion, if stored between 2°C and 25°C (36°F and 77°F), is stable for 4 hours. Docetaxel Injection final dilution for infusion (in either 0.9% Sodium Chloride solution or 5% Dextrose solution) should be used within 4 hours (including the 1 hour intravenous administration).
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Docetaxel Injection is contraindicated in patients with:
- neutrophil counts of <1500 cells/mm3 [see Warnings and Precautions (5.3)].
- a history of severe hypersensitivity reactions to docetaxel or to other drugs formulated with polysorbate 80. Severe reactions, including anaphylaxis, have occurred [see Warnings and Precautions (5.5)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Toxic Deaths
Breast Cancer
Docetaxel administered at 100 mg/m2 was associated with deaths considered possibly or probably related to treatment in 2.0% (19/965) of metastatic breast cancer patients, both previously treated and untreated, with normal baseline liver function and in 11.5% (7/61) of patients with various tumor types who had abnormal baseline liver function (AST and/or ALT >1.5 times ULN together with AP >2.5 times ULN). Among patients dosed at 60 mg/m2, mortality related to treatment occurred in 0.6% (3/481) of patients with normal liver function, and in 3 of 7 patients with abnormal liver function. Approximately half of these deaths occurred during the first cycle. Sepsis accounted for the majority of the deaths.
Non-small Cell Lung Cancer
Docetaxel administered at a dose of 100 mg/m2 in patients with locally advanced or metastatic non-small cell lung cancer who had a history of prior platinum-based chemotherapy was associated with increased treatment-related mortality (14% and 5% in two randomized, controlled studies). There were 2.8% treatment-related deaths among the 176 patients treated at the 75 mg/m2 dose in the randomized trials. Among patients who experienced treatment-related mortality at the 75 mg/m2 dose level, 3 of 5 patients had an ECOG PS of 2 at study entry [see Dosage and Administration (2.2), Clinical Studies (14)].
5.2 Hepatic Impairment
Patients with elevations of bilirubin or abnormalities of transaminase concurrent with alkaline phosphatase are at increased risk for the development of severe neutropenia, febrile neutropenia, infections, severe thrombocytopenia, severe stomatitis, severe skin toxicity, and toxic death.
Avoid Docetaxel Injection in patients with bilirubin > upper limit of normal (ULN), or to patients with AST and/or ALT >1.5 × ULN concomitant with alkaline phosphatase >2.5 × ULN [see Warnings and Precautions (5.1)].
For patients with isolated elevations of transaminase >1.5 × ULN, consider Docetaxel Injection dose modifications [see Dosage and Administration (2.7)].
Measure bilirubin, AST or ALT, and alkaline phosphatase prior to each cycle of Docetaxel Injection therapy.
5.3 Hematologic Effects
Perform frequent peripheral blood cell counts on all patients receiving Docetaxel Injection. Do not retreat patients with subsequent cycles of Docetaxel Injection until neutrophils recover to a level >1500 cells/mm3 [see Contraindications (4)]. Avoid retreating patients until platelets recover to a level >100,000 cells/mm3.
A 25% reduction in the dose of Docetaxel Injection is recommended during subsequent cycles following severe neutropenia (<500 cells/mm3) lasting 7 days or more, febrile neutropenia, or a grade 4 infection in a Docetaxel Injection cycle [see Dosage and Administration (2.7)].
Neutropenia (<2000 neutrophils/mm3) occurs in virtually all patients given 60 mg/m2 to 100 mg/m2 of docetaxel and grade 4 neutropenia (<500 cells/mm3) occurs in 85% of patients given 100 mg/m2 and 75% of patients given 60 mg/m2. Frequent monitoring of blood counts is, therefore, essential so that dose can be adjusted. Docetaxel Injection should not be administered to patients with neutrophils <1500 cells/mm3.
Febrile neutropenia occurred in about 12% of patients given 100 mg/m2 but was very uncommon in patients given 60 mg/m2.
Hematologic responses, febrile reactions and infections, and rates of septic death for different regimens are dose related [see Adverse Reactions (6.1), Clinical Studies (14)].
Three breast cancer patients with severe liver impairment (bilirubin >1.7 times ULN) developed fatal gastrointestinal bleeding associated with severe drug-induced thrombocytopenia. In gastric cancer patients treated with docetaxel in combination with cisplatin and fluorouracil (TCF), febrile neutropenia and/or neutropenic infection occurred in 12% of patients receiving G-CSF compared to 28% who did not. Patients receiving TCF should be closely monitored during the first and subsequent cycles for febrile neutropenia and neutropenic infection [see Dosage and Administration (2.7), Adverse Reactions (6)].
5.4 Enterocolitis and Neutropenic Colitis
Enterocolitis and neutropenic colitis (typhlitis) have occurred in patients treated with Docetaxel Injection alone and in combination with other chemotherapeutic agents, despite the coadministration of G-CSF. Caution is recommended for patients with neutropenia, particularly at risk for developing gastrointestinal complications. Enterocolitis and neutropenic enterocolitis may develop at any time, and could lead to death as early as the first day of symptom onset.
Monitor patients closely from onset of any symptoms of gastrointestinal toxicity. Inform patients to contact their healthcare provider with new, or worsening symptoms of gastrointestinal toxicity [see Dosage and Administration (2), Warnings and Precautions (5.3), Adverse Reactions (6.2)].
5.5 Hypersensitivity Reactions
Monitor patients closely for hypersensitivity reactions, especially during the first and second infusions. Severe hypersensitivity reactions characterized by generalized rash/erythema, hypotension and/or bronchospasm, or fatal anaphylaxis, have been reported in patients premedicated with 3 days of corticosteroids. Severe hypersensitivity reactions require immediate discontinuation of the Docetaxel Injection infusion and aggressive therapy. Do not rechallenge patients with a history of severe hypersensitivity reactions with Docetaxel Injection [see Contraindications (4)].
Patients who have previously experienced a hypersensitivity reaction to paclitaxel may develop a hypersensitivity reaction to docetaxel that may include severe or fatal reactions such as anaphylaxis. Monitor patients with a previous history of hypersensitivity to paclitaxel closely during initiation of docetaxel therapy. Hypersensitivity reactions may occur within a few minutes following initiation of a Docetaxel Injection infusion. If minor reactions such as flushing or localized skin reactions occur, interruption of therapy is not required. All patients should be premedicated with an oral corticosteroid prior to the initiation of the infusion of Docetaxel Injection [see Dosage and Administration (2.6)].
5.6 Fluid Retention
Severe fluid retention has been reported following docetaxel therapy. Patients should be premedicated with oral corticosteroids prior to each Docetaxel Injection administration to reduce the incidence and severity of fluid retention [see Dosage and Administration (2.6)]. Patients with pre-existing effusions should be closely monitored from the first dose for the possible exacerbation of the effusions.
When fluid retention occurs, peripheral edema usually starts in the lower extremities and may become generalized with a median weight gain of 2 kg.
Among 92 breast cancer patients premedicated with 3-day corticosteroids, moderate fluid retention occurred in 27.2% and severe fluid retention in 6.5%. The median cumulative dose to onset of moderate or severe fluid retention was 819 mg/m2. Nine of 92 patients (9.8%) of patients discontinued treatment due to fluid retention: 4 patients discontinued with severe fluid retention; the remaining 5 had mild or moderate fluid retention. The median cumulative dose to treatment discontinuation due to fluid retention was 1021 mg/m2. Fluid retention was completely, but sometimes slowly, reversible with a median of 16 weeks from the last infusion of docetaxel to resolution (range: 0 to 42+ weeks). Patients developing peripheral edema may be treated with standard measures, e.g., salt restriction, oral diuretic(s).
5.7 Second Primary Malignancies
Second primary malignancies, notably acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), Non-Hodgkin's Lymphoma (NHL), and renal cancer, have been reported in patients treated with docetaxel-containing regimens. These adverse reactions may occur several months or years after docetaxel-containing therapy.
Treatment-related AML or MDS has occurred in patients given anthracyclines and/or cyclophosphamide, including use in adjuvant therapy for breast cancer. In the adjuvant breast cancer trial (TAX316) AML occurred in 3 of 744 patients who received docetaxel, doxorubicin, and cyclophosphamide (TAC) and in 1 of 736 patients who received fluorouracil, doxorubicin, and cyclophosphamide [see Clinical Studies (14.2)]. In TAC-treated patients, the risk of delayed myelodysplasia or myeloid leukemia requires hematological follow-up. Monitor patients for second primary malignancies [see Adverse Reactions (6.1)].
5.8 Cutaneous Reactions
Localized erythema of the extremities with edema followed by desquamation has been observed. In case of severe skin toxicity, an adjustment in dosage is recommended [see Dosage and Administration (2.7)]. The discontinuation rate due to skin toxicity was 1.6% (15/965) for metastatic breast cancer patients. Among 92 breast cancer patients premedicated with 3-day corticosteroids, there were no cases of severe skin toxicity reported and no patient discontinued docetaxel due to skin toxicity.
Severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and acute generalized exanthematous pustulosis (AGEP) have been reported in association with docetaxel treatment. Patients should be informed about the signs and symptoms of serious skin manifestations and monitored closely. Permanent treatment discontinuation should be considered in patients who experience SCARs.
5.9 Neurologic Reactions
Severe neurosensory symptoms (e.g., paresthesia, dysesthesia, pain) were observed in 5.5% (53/965) of metastatic breast cancer patients, and resulted in treatment discontinuation in 6.1%. When these symptoms occur, dosage must be adjusted. If symptoms persist, treatment should be discontinued [see Dosage and Administration (2.7)]. Patients who experienced neurotoxicity in clinical trials and for whom follow-up information on the complete resolution of the event was available had spontaneous reversal of symptoms with a median of 9 weeks from onset (range: 0 to 106 weeks). Severe peripheral motor neuropathy mainly manifested as distal extremity weakness occurred in 4.4% (42/965).
5.10 Eye Disorders
Cystoid macular edema (CME) has been reported in patients treated with Docetaxel Injection. Patients with impaired vision should undergo a prompt and comprehensive ophthalmologic examination. If CME is diagnosed, Docetaxel Injection treatment should be discontinued and appropriate treatment initiated. Alternative non-taxane cancer treatment should be considered.
5.11 Asthenia
Severe asthenia has been reported in 14.9% (144/965) of metastatic breast cancer patients but has led to treatment discontinuation in only 1.8%. Symptoms of fatigue and weakness may last a few days up to several weeks and may be associated with deterioration of performance status in patients with progressive disease.
5.12 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies and its mechanism of action, Docetaxel Injection can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Available data from case reports in the literature and pharmacovigilance with docetaxel use in pregnant women are not sufficient to inform the drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, administration of docetaxel to pregnant rats and rabbits during the period of organogenesis caused embryo-fetal toxicities, including intrauterine mortality, at doses as low as 0.02 and 0.003 times the recommended human dose based on body surface area, respectively.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Verify pregnancy status in females of reproductive potential prior to initiating Docetaxel Injection. Advise females of reproductive potential to use effective contraception during treatment and for 2 months after the last dose of Docetaxel Injection. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 4 months after the last dose of Docetaxel Injection [see Use in Specific Populations (8.1, 8.3)].
5.13 Alcohol Content
Cases of intoxication have been reported with some formulations of docetaxel due to the alcohol content. The alcohol content in a dose of Docetaxel Injection may affect the central nervous system and should be taken into account for patients in whom alcohol intake should be avoided or minimized. Consideration should be given to the alcohol content in Docetaxel Injection on the ability to drive or use machines immediately after the infusion. Each administration of Docetaxel Injection at 100 mg/m2 delivers 1.8 g/m2 of ethanol. For a patient with a BSA of 2.0 m2, this would deliver 3.6 grams of ethanol [see Description (11)]. Other docetaxel products may have a different amount of alcohol.
5.14 Tumor Lysis Syndrome
Tumor lysis syndrome has been reported with docetaxel [see Adverse Reactions (6.2)]. Patients at risk of tumor lysis syndrome (e.g., with renal impairment, hyperuricemia, bulky tumor) should be closely monitored prior to initiating Docetaxel Injection and periodically during treatment. Correction of dehydration and treatment of high uric acid levels are recommended prior to initiation of treatment.
-
6 ADVERSE REACTIONS
The most serious adverse reactions from docetaxel are:
- Toxic Deaths [see Boxed Warning, Warnings and Precautions (5.1)]
- Hepatic Impairment [see Boxed Warning, Warnings and Precautions (5.2)]
- Hematologic Effects [see Boxed Warning, Warnings and Precautions (5.3)]
- Enterocolitis and Neutropenic Colitis [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Boxed Warning, Warnings and Precautions (5.5)]
- Fluid Retention [see Boxed Warning, Warnings and Precautions (5.6)]
- Second Primary Malignancies [see Warnings and Precautions (5.7)]
- Cutaneous Reactions [see Warnings and Precautions (5.8)]
- Neurologic Reactions [see Warnings and Precautions (5.9)]
- Eye Disorders [see Warnings and Precautions (5.10)]
- Asthenia [see Warnings and Precautions (5.11)]
- Alcohol Content [see Warnings and Precautions (5.13)]
The most common adverse reactions across all docetaxel indications are infections, neutropenia, anemia, febrile neutropenia, hypersensitivity, thrombocytopenia, neuropathy, dysgeusia, dyspnea, constipation, anorexia, nail disorders, fluid retention, asthenia, pain, nausea, diarrhea, vomiting, mucositis, alopecia, skin reactions, and myalgia. Incidence varies depending on the indication.
Adverse reactions are described according to indication. Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Responding patients may not experience an improvement in performance status on therapy and may experience worsening. The relationship between changes in performance status, response to therapy, and treatment-related side effects has not been established.
6.1 Clinical Trials Experience
Breast Cancer
Monotherapy with Docetaxel for Locally Advanced or Metastatic Breast Cancer after Failure of Prior Chemotherapy
Docetaxel 100 mg/m2: Adverse drug reactions occurring in at least 5% of patients are compared for three populations who received docetaxel administered at 100 mg/m2 as a 1-hour infusion every 3 weeks: 2045 patients with various tumor types and normal baseline liver function tests; the subset of 965 patients with locally advanced or metastatic breast cancer, both previously treated and untreated with chemotherapy, who had normal baseline liver function tests; and an additional 61 patients with various tumor types who had abnormal liver function tests at baseline. These reactions were described using COSTART terms and were considered possibly or probably related to docetaxel. At least 95% of these patients did not receive hematopoietic support. The safety profile is generally similar in patients receiving docetaxel for the treatment of breast cancer and in patients with other tumor types (see Table 3).
Table 3: Summary of Adverse Reactions in Patients Receiving Docetaxel at 100 mg/m2 - * Normal Baseline LFTs: Transaminases ≤1.5 times ULN or alkaline phosphatase ≤2.5 times ULN or isolated elevations of transaminases or alkaline phosphatase up to 5 times ULN
- † Elevated Baseline LFTs: AST and/or ALT >1.5 times ULN concurrent with alkaline phosphatase >2.5 times ULN
- ‡ Febrile Neutropenia: ANC grade 4 with fever >38°C with intravenous antibiotics and/or hospitalization
Adverse Reaction
All Tumor Types
Normal LFTs*
n=2045
%All Tumor Types
Elevated LFTs†
n=61
%Breast Cancer
Normal LFTs*
n=965
%Hematologic
Neutropenia
<2000 cells/mm3
96
96
99
<500 cells/mm3
75
88
86
Leukopenia
<4000 cells/mm3
96
98
99
<1000 cells/mm3
32
47
44
Thrombocytopenia
<100,000 cells/mm3
8
25
9
Anemia
<11 g/dL
90
92
94
<8 g/dL
9
31
8
Febrile Neutropenia‡
11
26
12
Septic Death
2
5
1
Non-Septic Death
1
7
1
Infections
Any
22
33
22
Severe
6
16
6
Fever in Absence of Infection
Any
31
41
35
Severe
2
8
2
Hypersensitivity Reactions
Regardless of Premedication
Any
21
20
18
Severe
4
10
3
With 3-day Premedication
n=92
n=3
n=92
Any
15
33
15
Severe
2
0
2
Fluid Retention
Regardless of Premedication
Any
47
39
60
Severe
7
8
9
With 3-day Premedication
n=92
n=3
n=92
Any
64
67
64
Severe
7
33
7
Neurosensory
Any
49
34
58
Severe
4
0
6
Cutaneous
Any
48
54
47
Severe
5
10
5
Nail Changes
Any
31
23
41
Severe
3
5
4
Gastrointestinal
Nausea
39
38
42
Vomiting
22
23
23
Diarrhea
39
33
43
Severe
5
5
6
Stomatitis
Any
42
49
52
Severe
6
13
7
Alopecia
76
62
74
Asthenia
Any
62
53
66
Severe
13
25
15
Myalgia
Any
19
16
21
Severe
2
2
2
Arthralgia
9
7
8
Infusion Site Reactions
4
3
4
Hematologic Reactions
Reversible marrow suppression was the major dose-limiting toxicity of docetaxel [see Warnings and Precautions (5.3)]. The median time to nadir was 7 days, while the median duration of severe neutropenia (<500 cells/mm3) was 7 days. Among 2045 patients with solid tumors and normal baseline LFTs, severe neutropenia occurred in 75.4% and lasted for more than 7 days in 2.9% of cycles.
Febrile neutropenia (<500 cells/mm3 with fever >38°C with intravenous antibiotics and/or hospitalization) occurred in 11% of patients with solid tumors, in 12.3% of patients with metastatic breast cancer, and in 9.8% of 92 breast cancer patients premedicated with 3-day corticosteroids.
Severe infectious episodes occurred in 6.1% of patients with solid tumors, in 6.4% of patients with metastatic breast cancer, and in 5.4% of 92 breast cancer patients premedicated with 3-day corticosteroids.
Thrombocytopenia (<100,000 cells/mm3) associated with fatal gastrointestinal hemorrhage has been reported.
Hypersensitivity Reactions
Severe hypersensitivity reactions have been reported [see Boxed Warning, Warnings and Precautions (5.5)]. Minor events, including flushing, rash with or without pruritus, chest tightness, back pain, dyspnea, drug fever, or chills, have been reported and resolved after discontinuing the infusion and instituting appropriate therapy.
Fluid Retention
Fluid retention can occur with the use of docetaxel [see Boxed Warning, Dosage and Administration (2.6), Warnings and Precautions (5.6)].
Cutaneous Reactions
Severe skin toxicity is discussed elsewhere in the label [see Warnings and Precautions (5.8)]. Reversible cutaneous reactions characterized by a rash including localized eruptions, mainly on the feet and/or hands, but also on the arms, face, or thorax, usually associated with pruritus, have been observed. Eruptions generally occurred within 1 week after docetaxel infusion, recovered before the next infusion, and were not disabling.
Severe nail disorders were characterized by hypo- or hyperpigmentation, and occasionally by onycholysis (in 0.8% of patients with solid tumors) and pain.
Neurologic Reactions
Neurologic reactions are discussed elsewhere in the label [see Warnings and Precautions (5.9)].
Gastrointestinal Reactions
Nausea, vomiting, and diarrhea were generally mild to moderate. Severe reactions occurred in 3%–5% of patients with solid tumors and to a similar extent among metastatic breast cancer patients. The incidence of severe reactions was 1% or less for the 92 breast cancer patients premedicated with 3-day corticosteroids.
Severe stomatitis occurred in 5.5% of patients with solid tumors, in 7.4% of patients with metastatic breast cancer, and in 1.1% of the 92 breast cancer patients premedicated with 3-day corticosteroids.
Cardiovascular Reactions
Hypotension occurred in 2.8% of patients with solid tumors; 1.2% required treatment. Clinically meaningful events such as heart failure, sinus tachycardia, atrial flutter, dysrhythmia, unstable angina, pulmonary edema, and hypertension have occurred. Seven of 86 (8.1%) of metastatic breast cancer patients receiving docetaxel 100 mg/m2 in a randomized trial and who had serial left ventricular ejection fractions assessed developed deterioration of LVEF by ≥10% associated with a drop below the institutional lower limit of normal.
Infusion Site Reactions
Infusion site reactions were generally mild and consisted of hyperpigmentation, inflammation, redness or dryness of the skin, phlebitis, extravasation, or swelling of the vein.
Hepatic Reactions
In patients with normal LFTs at baseline, bilirubin values greater than the ULN occurred in 8.9% of patients. Increases in AST or ALT >1.5 times the ULN, or alkaline phosphatase >2.5 times ULN, were observed in 18.9% and 7.3% of patients, respectively. While on docetaxel, increases in AST and/or ALT >1.5 times ULN concomitant with alkaline phosphatase >2.5 times ULN occurred in 4.3% of patients with normal LFTs at baseline. Whether these changes were related to the drug or underlying disease has not been established.
Hematologic and Other Toxicity: Relation to Dose and Baseline Liver Chemistry Abnormalities
Hematologic and other toxicity is increased at higher doses and in patients with elevated baseline liver function tests (LFTs). In the following tables, adverse drug reactions are compared for three populations: 730 patients with normal LFTs given docetaxel at 100 mg/m2 in the randomized and single arm studies of metastatic breast cancer after failure of previous chemotherapy; 18 patients in these studies who had abnormal baseline LFTs (defined as AST and/or ALT >1.5 times ULN concurrent with alkaline phosphatase >2.5 times ULN); and 174 patients in Japanese studies given docetaxel at 60 mg/m2 who had normal LFTs (see Tables 4 and 5).
Table 4: Hematologic Adverse Reactions in Breast Cancer Patients Previously Treated with Chemotherapy Treated at Docetaxel 100 mg/m2 with Normal or Elevated Liver Function Tests or 60 mg/m2 with Normal Liver Function Tests - * Normal Baseline LFTs: Transaminases ≤1.5 times ULN or alkaline phosphatase ≤2.5 times ULN or isolated elevations of transaminases or alkaline phosphatase up to 5 times ULN
- † Elevated Baseline LFTs: AST and/or ALT >1.5 times ULN concurrent with alkaline phosphatase >2.5 times ULN
- ‡ Incidence of infection requiring hospitalization and/or intravenous antibiotics was 8.5% (n=62) among the 730 patients with normal LFTs at baseline; 7 patients had concurrent grade 3 neutropenia, and 46 patients had grade 4 neutropenia.
- § Febrile Neutropenia: For 100 mg/m2, ANC grade 4 and fever >38°C with intravenous antibiotics and/or hospitalization; for 60 mg/m2, ANC grade 3/4 and fever >38.1°C
Adverse Reaction
Docetaxel
100 mg/m2Docetaxel
60 mg/m2Normal LFTs*
n=730
%Elevated LFTs†
n=18
%Normal LFTs*
n=174
%Neutropenia
Any <2000 cells/mm3
98
100
95
Grade 4 <500 cells/mm3
84
94
75
Thrombocytopenia
Any <100,000 cells/mm3
11
44
14
Grade 4 <20,000 cells/mm3
1
17
1
Anemia <11 g/dL
95
94
65
Infection‡
Any
23
39
1
Grade 3 and 4
7
33
0
Febrile Neutropenia§
By Patient
12
33
0
By Course
2
9
0
Septic Death
2
6
1
Non-Septic Death
1
11
0
Table 5: Non-Hematologic Adverse Reactions in Breast Cancer Patients Previously Treated with Chemotherapy Treated at Docetaxel 100 mg/m2 with Normal or Elevated Liver Function Tests or 60 mg/m2 with Normal Liver Function Tests NA = not available - * Normal Baseline LFTs: Transaminases ≤1.5 times ULN or alkaline phosphatase ≤2.5 times ULN or isolated elevations of transaminases or alkaline phosphatase up to 5 times ULN
- † Elevated Baseline Liver Function: AST and/or ALT >1.5 times ULN concurrent with alkaline phosphatase >2.5 times ULN
- ‡ Fluid Retention includes (by COSTART): edema (peripheral, localized, generalized, lymphedema, pulmonary edema, and edema otherwise not specified) and effusion (pleural, pericardial, and ascites); no premedication given with the 60 mg/m2 dose
Adverse Reaction
Docetaxel
100 mg/m2Docetaxel
60 mg/m2Normal LFTs*
n=730
%Elevated LFTs†
n=18
%Normal LFTs*
n=174
%Acute Hypersensitivity Reaction Regardless of Premedication
Any
13
6
1
Severe
1
0
0
Fluid Retention‡ Regardless of Premedication
Any
56
61
13
Severe
8
17
0
Neurosensory
Any
57
50
20
Severe
6
0
0
Myalgia
23
33
3
Cutaneous
Any
45
61
31
Severe
5
17
0
Asthenia
Any
65
44
66
Severe
17
22
0
Diarrhea
Any
42
28
NA
Severe
6
11
Stomatitis
Any
53
67
19
Severe
8
39
1
In the three-arm monotherapy trial, TAX313, which compared docetaxel 60 mg/m2, 75 mg/m2 and 100 mg/m2 in advanced breast cancer, grade 3/4 or severe adverse reactions occurred in 49.0% of patients treated with docetaxel 60 mg/m2 compared to 55.3% and 65.9% treated with 75 mg/m2 and 100 mg/m2, respectively. Discontinuation due to adverse reactions was reported in 5.3% of patients treated with 60 mg/m2 versus 6.9% and 16.5% for patients treated at 75 mg/m2 and 100 mg/m2, respectively. Deaths within 30 days of last treatment occurred in 4.0% of patients treated with 60 mg/m2 compared to 5.3% and 1.6% for patients treated at 75 mg/m2 and 100 mg/m2, respectively.
The following adverse reactions were associated with increasing docetaxel doses: fluid retention (26%, 38%, and 46% at 60 mg/m2, 75 mg/m2, and 100 mg/m2, respectively), thrombocytopenia (7%, 11% and 12%, respectively), neutropenia (92%, 94%, and 97%, respectively), febrile neutropenia (5%, 7%, and 14%, respectively), treatment-related grade 3/4 infection (2%, 3%, and 7%, respectively) and anemia (87%, 94%, and 97%, respectively).
Combination Therapy with Docetaxel in the Adjuvant Treatment of Breast Cancer
The following table presents treatment-emergent adverse reactions observed in 744 patients, who were treated with docetaxel 75 mg/m2 every 3 weeks in combination with doxorubicin and cyclophosphamide (see Table 6).
Table 6: Clinically Important Treatment-Emergent Adverse Reactions Regardless of Causal Relationship in Patients Receiving Docetaxel in Combination with Doxorubicin and Cyclophosphamide (TAX316) - * COSTART term and grading system for events related to treatment.
Docetaxel 75 mg/m2 + Doxorubicin 50 mg/m2 + Cyclophosphamide 500 mg/m2 (TAC)
n=744
%Fluorouracil 500 mg/m2 + Doxorubicin 50 mg/m2 + Cyclophosphamide 500 mg/m2 (FAC)
n=736
%Adverse Reaction
Any
Grade 3/4
Any
Grade 3/4
Anemia
92
4
72
2
Neutropenia
71
66
82
49
Fever in absence of infection
47
1
17
0
Infection
39
4
36
2
Thrombocytopenia
39
2
28
1
Febrile neutropenia
25
N/A
3
N/A
Neutropenic infection
12
N/A
6
N/A
Hypersensitivity reactions
13
1
4
0
Lymphedema
4
0
1
0
Fluid Retention*
35
1
15
0
Peripheral edema
27
0
7
0
Weight gain
13
0
9
0
Neuropathy sensory
26
0
10
0
Neuro-cortical
5
1
6
1
Neuropathy motor
4
0
2
0
Neuro-cerebellar
2
0
2
0
Syncope
2
1
1
0
Alopecia
98
N/A
97
N/A
Skin toxicity
27
1
18
0
Nail disorders
19
0
14
0
Nausea
81
5
88
10
Stomatitis
69
7
53
2
Vomiting
45
4
59
7
Diarrhea
35
4
28
2
Constipation
34
1
32
1
Taste perversion
28
1
15
0
Anorexia
22
2
18
1
Abdominal Pain
11
1
5
0
Amenorrhea
62
N/A
52
N/A
Cough
14
0
10
0
Cardiac dysrhythmias
8
0
6
0
Vasodilatation
27
1
21
1
Hypotension
2
0
1
0
Phlebitis
1
0
1
0
Asthenia
81
11
71
6
Myalgia
27
1
10
0
Arthralgia
19
1
9
0
Lacrimation disorder
11
0
7
0
Conjunctivitis
5
0
7
0
Of the 744 patients treated with TAC, 36.3% experienced severe treatment-emergent adverse reactions compared to 26.6% of the 736 patients treated with FAC. Dose reductions due to hematologic toxicity occurred in 1% of cycles in the TAC arm versus 0.1% of cycles in the FAC arm. Six percent of patients treated with TAC discontinued treatment due to adverse reactions, compared to 1.1% treated with FAC; fever in the absence of infection and allergy being the most common reasons for withdrawal among TAC-treated patients. Two patients died in each arm within 30 days of their last study treatment; 1 death per arm was attributed to study drugs.
Fever and Infection
During the treatment period, fever in the absence of infection was seen in 46.5% of TAC-treated patients and in 17.1% of FAC-treated patients. Grade 3/4 fever in the absence of infection was seen in 1.3% and 0% of TAC- and FAC-treated patients, respectively. Infection was seen in 39.4% of TAC-treated patients compared to 36.3% of FAC-treated patients. Grade 3/4 infection was seen in 3.9% and 2.2% of TAC-treated and FAC-treated patients, respectively. There were no septic deaths in either treatment arm during the treatment period.
Gastrointestinal Reactions
In addition to gastrointestinal reactions reflected in the table above, 7 patients in the TAC arm were reported to have colitis/enteritis/large intestine perforation versus one patient in the FAC arm. Five of the 7 TAC-treated patients required treatment discontinuation; no deaths due to these events occurred during the treatment period.
Cardiovascular Reactions
More cardiovascular reactions were reported in the TAC arm versus the FAC arm during the treatment period: arrhythmias, all grades (6.2% vs. 4.9%), and hypotension, all grades (1.9% vs. 0.8%). Twenty-six (26) patients (3.5%) in the TAC arm and 17 patients (2.3%) in the FAC arm developed CHF during the study period. All except one patient in each arm were diagnosed with CHF during the follow-up period. Two (2) patients in TAC arm and 4 patients in FAC arm died due to CHF. The risk of CHF was higher in the TAC arm in the first year, and then was similar in both treatment arms.
Adverse Reactions during the Follow-Up Period (Median Follow-Up Time of 8 Years)
In study TAX316, the most common adverse reactions that started during the treatment period and persisted into the follow-up period in TAC and FAC patients are described below (median follow-up time of 8 years).
Nervous System Disorders: In study TAX316, peripheral sensory neuropathy started during the treatment period and persisted into the follow-up period in 84 patients (11.3%) in TAC arm and 15 patients (2%) in FAC arm. At the end of the follow-up period (median follow-up time of 8 years), peripheral sensory neuropathy was observed to be ongoing in 10 patients (1.3%) in TAC arm, and in 2 patients (0.3%) in FAC arm.
Skin and Subcutaneous Tissue Disorders: In study TAX316, alopecia persisting into the follow-up period after the end of chemotherapy was reported in 687 of 744 TAC patients (92.3%) and 645 of 736 FAC patients (87.6%). At the end of the follow-up period (actual median follow-up time of 8 years), alopecia was observed to be ongoing in 29 TAC patients (3.9%) and 16 FAC patients (2.2%).
Reproductive System and Breast Disorders: In study TAX316, amenorrhea that started during the treatment period and persisted into the follow-up period after the end of chemotherapy was reported in 202 of 744 TAC patients (27.2%) and 125 of 736 FAC patients (17.0%). Amenorrhea was observed to be ongoing at the end of the follow-up period (median follow-up time of 8 years) in 121 of 744 TAC patients (16.3%) and 86 FAC patients (11.7%).
General Disorders and Administration Site Conditions: In study TAX316, peripheral edema that started during the treatment period and persisted into the follow-up period after the end of chemotherapy was observed in 119 of 744 TAC patients (16.0%) and 23 of 736 FAC patients (3.1%). At the end of the follow-up period (actual median follow-up time of 8 years), peripheral edema was ongoing in 19 TAC patients (2.6%) and 4 FAC patients (0.5%).
In study TAX316, lymphedema that started during the treatment period and persisted into the follow-up period after the end of chemotherapy was reported in 11 of 744 TAC patients (1.5%) and 1 of 736 FAC patients (0.1%). At the end of the follow-up period (actual median follow-up time of 8 years), lymphedema was observed to be ongoing in 6 TAC patients (0.8%) and 1 FAC patient (0.1%).
In study TAX316, asthenia that started during the treatment period and persisted into the follow-up period after the end of chemotherapy was reported in 236 of 744 TAC patients (31.7%) and 180 of 736 FAC patients (24.5%). At the end of the follow-up period (actual median follow-up time of 8 years), asthenia was observed to be ongoing in 29 TAC patients (3.9%) and 16 FAC patients (2.2%).
Acute Myeloid Leukemia (AML)/Myelodysplastic Syndrome (MDS): AML occurred in the adjuvant breast cancer trial (TAX316). The cumulative risk of developing treatment-related AML at median follow-up time of 8 years in TAX316 was 0.4% for TAC-treated patients and 0.1% for FAC-treated patients. One TAC patient (0.1%) and 1 FAC patient (0.1%) died due to AML during the follow-up period (median follow-up time of 8 years).
Myelodysplastic syndrome occurred in 2 of 744 (0.3%) patients who received TAC and in 1 of 736 (0.1%) patients who received FAC. AML occurs at a higher frequency when these agents are given in combination with radiation therapy.
Lung Cancer
Monotherapy with Docetaxel for Unresectable, Locally Advanced or Metastatic NSCLC Previously Treated with Platinum-Based Chemotherapy
Docetaxel 75 mg/m2: Treatment-emergent adverse drug reactions are shown in Table 7. Included in this table are safety data for a total of 176 patients with non-small cell lung carcinoma and a history of prior treatment with platinum-based chemotherapy who were treated in two randomized, controlled trials. These reactions were described using NCI Common Toxicity Criteria regardless of relationship to study treatment, except for the hematologic toxicities or where otherwise noted.
Table 7: Treatment-Emergent Adverse Reactions Regardless of Relationship to Treatment in Patients Receiving Docetaxel as Monotherapy for Non-small Cell Lung Cancer Previously Treated with Platinum-Based Chemotherapy* - * Normal Baseline LFTs: Transaminases ≤1.5 times ULN or alkaline phosphatase ≤2.5 times ULN or isolated elevations of transaminases or alkaline phosphatase up to 5 times ULN
- † Febrile Neutropenia: ANC grade 4 with fever >38°C with intravenous antibiotics and/or hospitalization
- ‡ Not Applicable
- § Not Done
- ¶ COSTART term and grading system
Adverse Reaction
Docetaxel 75 mg/m2
n=176
%Best Supportive Care
n=49
%Vinorelbine/Ifosfamide
n=119
%Neutropenia
Any
84
14
83
Grade 3/4
65
12
57
Leukopenia
Any
84
6
89
Grade 3/4
49
0
43
Thrombocytopenia
Any
8
0
8
Grade 3/4
3
0
2
Anemia
Any
91
55
91
Grade 3/4
9
12
14
Febrile Neutropenia†
6
NA‡
1
Infection
Any
34
29
30
Grade 3/4
10
6
9
Treatment Related Mortality
3
NA‡
3
Hypersensitivity Reactions
Any
6
0
1
Grade 3/4
3
0
0
Fluid Retention
Any
34
ND§
23
Severe
3
3
Neurosensory
Any
23
14
29
Grade 3/4
2
6
5
Neuromotor
Any
16
8
10
Grade 3/4
5
6
3
Skin
Any
20
6
17
Grade 3/4
1
2
1
Gastrointestinal
Nausea
Any
34
31
31
Grade 3/4
5
4
8
Vomiting
Any
22
27
22
Grade 3/4
3
2
6
Diarrhea
Any
23
6
12
Grade 3/4
3
0
4
Alopecia
56
35
50
Asthenia
Any
53
57
54
Severe¶
18
39
23
Stomatitis
Any
26
6
8
Grade 3/4
2
0
1
Pulmonary
Any
41
49
45
Grade 3/4
21
29
19
Nail Disorder
Any
11
0
2
Severe¶
1
0
0
Myalgia
Any
6
0
3
Severe¶
0
0
0
Arthralgia
Any
3
2
2
Severe¶
0
0
1
Taste Perversion
Any
6
0
0
Severe¶
1
0
0
Combination Therapy with Docetaxel in Chemotherapy-Naïve Advanced Unresectable or Metastatic NSCLC
Table 8 presents safety data from two arms of an open label, randomized controlled trial (TAX326) that enrolled patients with unresectable stage IIIB or IV non-small cell lung cancer and no history of prior chemotherapy. Adverse reactions were described using the NCI Common Toxicity Criteria except where otherwise noted.
Table 8: Adverse Reactions Regardless of Relationship to Treatment in Chemotherapy-Naïve Advanced Non-small Cell Lung Cancer Patients Receiving Docetaxel in Combination with Cisplatin - * Replaces NCI term "Allergy"
- † COSTART term and grading system
Adverse Reaction
Docetaxel 75 mg/m2 + Cisplatin 75 mg/m2
n=406
%Vinorelbine 25 mg/m2 + Cisplatin 100 mg/m2
n=396
%Neutropenia
Any
91
90
Grade 3/4
74
78
Febrile Neutropenia
5
5
Thrombocytopenia
Any
15
15
Grade 3/4
3
4
Anemia
Any
89
94
Grade 3/4
7
25
Infection
Any
35
37
Grade 3/4
8
8
Fever in absence of infection
Any
33
29
Grade 3/4
<1
1
Hypersensitivity Reaction*
Any
12
4
Grade 3/4
3
<1
Fluid Retention†
Any
54
42
All severe or life-threatening events
Pleural effusion2
2
Any
23
22
All severe or life-threatening events
Peripheral edema2
2
Any
34
18
All severe or life-threatening events
Weight gain<1
<1
Any
15
9
All severe or life-threatening events
<1
<1
Neurosensory
Any
47
42
Grade 3/4
4
4
Neuromotor
Any
19
17
Grade 3/4
3
6
Skin
Any
16
14
Grade 3/4
<1
1
Nausea
Any
72
76
Grade 3/4
10
17
Vomiting
Any
55
61
Grade 3/4
8
16
Diarrhea
Any
47
25
Grade 3/4
7
3
Anorexia†
Any
42
40
All severe or life-threatening events
5
5
Stomatitis
Any
24
21
Grade 3/4
2
1
Alopecia
Any
75
42
Grade 3
<1
0
Asthenia†
Any
74
75
All severe or life-threatening events
12
14
Nail Disorder†
Any
14
<1
All severe events
<1
0
Myalgia†
Any
18
12
All severe events
<1
<1
Deaths within 30 days of last study treatment occurred in 31 patients (7.6%) in the docetaxel+cisplatin arm and 37 patients (9.3%) in the vinorelbine+cisplatin arm. Deaths within 30 days of last study treatment attributed to study drug occurred in 9 patients (2.2%) in the docetaxel+cisplatin arm and 8 patients (2.0%) in the vinorelbine+cisplatin arm.
The second comparison in the study, vinorelbine+cisplatin versus docetaxel+carboplatin (which did not demonstrate a superior survival associated with docetaxel, [see Clinical Studies (14.3)]) demonstrated a higher incidence of thrombocytopenia, diarrhea, fluid retention, hypersensitivity reactions, skin toxicity, alopecia and nail changes on the docetaxel+carboplatin arm, while a higher incidence of anemia, neurosensory toxicity, nausea, vomiting, anorexia and asthenia was observed on the vinorelbine+cisplatin arm.
Prostate Cancer
Combination Therapy with Docetaxel in Patients with Prostate Cancer
The following data are based on the experience of 332 patients, who were treated with docetaxel 75 mg/m2 every 3 weeks in combination with prednisone 5 mg orally twice daily (see Table 9).
Table 9: Clinically Important Treatment-Emergent Adverse Reactions (Regardless of Relationship) in Patients with Prostate Cancer who Received Docetaxel in Combination with Prednisone (TAX327) - * Related to treatment
Docetaxel 75 mg/m2
every 3 weeks + prednisone
5 mg twice daily
n=332
%Mitoxantrone 12 mg/m2
every 3 weeks + prednisone
5 mg twice daily
n=335
%Adverse Reaction
Any
Grade 3/4
Any
Grade 3/4
Anemia
67
5
58
2
Neutropenia
41
32
48
22
Thrombocytopenia
3
1
8
1
Febrile Neutropenia
3
N/A
2
N/A
Infection
32
6
20
4
Epistaxis
6
0
2
0
Allergic Reactions
8
1
1
0
Fluid Retention*
24
1
5
0
Weight Gain*
8
0
3
0
Peripheral Edema*
18
0
2
0
Neuropathy Sensory
30
2
7
0
Neuropathy Motor
7
2
3
1
Rash/Desquamation
6
0
3
1
Alopecia
65
N/A
13
N/A
Nail Changes
30
0
8
0
Nausea
41
3
36
2
Diarrhea
32
2
10
1
Stomatitis/Pharyngitis
20
1
8
0
Taste Disturbance
18
0
7
0
Vomiting
17
2
14
2
Anorexia
17
1
14
0
Cough
12
0
8
0
Dyspnea
15
3
9
1
Cardiac left ventricular function
10
0
22
1
Fatigue
53
5
35
5
Myalgia
15
0
13
1
Tearing
10
1
2
0
Arthralgia
8
1
5
1
Gastric Cancer
Combination Therapy with Docetaxel Injection in Gastric Adenocarcinoma
Data in the following table are based on the experience of 221 patients with advanced gastric adenocarcinoma and no history of prior chemotherapy for advanced disease who were treated with Docetaxel Injection 75 mg/m2 in combination with cisplatin and fluorouracil (see Table 10).
Table 10: Clinically Important Treatment-Emergent Adverse Reactions Regardless of Relationship to Treatment in the Gastric Cancer Study Clinically important treatment-emergent adverse reactions were determined based upon frequency, severity, and clinical impact of the adverse reaction. - * Related to treatment
Docetaxel Injection 75 mg/m2 +
cisplatin 75 mg/m2 +
fluorouracil 750 mg/m2
n=221Cisplatin 100 mg/m2 + fluorouracil 1000 mg/m2
n=224Adverse Reaction
Any %
Grade 3/4 %
Any %
Grade 3/4 %
Anemia
97
18
93
26
Neutropenia
96
82
83
57
Fever in the absence of infection
36
2
23
1
Thrombocytopenia
26
8
39
14
Infection
29
16
23
10
Febrile neutropenia
16
N/A
5
N/A
Neutropenic infection
16
N/A
10
N/A
Allergic reactions
10
2
6
0
Fluid retention*
15
0
4
0
Edema*
13
0
3
0
Lethargy
63
21
58
18
Neurosensory
38
8
25
3
Neuromotor
9
3
8
3
Dizziness
16
5
8
2
Alopecia
67
5
41
1
Rash/itch
12
1
9
0
Nail changes
8
0
0
0
Skin desquamation
2
0
0
0
Nausea
73
16
76
19
Vomiting
67
15
73
19
Anorexia
51
13
54
12
Stomatitis
59
21
61
27
Diarrhea
78
20
50
8
Constipation
25
2
34
3
Esophagitis/dysphagia/ odynophagia
16
2
14
5
Gastrointestinal pain/cramping
11
2
7
3
Cardiac dysrhythmias
5
2
2
1
Myocardial ischemia
1
0
3
2
Tearing
8
0
2
0
Altered hearing
6
0
13
2
Head and Neck Cancer
Combination Therapy with Docetaxel Injection in Head and Neck Cancer
Table 11 summarizes the safety data obtained from patients that received induction chemotherapy with Docetaxel Injection 75 mg/m2 in combination with cisplatin and fluorouracil followed by radiotherapy (TAX323; 174 patients) or chemoradiotherapy (TAX324; 251 patients). The treatment regimens are described in Section 14.6.
Table 11: Clinically Important Treatment-Emergent Adverse Reactions (Regardless of Relationship) in Patients with SCCHN Receiving Induction Chemotherapy with Docetaxel Injection in Combination with Cisplatin and Fluorouracil Followed by Radiotherapy (TAX323) or Chemoradiotherapy (TAX324) Clinically important treatment-emergent adverse reactions based upon frequency, severity, and clinical impact. - * Febrile neutropenia: grade ≥2 fever concomitant with grade 4 neutropenia requiring intravenous antibiotics and/or hospitalization
- † Related to treatment
- ‡ Includes superficial and deep vein thrombosis and pulmonary embolism
TAX323 (n=355)
TAX324 (n=494)
Docetaxel Injection arm (n=174)
Comparator arm (n=181)
Docetaxel Injection arm (n=251)
Comparator arm (n=243)
Adverse Reaction (by Body System)
Any
%Grade 3/4
%Any
%Grade 3/4
%Any
%Grade 3/4
%Any
%Grade 3/4
%Neutropenia
93
76
87
53
95
84
84
56
Anemia
89
9
88
14
90
12
86
10
Thrombocytopenia
24
5
47
18
28
4
31
11
Infection
27
9
26
8
23
6
28
5
Febrile neutropenia*
5
N/A
2
N/A
12
N/A
7
N/A
Neutropenic infection
14
N/A
8
N/A
12
N/A
8
N/A
Cancer pain
21
5
16
3
17
9
20
11
Lethargy
41
3
38
3
61
5
56
10
Fever in the absence of infection
32
1
37
0
30
4
28
3
Myalgia
10
1
7
0
7
0
7
2
Weight loss
21
1
27
1
14
2
14
2
Allergy
6
0
3
0
2
0
0
0
Fluid retention†
20
0
14
1
13
1
7
2
Edema only
13
0
7
0
12
1
6
1
Weight gain only
6
0
6
0
0
0
1
0
Dizziness
2
0
5
1
16
4
15
2
Neurosensory
18
1
11
1
14
1
14
0
Altered hearing
6
0
10
3
13
1
19
3
Neuromotor
2
1
4
1
9
0
10
2
Alopecia
81
11
43
0
68
4
44
1
Rash/itch
12
0
6
0
20
0
16
1
Dry skin
6
0
2
0
5
0
3
0
Desquamation
4
1
6
0
2
0
5
0
Nausea
47
1
51
7
77
14
80
14
Stomatitis
43
4
47
11
66
21
68
27
Vomiting
26
1
39
5
56
8
63
10
Diarrhea
33
3
24
4
48
7
40
3
Constipation
17
1
16
1
27
1
38
1
Anorexia
16
1
25
3
40
12
34
12
Esophagitis/dysphagia/ Odynophagia
13
1
18
3
25
13
26
10
Taste, sense of smell altered
10
0
5
0
20
0
17
1
Gastrointestinal pain/cramping
8
1
9
1
15
5
10
2
Heartburn
6
0
6
0
13
2
13
1
Gastrointestinal bleeding
4
2
0
0
5
1
2
1
Cardiac dysrhythmia
2
2
2
1
6
3
5
3
Venous‡
3
2
6
2
4
2
5
4
Ischemia myocardial
2
2
1
0
2
1
1
1
Tearing
2
0
1
0
2
0
2
0
Conjunctivitis
1
0
1
0
1
0
0.4
0
6.2 Postmarketing Experience
The following adverse reactions have been identified from clinical trials and/or postmarketing surveillance. Because these reactions are reported from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a whole: diffuse pain, chest pain, radiation recall phenomenon, injection site recall reaction (recurrence of skin reaction at a site of previous extravasation following administration of docetaxel at a different site) at the site of previous extravasation.
Cardiovascular: atrial fibrillation, deep vein thrombosis, ECG abnormalities, thrombophlebitis, pulmonary embolism, syncope, tachycardia, myocardial infarction. Ventricular arrhythmia, including ventricular tachycardia, in patients treated with docetaxel in combination regimens including doxorubicin, 5-fluorouracil and/or cyclophosphamide may be associated with fatal outcome.
Cutaneous: cutaneous lupus erythematosus, bullous eruptions such as erythema multiforme and severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome, toxic epidermal necrolysis and acute generalized exanthematous pustulosis, scleroderma-like changes (usually preceded by peripheral lymphedema), severe palmar-plantar erythrodysesthesia, and permanent alopecia.
Gastrointestinal: enterocolitis, including colitis, ischemic colitis, and neutropenic enterocolitis, which may be fatal. Abdominal pain, anorexia, constipation, duodenal ulcer, esophagitis, gastrointestinal hemorrhage, gastrointestinal perforation, intestinal obstruction, ileus, and dehydration as a consequence of gastrointestinal events.
Hearing: ototoxicity, hearing disorders and/or hearing loss, including during use with other ototoxic drugs.
Hematologic: bleeding episodes, disseminated intravascular coagulation (DIC), often in association with sepsis or multiorgan failure.
Hepatic: hepatitis, sometimes fatal, primarily in patients with pre-existing liver disorders.
Hypersensitivity: anaphylactic shock with fatal outcome in patients who received premedication. Severe hypersensitivity reactions with fatal outcome with docetaxel in patients who previously experienced hypersensitivity reactions to paclitaxel.
Metabolism and nutrition disorders: electrolyte imbalance, including hyponatremia, hypokalemia, hypomagnesemia, and hypocalcemia. Tumor lysis syndrome, sometimes fatal.
Neurologic: confusion, seizures or transient loss of consciousness, sometimes appearing during the infusion of the drug.
Ophthalmologic: conjunctivitis, lacrimation or lacrimation with or without conjunctivitis, cystoid macular edema (CME). Excessive tearing which may be attributable to lacrimal duct obstruction. Transient visual disturbances (flashes, flashing lights, scotomata), typically occurring during drug infusion and reversible upon discontinuation of the infusion, in association with hypersensitivity reactions.
Respiratory: dyspnea, acute pulmonary edema, acute respiratory distress syndrome/pneumonitis, interstitial lung disease, interstitial pneumonia, respiratory failure, and pulmonary fibrosis, which may be fatal. Radiation pneumonitis in patients receiving concomitant radiotherapy.
Renal: renal insufficiency and renal failure, the majority of cases were associated with concomitant nephrotoxic drugs.
Second primary malignancies: second primary malignancies, including AML, MDS, NHL, and renal cancer [see Warnings and Precautions (5.7)].
Musculoskeletal Disorder: myositis.
-
7 DRUG INTERACTIONS
Docetaxel is a CYP3A4 substrate. In vitro studies have shown that the metabolism of docetaxel may be modified by the concomitant administration of compounds that induce, inhibit, or are metabolized by cytochrome P450 3A4.
In vivo studies showed that the exposure of docetaxel increased 2.2-fold when it was coadministered with ketoconazole, a potent inhibitor of CYP3A4. Protease inhibitors, particularly ritonavir, may increase the exposure of docetaxel. Concomitant use of Docetaxel Injection and drugs that inhibit CYP3A4 may increase exposure to docetaxel and should be avoided. In patients receiving treatment with Docetaxel Injection close monitoring for toxicity and a Docetaxel Injection dose reduction could be considered if systemic administration of a potent CYP3A4 inhibitor cannot be avoided [see Dosage and Administration (2.7), Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal reproduction studies and its mechanism of action, Docetaxel Injection can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Available data from case reports in the literature and pharmacovigilance with docetaxel use in pregnant women are not sufficient to inform the drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Docetaxel Injection contains alcohol which can interfere with neurobehavioral development (see Clinical Considerations). In animal reproductive studies, administration of docetaxel to pregnant rats and rabbits during the period of organogenesis caused an increased incidence of embryo-fetal toxicities, including intrauterine mortality, at doses as low as 0.02 and 0.003 times the recommended human dose based on body surface area, respectively (see Data). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Docetaxel Injection contains alcohol [see Warnings and Precautions (5.13)]. Published studies have demonstrated that alcohol is associated with fetal harm including central nervous system abnormalities, behavioral disorders, and impaired intellectual development.
Animal data
Intravenous administration of ≥0.3 and 0.03 mg/kg/day docetaxel to pregnant rats and rabbits, respectively, during the period of organogenesis caused an increased incidence of intrauterine mortality, resorptions, reduced fetal weights, and fetal ossification delays. Maternal toxicity was also observed at these doses, which were approximately 0.02 and 0.003 times the daily maximum recommended human dose based on body surface area, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of docetaxel in human milk, or on its effects on milk production or the breastfed child. No lactation studies in animals have been conducted. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with Docetaxel Injection and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on findings in animals, Docetaxel Injection can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating Docetaxel Injection.
Contraception
Infertility
Based on findings in animal studies, Docetaxel Injection may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The alcohol content of Docetaxel Injection should be taken into account when given to pediatric patients [see Warnings and Precautions (5.13)].
The efficacy of docetaxel in pediatric patients as monotherapy or in combination has not been established. The overall safety profile of docetaxel in pediatric patients receiving monotherapy or TCF was consistent with the known safety profile in adults.
Docetaxel has been studied in a total of 289 pediatric patients: 239 in 2 trials with monotherapy and 50 in combination treatment with cisplatin and 5-fluorouracil (TCF).
Docetaxel Monotherapy
Docetaxel monotherapy was evaluated in a dose-finding phase 1 trial in 61 pediatric patients (median age 12.5 years, range 1–22 years) with a variety of refractory solid tumors. The recommended dose was 125 mg/m2 as a 1-hour intravenous infusion every 21 days. The primary dose limiting toxicity was neutropenia.
The recommended dose for docetaxel monotherapy was evaluated in a phase 2 single-arm trial in 178 pediatric patients (median age 12 years, range 1–26 years) with a variety of recurrent/refractory solid tumors. Efficacy was not established with tumor response rates ranging from one complete response (CR) (0.6%) in a patient with undifferentiated sarcoma to four partial responses (2.2%) seen in one patient each with Ewing Sarcoma, neuroblastoma, osteosarcoma, and squamous cell carcinoma.
Docetaxel in Combination
Docetaxel was studied in combination with cisplatin and 5-fluorouracil (TCF) versus cisplatin and 5-fluorouracil (CF) for the induction treatment of nasopharyngeal carcinoma (NPC) in pediatric patients prior to chemoradiation consolidation. Seventy-five patients (median age 16 years, range 9 to 21 years) were randomized (2:1) to docetaxel (75 mg/m2) in combination with cisplatin (75 mg/m2) and 5-fluorouracil (750 mg/m2) (TCF) or to cisplatin (80 mg/m2) and 5-fluorouracil (1000 mg/m2/day) (CF). The primary endpoint was the CR rate following induction treatment of NPC. One patient out of 50 in the TCF group (2%) had a complete response while none of the 25 patients in the CF group had a complete response.
Pharmacokinetics
Pharmacokinetic parameters for docetaxel were determined in 2 pediatric solid tumor trials. Following docetaxel administration at 55 mg/m2 to 235 mg/m2 in a 1-hour intravenous infusion every 3 weeks in 25 patients aged 1 to 20 years (median 11 years), docetaxel clearance was 17.3±10.9 L/h/m2.
Docetaxel was administered in combination with cisplatin and 5-fluorouracil (TCF), at dose levels of 75 mg/m2 in a 1-hour intravenous infusion day 1 in 28 patients aged 10 to 21 years (median 16 years, 17 patients were older than 16). Docetaxel clearance was 17.9±8.75 L/h/m2, corresponding to an AUC of 4.20±2.57 μg∙h/mL.
In summary, the body surface area adjusted clearance of docetaxel monotherapy and TCF combination in children were comparable to those in adults [see Clinical Pharmacology (12.3)].
8.5 Geriatric Use
In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy in elderly patients.
Non-small Cell Lung Cancer
In a study conducted in chemotherapy-naïve patients with NSCLC (TAX326), 148 patients (36%) in the docetaxel+cisplatin group were 65 years of age or greater. There were 128 patients (32%) in the vinorelbine+cisplatin group 65 years of age or greater. In the docetaxel+cisplatin group, patients less than 65 years of age had a median survival of 10.3 months (95% CI: 9.1 months, 11.8 months) and patients 65 years or older had a median survival of 12.1 months (95% CI: 9.3 months, 14 months). In patients 65 years of age or greater treated with docetaxel+cisplatin, diarrhea (55%), peripheral edema (39%) and stomatitis (28%) were observed more frequently than in the vinorelbine+cisplatin group (diarrhea 24%, peripheral edema 20%, stomatitis 20%). Patients treated with docetaxel+cisplatin who were 65 years of age or greater were more likely to experience diarrhea (55%), infections (42%), peripheral edema (39%) and stomatitis (28%) compared to patients less than the age of 65 administered the same treatment (43%, 31%, 31% and 21%, respectively).
When docetaxel was combined with carboplatin for the treatment of chemotherapy-naïve, advanced non-small cell lung carcinoma, patients 65 years of age or greater (28%) experienced higher frequency of infection compared to similar patients treated with docetaxel+cisplatin, and a higher frequency of diarrhea, infection and peripheral edema than elderly patients treated with vinorelbine+cisplatin.
Prostate Cancer
Of the 333 patients treated with docetaxel every three weeks plus prednisone in the prostate cancer study (TAX327), 209 patients were 65 years of age or greater and 68 patients were older than 75 years. In patients treated with docetaxel every three weeks, the following treatment-emergent adverse reactions occurred at rates ≥10% higher in patients 65 years of age or greater compared to younger patients: anemia (71% vs. 59%), infection (37% vs. 24%), nail changes (34% vs. 23%), anorexia (21% vs. 10%), weight loss (15% vs. 5%), respectively.
Breast Cancer
In the adjuvant breast cancer trial (TAX316), docetaxel in combination with doxorubicin and cyclophosphamide was administered to 744 patients of whom 48 (6%) were 65 years of age or greater. The number of elderly patients who received this regimen was not sufficient to determine whether there were differences in safety and efficacy between elderly and younger patients.
Gastric Cancer
Among the 221 patients treated with Docetaxel Injection in combination with cisplatin and fluorouracil in the gastric cancer study, 54 were 65 years of age or older and 2 patients were older than 75 years. In this study, the number of patients who were 65 years of age or older was insufficient to determine whether they respond differently from younger patients. However, the incidence of serious adverse reactions was higher in the elderly patients compared to younger patients. The incidence of the following adverse reactions (all grades, regardless of relationship): lethargy, stomatitis, diarrhea, dizziness, edema, febrile neutropenia/neutropenic infection occurred at rates ≥10% higher in patients who were 65 years of age or older compared to younger patients. Elderly patients treated with TCF should be closely monitored.
Head and Neck Cancer
Among the 174 and 251 patients who received the induction treatment with Docetaxel Injection in combination with cisplatin and fluorouracil (TPF) for SCCHN in the TAX323 and TAX324 studies, 18 (10%) and 32 (13%) of the patients were 65 years of age or older, respectively.
These clinical studies of Docetaxel Injection in combination with cisplatin and fluorouracil in patients with SCCHN did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience with this treatment regimen has not identified differences in responses between elderly and younger patients.
8.6 Hepatic Impairment
Avoid Docetaxel Injection in patients with bilirubin > ULN and patients with AST and/or ALT >1.5 × ULN concomitant with alkaline phosphatase >2.5 × ULN [see Boxed Warning, Warnings and Precautions (5.2), Clinical Pharmacology (12.3)].
The alcohol content of Docetaxel Injection should be taken into account when given to patients with hepatic impairment [see Warnings and Precautions (5.13)].
-
10 OVERDOSAGE
There is no known antidote for Docetaxel Injection overdosage. In case of overdosage, the patient should be kept in a specialized unit where vital functions can be closely monitored. Anticipated complications of overdosage include: bone marrow suppression, peripheral neurotoxicity, and mucositis. Patients should receive therapeutic G-CSF as soon as possible after discovery of overdose. Other appropriate symptomatic measures should be taken, as needed.
In two reports of overdose, one patient received 150 mg/m2 and the other received 200 mg/m2 as 1-hour infusions. Both patients experienced severe neutropenia, mild asthenia, cutaneous reactions, and mild paresthesia, and recovered without incident. In mice, lethality was observed following single intravenous doses that were ≥154 mg/kg (about 4.5 times the human dose of 100 mg/m2 on a mg/m2 basis); neurotoxicity associated with paralysis, non-extension of hind limbs, and myelin degeneration was observed in mice at 48 mg/kg (about 1.5 times the human dose of 100 mg/m2 basis). In male and female rats, lethality was observed at a dose of 20 mg/kg (comparable to the human dose of 100 mg/m2 on a mg/m2 basis) and was associated with abnormal mitosis and necrosis of multiple organs.
-
11 DESCRIPTION
Docetaxel is an antineoplastic agent belonging to the taxoid family. It is prepared by semisynthesis beginning with a precursor extracted from the renewable needle biomass of yew plants. The chemical name for docetaxel is (2R,3S)-N-carboxy-3-phenylisoserine,N-tert-butyl ester, 13-ester with 5β-20-epoxy-1,2α,4,7β,10β,13α-hexahydroxytax-11-en-9-one 4-acetate 2-benzoate. Docetaxel (anhydrous) has the following structural formula:
Docetaxel is a white to almost-white powder with an empirical formula of C43H53NO14, and a molecular weight of 807.88. It is highly lipophilic and practically insoluble in water.
Docetaxel Injection, USP is a sterile, non-pyrogenic, clear, colorless to pale yellow solution at 10 mg/mL concentration.
Each mL contains 10 mg docetaxel (anhydrous), 260 mg polysorbate 80 NF, 4 mg anhydrous citric acid USP, 23% v/v dehydrated alcohol USP, and polyethylene glycol 300 NF.
Docetaxel Injection is available in single-dose vials containing 20 mg (2 mL) docetaxel (anhydrous) and multiple-dose vials containing 160 mg (16 mL) docetaxel (anhydrous).
Docetaxel Injection requires NO prior dilution with a diluent and is ready to add to the infusion solution.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Docetaxel is an antineoplastic agent that acts by disrupting the microtubular network in cells that is essential for mitotic and interphase cellular functions. Docetaxel binds to free tubulin and promotes the assembly of tubulin into stable microtubules while simultaneously inhibiting their disassembly. This leads to the production of microtubule bundles without normal function and to the stabilization of microtubules, which results in the inhibition of mitosis in cells. Docetaxel's binding to microtubules does not alter the number of protofilaments in the bound microtubules, a feature which differs from most spindle poisons currently in clinical use.
12.3 Pharmacokinetics
Absorption
The pharmacokinetics of docetaxel has been evaluated in cancer patients after administration of 20 mg/m2 to 115 mg/m2 in phase 1 studies. The area under the curve (AUC) was dose proportional following doses of 70 mg/m2 to 115 mg/m2 with infusion times of 1 to 2 hours.
Docetaxel’s pharmacokinetic profile is consistent with a three-compartment pharmacokinetic model, with initial rapid distribution phase and the late (terminal) phase.
Distribution
Mean steady state volume of distribution was 113 L. Docetaxel is approximately 94% protein bound in vitro, mainly to α1-acid glycoprotein, albumin, and lipoproteins. In three cancer patients, the in vitro binding to plasma proteins was approximately 97%. Dexamethasone does not affect the protein binding of docetaxel.
Elimination
With extended plasma sampling up to 8 to 22 days post infusion, the estimated mean total body clearance was 18 L/h/m2 (range of means: 14 to 23) and mean terminal elimination half-life was 116 hours (range of means: 92 to 135).
Metabolism
Docetaxel is metabolized by the CYP3A4 isoenzyme in vitro [see Drug Interactions (7)].
Excretion
In three cancer patients urinary and fecal excretion accounted for approximately 6% and 75% of the administered radioactivity, respectively, within 7 days. About 80% of the radioactivity recovered in feces was excreted during the first 48 hours as 1 major and 3 minor metabolites with less than 8% as unchanged drug.
Specific Populations
Effect of Age
A population pharmacokinetic analysis was carried out after docetaxel treatment of 535 patients dosed at 100 mg/m2. Pharmacokinetic parameters estimated by this analysis were very close to those estimated from phase 1 studies. The pharmacokinetics of docetaxel was not influenced by age.
Effect of Gender
The population pharmacokinetics analysis described above also indicated that gender did not influence the pharmacokinetics of docetaxel.
Hepatic Impairment
The population pharmacokinetic analysis described above indicated that in patients with clinical chemistry data suggestive of mild to moderate liver impairment (AST and/or ALT >1.5 times ULN concomitant with alkaline phosphatase >2.5 times ULN), total body clearance was lowered by an average of 27%, resulting in a 38% increase in systemic exposure (AUC). This average, however, includes a substantial range and there is, at present, no measurement that would allow recommendation for dose adjustment in such patients. Patients with combined abnormalities of transaminase and alkaline phosphatase should not be treated with Docetaxel Injection. Patients with severe hepatic impairment have not been studied [see Warnings and Precautions (5.2), Use in Specific Populations (8.6)].
Drug Interaction Studies
Effect of Ketoconazole
The effect of ketoconazole (a strong CYP3A4 inhibitor) on the pharmacokinetics of docetaxel was investigated in 7 cancer patients. Patients were randomized to receive either docetaxel (100 mg/m2 intravenous) alone or docetaxel (10 mg/m2 intravenous) in combination with ketoconazole (200 mg orally once daily for 3 days) in a crossover design with a 3-week washout period. The results of this study indicated that the mean dose-normalized AUC of docetaxel was increased 2.2-fold and its clearance was reduced by 49% when docetaxel was coadministered with ketoconazole [see Dosage and Administration (2.7), Drug Interactions (7)].
Effect of Combination Therapies
- Dexamethasone: Docetaxel total body clearance was not modified by pretreatment with dexamethasone.
- Cisplatin: Clearance of docetaxel in combination therapy with cisplatin was similar to that previously observed following monotherapy with docetaxel. The pharmacokinetic profile of cisplatin in combination therapy with docetaxel was similar to that observed with cisplatin alone.
- Cisplatin and Fluorouracil: The combined administration of docetaxel, cisplatin and fluorouracil in 12 patients with solid tumors had no influence on the pharmacokinetics of each individual drug.
- Prednisone: A population pharmacokinetic analysis of plasma data from 40 patients with metastatic castration-resistant prostate cancer indicated that docetaxel systemic clearance in combination with prednisone is similar to that observed following administration of docetaxel alone.
- Cyclophosphamide and Doxorubicin: A study was conducted in 30 patients with advanced breast cancer to determine the potential for drug-drug interactions between docetaxel (75 mg/m2), doxorubicin (50 mg/m2), and cyclophosphamide (500 mg/m2) when administered in combination. The coadministration of docetaxel had no effect on the pharmacokinetics of doxorubicin and cyclophosphamide when the three drugs were given in combination compared to coadministration of doxorubicin and cyclophosphamide only. In addition, doxorubicin and cyclophosphamide had no effect on docetaxel plasma clearance when the three drugs were given in combination compared to historical data for docetaxel monotherapy.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with docetaxel have not been performed.
Docetaxel was genotoxic by an aneugenic mechanism in the in vitro chromosome aberration test in CHO-K1 cells and in the in vivo micronucleus test in mice administered doses of 0.39 to 1.56 mg/kg (about 1/60th to 1/15th the recommended human dose on a mg/m2 basis). Docetaxel was not mutagenic in the Ames test or the CHO/HGPRT gene mutation assays.
Docetaxel did not reduce fertility in rats when administered in multiple intravenous doses of up to 0.3 mg/kg (about 1/50th the recommended human dose on a mg/m2 basis), but decreased testicular weights were reported. This correlates with findings of a 10-cycle toxicity study (dosing once every 21 days for 6 months) in rats and dogs in which testicular atrophy or degeneration was observed at intravenous doses of 5 mg/kg in rats and 0.375 mg/kg in dogs (about 1/3rd and 1/15th the recommended human dose on a mg/m2 basis, respectively). An increased frequency of dosing in rats produced similar effects at lower dose levels.
-
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic Breast Cancer
The efficacy and safety of docetaxel have been evaluated in locally advanced or metastatic breast cancer after failure of previous chemotherapy (alkylating agent-containing regimens or anthracycline-containing regimens).
Randomized Trials
In one randomized trial, patients with a history of prior treatment with an anthracycline-containing regimen were assigned to treatment with docetaxel (100 mg/m2 every 3 weeks) or the combination of mitomycin (12 mg/m2 every 6 weeks) and vinblastine (6 mg/m2 every 3 weeks). Two hundred three patients were randomized to docetaxel and 189 to the comparator arm. Most patients had received prior chemotherapy for metastatic disease; only 27 patients on the docetaxel arm and 33 patients on the comparator arm entered the study following relapse after adjuvant therapy. Three-quarters of patients had measurable, visceral metastases. The primary endpoint was time to progression. The following table summarizes the study results (see Table 12).
Table 12: Efficacy of Docetaxel in the Treatment of Breast Cancer Patients Previously Treated with an Anthracycline-Containing Regimen (Intent-to-Treat Analysis) - * For the risk ratio, a value less than 1.00 favors docetaxel.
Efficacy Parameter
Docetaxel
(n=203)Mitomycin/Vinblastine
(n=189)p-value
Median Survival
11.4 months
8.7 months
p=0.01
Log RankRisk Ratio*, Mortality
(Docetaxel: Control)95% CI (Risk Ratio)
0.730.58–0.93
Median Time to Progression
4.3 months
2.5 months
p=0.01
Log RankRisk Ratio*,
Progression
(Docetaxel: Control)95% CI (Risk Ratio)
0.75
0.61–0.94Overall Response Rate
Complete Response Rate
28.1%
3.4%
9.5%
1.6%
p<0.0001
Chi Square
In a second randomized trial, patients previously treated with an alkylating-containing regimen were assigned to treatment with docetaxel (100 mg/m2) or doxorubicin (75 mg/m2) every 3 weeks. One hundred sixty-one patients were randomized to docetaxel and 165 patients to doxorubicin. Approximately one-half of patients had received prior chemotherapy for metastatic disease, and one-half entered the study following relapse after adjuvant therapy. Three-quarters of patients had measurable, visceral metastases. The primary endpoint was time to progression. The study results are summarized below (see Table 13).
Table 13: Efficacy of Docetaxel in the Treatment of Breast Cancer Patients Previously Treated with an Alkylating-Containing Regimen (Intent-to-Treat Analysis) - * For the risk ratio, a value less than 1.00 favors docetaxel.
Efficacy Parameter
Docetaxel
(n=161)Doxorubicin
(n=165)p-value
Median Survival
14.7 months
14.3 months
p=0.39
Log RankRisk Ratio*, Mortality
(Docetaxel: Control)95% CI (Risk Ratio)
0.890.68–1.16
Median Time to Progression
6.5 months
5.3 months
p=0.45
Log RankRisk Ratio*, Progression
(Docetaxel: Control)95% CI (Risk Ratio)
0.930.71–1.16
Overall Response Rate
Complete Response Rate
45.3%
6.8%
29.7%
4.2%
p=0.004
Chi Square
In another multicenter open-label, randomized trial (TAX313), in the treatment of patients with advanced breast cancer who progressed or relapsed after one prior chemotherapy regimen, 527 patients were randomized to receive docetaxel monotherapy 60 mg/m2 (n=151), 75 mg/m2 (n=188) or 100 mg/m2 (n=188). In this trial, 94% of patients had metastatic disease and 79% had received prior anthracycline therapy. Response rate was the primary endpoint. Response rates increased with docetaxel dose: 19.9% for the 60 mg/m2 group compared to 22.3% for the 75 mg/m2 and 29.8% for the 100 mg/m2 group; pair-wise comparison between the 60 mg/m2 and 100 mg/m2 groups was statistically significant (p=0.037).
Single Arm Studies
Docetaxel at a dose of 100 mg/m2 was studied in six single arm studies involving a total of 309 patients with metastatic breast cancer in whom previous chemotherapy had failed. Among these, 190 patients had anthracycline-resistant breast cancer, defined as progression during an anthracycline-containing chemotherapy regimen for metastatic disease, or relapse during an anthracycline-containing adjuvant regimen. In anthracycline-resistant patients, the overall response rate was 37.9% (72/190; 95% CI: 31.0–44.8) and the complete response rate was 2.1%.
Docetaxel was also studied in three single arm Japanese studies at a dose of 60 mg/m2, in 174 patients who had received prior chemotherapy for locally advanced or metastatic breast cancer. Among 26 patients whose best response to an anthracycline had been progression, the response rate was 34.6% (95% CI: 17.2–55.7), similar to the response rate in single arm studies of 100 mg/m2.
14.2 Adjuvant Treatment of Breast Cancer
A multicenter, open-label, randomized trial (TAX316) evaluated the efficacy and safety of docetaxel for the adjuvant treatment of patients with axillary-node-positive breast cancer and no evidence of distant metastatic disease. After stratification according to the number of positive lymph nodes (1–3, 4+), 1491 patients were randomized to receive either docetaxel 75 mg/m2 administered 1-hour after doxorubicin 50 mg/m2 and cyclophosphamide 500 mg/m2 (TAC arm), or doxorubicin 50 mg/m2 followed by fluorouracil 500 mg/m2 and cyclophosphamide 500 mg/m2 (FAC arm). Both regimens were administered every 3 weeks for 6 cycles. Docetaxel was administered as a 1-hour infusion; all other drugs were given as intravenous bolus on day 1. In both arms, after the last cycle of chemotherapy, patients with positive estrogen and/or progesterone receptors received tamoxifen 20 mg daily for up to 5 years. Adjuvant radiation therapy was prescribed according to guidelines in place at participating institutions and was given to 69% of patients who received TAC and 72% of patients who received FAC.
Results from a second interim analysis (median follow-up 55 months) are as follows: In study TAX316, the docetaxel-containing combination regimen TAC showed significantly longer disease-free survival (DFS) than FAC (hazard ratio=0.74; 2-sided 95% CI=0.60, 0.92, stratified log rank p=0.0047). The primary endpoint, disease-free survival, included local and distant recurrences, contralateral breast cancer and deaths from any cause. The overall reduction in risk of relapse was 25.7% for TAC-treated patients (see Figure 1).
At the time of this interim analysis, based on 219 deaths, overall survival was longer for TAC than FAC (hazard ratio=0.69, 2-sided 95% CI=0.53, 0.90) (see Figure 2). There will be further analysis at the time survival data mature.
The following table describes the results of subgroup analyses for DFS and OS (see Table 14).
Table 14: Subset Analyses-Adjuvant Breast Cancer Study - * A hazard ratio of less than 1 indicates that TAC is associated with a longer disease free survival or overall survival compared to FAC.
Disease Free Survival
Overall Survival
Patient subset
Number of patients
Hazard ratio*
95% CI
Hazard ratio*
95% CI
No. of positive nodes
Overall
744
0.74
(0.60, 0.92)
0.69
(0.53, 0.90)
1–3
467
0.64
(0.47, 0.87)
0.45
(0.29, 0.70)
4+
277
0.84
(0.63, 1.12)
0.93
(0.66, 1.32)
Receptor status
Positive
566
0.76
(0.59, 0.98)
0.69
(0.48, 0.99)
Negative
178
0.68
(0.48, 0.97)
0.66
(0.44, 0.98)
14.3 Non-small Cell Lung Cancer (NSCLC)
The efficacy and safety of docetaxel has been evaluated in patients with unresectable, locally advanced or metastatic non-small cell lung cancer whose disease has failed prior platinum-based chemotherapy or in patients who are chemotherapy-naïve.
Monotherapy with Docetaxel for NSCLC Previously Treated with Platinum-Based Chemotherapy
Two randomized, controlled trials established that a docetaxel dose of 75 mg/m2 was tolerable and yielded a favorable outcome in patients previously treated with platinum-based chemotherapy (see below). Docetaxel at a dose of 100 mg/m2, however, was associated with unacceptable hematologic toxicity, infections, and treatment-related mortality and this dose should not be used [see Boxed Warning, Dosage and Administration (2.7), Warnings and Precautions (5.3)].
One trial (TAX317), randomized patients with locally advanced or metastatic non-small cell lung cancer, a history of prior platinum-based chemotherapy, no history of taxane exposure, and an ECOG performance status ≤2 to docetaxel or best supportive care. The primary endpoint of the study was survival. Patients were initially randomized to docetaxel 100 mg/m2 or best supportive care, but early toxic deaths at this dose led to a dose reduction to docetaxel 75 mg/m2. A total of 104 patients were randomized in this amended study to either docetaxel 75 mg/m2 or best supportive care.
In a second randomized trial (TAX320), 373 patients with locally advanced or metastatic non-small cell lung cancer, a history of prior platinum-based chemotherapy, and an ECOG performance status ≤2 were randomized to docetaxel 75 mg/m2, docetaxel 100 mg/m2 and a treatment in which the investigator chose either vinorelbine 30 mg/m2 days 1, 8, and 15 repeated every 3 weeks or ifosfamide 2 g/m2 days 1–3 repeated every 3 weeks. Forty percent of the patients in this study had a history of prior paclitaxel exposure. The primary endpoint was survival in both trials. The efficacy data for the docetaxel 75 mg/m2 arm and the comparator arms are summarized in Table 15 and Figures 3 and 4 showing the survival curves for the two studies.
Table 15: Efficacy of Docetaxel in the Treatment of Non-small Cell Lung Cancer Patients Previously Treated with a Platinum-Based Chemotherapy Regimen (Intent-to-Treat Analysis) - * Vinorelbine/Ifosfamide
- † a value less than 1.00 favors docetaxel
- ‡ p≤0.05
- § uncorrected for multiple comparisons
TAX317
TAX320
Docetaxel
75 mg/m2
n=55Best Supportive Care
n=49Docetaxel
75 mg/m2
n=125Control (V/I*)
n=123Overall Survival
Log-rank Testp=0.01
p=0.13
Risk Ratio†, Mortality
(Docetaxel: Control)
95% CI (Risk Ratio)
0.56
(0.35, 0.88)
0.82
(0.63, 1.06)Median Survival
95% CI7.5 months‡
(5.5, 12.8)4.6 months
(3.7, 6.1)5.7 months
(5.1, 7.1)5.6 months
(4.4, 7.9)% 1-year Survival
95% CI12%
(2, 23)20%
(13, 27)Time to Progression
95% CI12.3 weeks‡
(9.0, 18.3)7.0 weeks
(6.0, 9.3)8.3 weeks
(7.0, 11.7)7.6 weeks
(6.7, 10.1)Response Rate
95% CI5.5%
(1.1, 15.1)Not Applicable
5.7%
(2.3, 11.3)0.8%
(0.0, 4.5)Only one of the two trials (TAX317) showed a clear effect on survival, the primary endpoint; that trial also showed an increased rate of survival to one year. In the second study (TAX320) the rate of survival at one year favored docetaxel 75 mg/m2.
Patients treated with docetaxel at a dose of 75 mg/m2 experienced no deterioration in performance status and body weight relative to the comparator arms used in these trials.
Combination Therapy with Docetaxel for Chemotherapy-Naïve NSCLC
In a randomized controlled trial (TAX326), 1218 patients with unresectable stage IIIB or IV NSCLC and no prior chemotherapy were randomized to receive one of three treatments: docetaxel 75 mg/m2 as a 1 hour infusion immediately followed by cisplatin 75 mg/m2 over 30 to 60 minutes every 3 weeks; vinorelbine 25 mg/m2 administered over 6 – 10 minutes on days 1, 8, 15, 22 followed by cisplatin 100 mg/m2 administered on day 1 of cycles repeated every 4 weeks; or a combination of docetaxel and carboplatin.
The primary efficacy endpoint was overall survival. Treatment with docetaxel+cisplatin did not result in a statistically significantly superior survival compared to vinorelbine+cisplatin (see table below). The 95% confidence interval of the hazard ratio (adjusted for interim analysis and multiple comparisons) shows that the addition of docetaxel to cisplatin results in an outcome ranging from a 6% inferior to a 26% superior survival compared to the addition of vinorelbine to cisplatin. The results of a further statistical analysis showed that at least (the lower bound of the 95% confidence interval) 62% of the known survival effect of vinorelbine when added to cisplatin (about a 2-month increase in median survival; Wozniak et al. JCO, 1998) was maintained. The efficacy data for the docetaxel+cisplatin arm and the comparator arm are summarized in Table 16.
Table 16: Survival Analysis of Docetaxel in Combination Therapy for Chemotherapy-Naïve NSCLC - * From the superiority test (stratified log rank) comparing docetaxel+cisplatin to vinorelbine+cisplatin
- † Hazard ratio of docetaxel+cisplatin versus vinorelbine+cisplatin. A hazard ratio of less than 1 indicates that docetaxel+cisplatin is associated with a longer survival.
- ‡ Adjusted for interim analysis and multiple comparisons.
Comparison
Docetaxel + Cisplatin
n=408Vinorelbine + Cisplatin n=405
Kaplan-Meier Estimate of Median Survival
10.9 months
10.0 months
p-value*
0.122
Estimated Hazard Ratio†
0.88
Adjusted 95% CI‡
(0.74, 1.06)
The second comparison in the same three-arm study, vinorelbine+cisplatin versus docetaxel+carboplatin, did not demonstrate superior survival associated with the docetaxel arm (Kaplan-Meier estimate of median survival was 9.1 months for docetaxel+carboplatin compared to 10.0 months on the vinorelbine+cisplatin arm) and the docetaxel+carboplatin arm did not demonstrate preservation of at least 50% of the survival effect of vinorelbine added to cisplatin. Secondary endpoints evaluated in the trial included objective response and time to progression. There was no statistically significant difference between docetaxel+cisplatin and vinorelbine+cisplatin with respect to objective response and time to progression (see Table 17).
Table 17: Response and TTP Analysis of Docetaxel in Combination Therapy for Chemotherapy-Naïve NSCLC - * Adjusted for multiple comparisons.
- † Kaplan-Meier estimates.
Endpoint
Docetaxel + Cisplatin
Vinorelbine + Cisplatin
p-value
Objective Response Rate
(95% CI)*31.6%
(26.5%, 36.8%)24.4%
(19.8%, 29.2%)Not Significant
21.4 weeks
(19.3, 24.6)22.1 weeks
(18.1, 25.6)Not Significant
14.4 Castration-Resistant Prostate Cancer
The safety and efficacy of docetaxel in combination with prednisone in patients with metastatic castration-resistant prostate cancer were evaluated in a randomized multicenter active control trial. A total of 1006 patients with Karnofsky Performance Status (KPS) ≥60 were randomized to the following treatment groups:
- Docetaxel 75 mg/m2 every 3 weeks for 10 cycles.
- Docetaxel 30 mg/m2 administered weekly for the first 5 weeks in a 6-week cycle for 5 cycles.
- Mitoxantrone 12 mg/m2 every 3 weeks for 10 cycles.
All 3 regimens were administered in combination with prednisone 5 mg twice daily, continuously.
In the docetaxel every three week arm, a statistically significant overall survival advantage was demonstrated compared to mitoxantrone. In the docetaxel weekly arm, no overall survival advantage was demonstrated compared to the mitoxantrone control arm. Efficacy results for the docetaxel every 3 week arm versus the control arm are summarized in Table 18 and Figure 5.
Table 18: Efficacy of Docetaxel in the Treatment of Patients with Metastatic Castration-Resistant Prostate Cancer (Intent-to-Treat Analysis) - * Stratified log-rank test. Threshold for statistical significance = 0.0175 because of 3 arms.
Docetaxel + Prednisone
every 3 weeksMitoxantrone + Prednisone
every 3 weeksNumber of patients
335
337
Median survival (months)
18.9
16.5
95% CI
(17.0–21.2)
(14.4–18.6)
Hazard ratio
0.761
—
95% CI
(0.619–0.936)
—
p-value*
0.0094
—
14.5 Gastric Adenocarcinoma
A multicenter, open-label, randomized trial was conducted to evaluate the safety and efficacy of Docetaxel Injection for the treatment of patients with advanced gastric adenocarcinoma, including adenocarcinoma of the gastroesophageal junction, who had not received prior chemotherapy for advanced disease. A total of 445 patients with KPS >70 were treated with either Docetaxel Injection (T) (75 mg/m2 on day 1) in combination with cisplatin (C) (75 mg/m2 on day 1) and fluorouracil (F) (750 mg/m2 per day for 5 days) or cisplatin (100 mg/m2 on day 1) and fluorouracil (1000 mg/m2 per day for 5 days). The length of a treatment cycle was 3 weeks for the TCF arm and 4 weeks for the CF arm. The demographic characteristics were balanced between the two treatment arms. The median age was 55 years, 71% were male, 71% were Caucasian, 24% were 65 years of age or older, 19% had a prior curative surgery and 12% had palliative surgery. The median number of cycles administered per patient was 6 (with a range of 1–16) for the TCF arm compared to 4 (with a range of 1–12) for the CF arm. Time to progression (TTP) was the primary endpoint and was defined as time from randomization to disease progression or death from any cause within 12 weeks of the last evaluable tumor assessment or within 12 weeks of the first infusion of study drugs for patients with no evaluable tumor assessment after randomization. The hazard ratio (HR) for TTP was 1.47 (CF/TCF, 95% CI: 1.19–1.83) with a significantly longer TTP (p=0.0004) in the TCF arm. Approximately 75% of patients had died at the time of this analysis. Overall survival was significantly longer (p=0.0201) in the TCF arm with a HR of 1.29 (95% CI: 1.04–1.61). Efficacy results are summarized in Table 19 and Figures 6 and 7.
Table 19: Efficacy of Docetaxel Injection in the Treatment of Patients with Gastric Adenocarcinoma - * For the hazard ratio (TCF/CF), values less than 1.00 favor the Docetaxel Injection arm.
- † Unstratified log-rank test
Endpoint
TCF
n=221CF
n=224Median TTP (months)
5.6
3.7
(95% CI)
(4.86–5.91)
(3.45–4.47)
Hazard ratio*
0.68
(95% CI)
(0.55–0.84)
†p-value
0.0004
Median survival (months)
9.2
8.6
(95% CI)
(8.38–10.58)
(7.16–9.46)
Hazard ratio*
0.77
(95% CI)
(0.62–0.96)
†p-value
0.0201
Overall Response Rate (CR+PR) (%)
36.7
25.4
p-value
0.0106
Subgroup analyses were consistent with the overall results across age, gender and race.
14.6 Head and Neck Cancer
Induction Chemotherapy Followed by Radiotherapy (TAX323)
The safety and efficacy of Docetaxel Injection in the induction treatment of patients with squamous cell carcinoma of the head and neck (SCCHN) was evaluated in a multicenter, open-label, randomized trial (TAX323). In this study, 358 patients with inoperable locally advanced SCCHN, and WHO performance status 0 or 1, were randomized to one of two treatment arms. Patients on the Docetaxel Injection arm received Docetaxel Injection (T) 75 mg/m2 followed by cisplatin (P) 75 mg/m2 on Day 1, followed by fluorouracil (F) 750 mg/m2 per day as a continuous infusion on Days 1–5. The cycles were repeated every three weeks for 4 cycles. Patients whose disease did not progress received radiotherapy (RT) according to institutional guidelines (TPF/RT). Patients on the comparator arm received cisplatin (P) 100 mg/m2 on Day 1, followed by fluorouracil (F) 1000 mg/m2/day as a continuous infusion on Days 1–5. The cycles were repeated every three weeks for 4 cycles. Patients whose disease did not progress received RT according to institutional guidelines (PF/RT). At the end of chemotherapy, with a minimal interval of 4 weeks and a maximal interval of 7 weeks, patients whose disease did not progress received radiotherapy (RT) according to institutional guidelines. Locoregional therapy with radiation was delivered either with a conventional fraction regimen (1.8 Gy–2.0 Gy once a day, 5 days per week for a total dose of 66 to 70 Gy) or with an accelerated/hyperfractionated regimen (twice a day, with a minimum interfraction interval of 6 hours, 5 days per week, for a total dose of 70 to 74 Gy, respectively). Surgical resection was allowed following chemotherapy, before or after radiotherapy.
The primary endpoint in this study, progression-free survival (PFS), was significantly longer in the TPF arm compared to the PF arm, p=0.0077 (median PFS: 11.4 vs. 8.3 months, respectively) with an overall median follow-up time of 33.7 months. Median overall survival with a median follow-up of 51.2 months was also significantly longer in favor of the TPF arm compared to the PF arm (median OS: 18.6 vs. 14.2 months, respectively). Efficacy results are presented in Table 20 and Figures 8 and 9.
Table 20: Efficacy of Docetaxel Injection in the Induction Treatment of Patients with Inoperable Locally Advanced SCCHN (Intent-to-Treat Analysis) A Hazard ratio of less than 1 favors Docetaxel Injection+cisplatin+fluorouracil - * Stratified log-rank test based on primary tumor site
- † Stratified log-rank test, not adjusted for multiple comparisons
- ‡ Chi square test, not adjusted for multiple comparisons
Endpoint
Docetaxel Injection + Cisplatin + Fluorouracil
n=177Cisplatin + Fluorouracil
n=181Median progression free survival (months)
11.4
8.3
(95% CI)
(10.1–14.0)
(7.4–9.1)
Adjusted Hazard ratio
0.71
(95% CI)
(0.56–0.91)
*p-value
0.0077
Median survival (months)
18.6
14.2
(95% CI)
(15.7–24.0)
(11.5–18.7)
Hazard ratio
0.71
(95% CI)
(0.56–0.90)
†p-value
0.0055
Best overall response (CR + PR) to chemotherapy (%)
(95% CI)
‡p-value67.8
(60.4–74.6)53.6
(46.0–61.0)0.006
Best overall response (CR + PR) to study treatment [chemotherapy +/- radiotherapy] (%)
(95% CI)
‡p-value72.3
(65.1–78.8)58.6
(51.0–65.8)0.006
Induction Chemotherapy Followed by Chemoradiotherapy (TAX324)
The safety and efficacy of Docetaxel Injection in the induction treatment of patients with locally advanced (unresectable, low surgical cure, or organ preservation) SCCHN was evaluated in a randomized, multicenter open-label trial (TAX324). In this study, 501 patients, with locally advanced SCCHN, and a WHO performance status of 0 or 1, were randomized to one of two treatment arms. Patients on the Docetaxel Injection arm received Docetaxel Injection (T) 75 mg/m2 by intravenous infusion on day 1 followed by cisplatin (P) 100 mg/m2 administered as a 30-minute to three-hour intravenous infusion, followed by the continuous intravenous infusion of fluorouracil (F) 1000 mg/m2/day from day 1 to day 4. The cycles were repeated every 3 weeks for 3 cycles. Patients on the comparator arm received cisplatin (P) 100 mg/m2 as a 30-minute to three-hour intravenous infusion on day 1 followed by the continuous intravenous infusion of fluorouracil (F) 1000 mg/m2/day from day 1 to day 5. The cycles were repeated every 3 weeks for 3 cycles.
All patients in both treatment arms who did not have progressive disease were to receive 7 weeks of chemoradiotherapy (CRT) following induction chemotherapy 3 to 8 weeks after the start of the last cycle. During radiotherapy, carboplatin (AUC 1.5) was given weekly as a one-hour intravenous infusion for a maximum of 7 doses. Radiation was delivered with megavoltage equipment using once daily fractionation (2 Gy per day, 5 days per week for 7 weeks for a total dose of 70–72 Gy). Surgery on the primary site of disease and/or neck could be considered at anytime following completion of CRT.
The primary efficacy endpoint, overall survival (OS), was significantly longer (log-rank test, p = 0.0058) with the Docetaxel Injection-containing regimen compared to PF [median OS: 70.6 vs. 30.1 months, respectively, hazard ratio (HR) = 0.70, 95% confidence interval (CI) = 0.54–0.90]. Overall survival results are presented in Table 21 and Figure 10.
Table 21: Efficacy of Docetaxel Injection in the Induction Treatment of Patients with Locally Advanced SCCHN (Intent-to-Treat Analysis) A Hazard ratio of less than 1 favors Docetaxel Injection+cisplatin+fluorouracil NE - not estimable - * unadjusted log-rank test
Endpoint
Docetaxel Injection +
Cisplatin + Fluorouracil
n=255Cisplatin +
Fluorouracil
n=246Median overall survival (months)
(95% CI)70.6
(49.0–NE)30.1
(20.9–51.5)Hazard ratio:
(95% CI)
*p-value0.70
(0.54–0.90)
0.0058 - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Docetaxel Injection, USP is supplied in single-dose or multiple-dose vials as a sterile, pyrogen-free, non-aqueous colorless to pale yellow solution. Discard unused portion of the single-dose vial.
The following strengths are available in a one-vial formulation:
Unit of Sale
Concentration
NDC: 0409-2026-01
Carton of 1 single-dose vial20 mg/2 mL
(10 mg/mL)NDC: 0409-0016-01
Carton of 1 multiple-dose vial160 mg/16 mL
(10 mg/mL)16.2 Storage
Store at 20°C to 25°C (68°F to 77°F). [See USP Controlled Room Temperature]. Retain in the original package to protect from light. Freezing does not adversely affect the product.
After first use and following multiple needle entries and product withdrawals, Docetaxel Injection multiple-dose vials are stable for up to 28 days when stored between 2°C and 8°C (36°F and 46°F) and protected from light.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Bone Marrow Suppression
Advise patients that periodic assessment of their blood count will be performed to detect neutropenia, thrombocytopenia, and/or anemia [see Contraindications (4), Warnings and Precautions (5.3)]. Instruct patients to monitor their temperature frequently and immediately report any occurrence of fever.
Enterocolitis and Neutropenic Colitis
Advise patients of the symptoms of colitis, such as abdominal pain or tenderness, and/or diarrhea, with or without fever, and instruct patients to promptly contact their healthcare provider if they experience these symptoms [see Dosage and Administration (2.7), Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Ask patients whether they have previously received paclitaxel therapy, and if they have experienced a hypersensitivity reaction to paclitaxel. Instruct patients to immediately report to their healthcare provider signs of a hypersensitivity reaction [see Contraindications (4), Warnings and Precautions (5.5)].
Fluid Retention
Advise patients to report signs of fluid retention such as peripheral edema in the lower extremities, weight gain, and dyspnea immediately to their healthcare provider [see Warnings and Precautions (5.6)].
Second Primary Malignancies
Advise patients on the risk of second primary malignancies during treatment with Docetaxel Injection [see Warnings and Precautions (5.7)].
Cutaneous Reactions
Advise patients that localized erythema of the extremities and severe skin toxicities may occur. Instruct patients to immediately report severe cutaneous reactions to their healthcare provider [see Dosage and Administration (2.7), Warnings and Precautions (5.8)].
Neurologic Reactions
Advise patients that neurosensory symptoms or peripheral neuropathy may occur. Instruct patients to immediately report neurologic reactions to their healthcare provider [see Dosage and Administration (2.7), Warnings and Precautions (5.9)].
Eye Disorders
Advise patients that vision disturbances and excessive tearing are associated with Docetaxel Injection administration. Instruct patients to immediately report any vision changes to their healthcare provider [see Warnings and Precautions (5.10)].
Gastrointestinal Reactions
Explain to patients that nausea, vomiting, diarrhea, and constipation are associated with Docetaxel Injection administration. Instruct patients to report any severe events to their healthcare provider [see Adverse Reactions (6)].
Cardiac Disorders
Advise patients to report any irregular and/or rapid heartbeat, severe shortness of breath, dizziness, and/or fainting immediately to their healthcare provider [see Adverse Reactions (6)].
Other Common Adverse Reactions
Advise patients that other common adverse reactions associated with Docetaxel Injection may include alopecia (cases of permanent hair loss have been reported), asthenia, anorexia, dysgeusia, mucositis, myalgia, nail disorders, or pain. Instruct patients to report these reactions to their healthcare provider if serious events occur [see Adverse Reactions (6)].
Importance of Corticosteroids
Explain the significance of oral corticosteroids such as dexamethasone administration to the patient to help facilitate compliance. Instruct patients to report to their healthcare provider if they were not compliant with the oral corticosteroid regimen [see Dosage and Administration (2.6)].
Embryo-Fetal Toxicity
Docetaxel Injection can cause fetal harm. Advise patients to inform their healthcare provider of a known or suspected pregnancy. Advise patients to avoid becoming pregnant while receiving this drug. Advise female patients of reproductive potential to use effective contraceptives during treatment and for 2 months after the last dose of Docetaxel Injection. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 4 months after the last dose of Docetaxel Injection [see Warnings and Precautions (5.12), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during Docetaxel Injection treatment and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential that Docetaxel Injection may impair fertility [see Nonclinical Toxicology (13.1)].
Alcohol Content in Docetaxel Injection
Explain to patients the possible effects of the alcohol content in Docetaxel Injection, including possible effects on the central nervous system [see Warnings and Precautions (5.13)].
Tumor Lysis Syndrome
Advise patients of the potential risk of tumor lysis syndrome and to immediately report any signs or symptoms associated with this event (nausea, vomiting, confusion, shortness of breath, seizure, irregular heartbeat, dark or cloudy urine, reduced amount of urine, unusual tiredness, muscle cramps) to their healthcare provider. Advise patients of the importance of keeping scheduled appointment for blood work or other laboratory tests and of drinking adequate fluids to avoid dehydration. [see Warnings and Precautions (5.14)].
Ability to Drive or Operate Machines
Explain to patients that Docetaxel Injection may impair their ability to drive or operate machines due to its side effects [see Adverse Reactions (6)] or due to the alcohol content of Docetaxel Injection [see Warnings and Precautions (5.13)]. Advise them not to drive or use machines if they experience these side effects during treatment.
Drug Interactions
Inform patients about the risk of drug interactions and the importance of providing a list of prescription and non-prescription drugs to their healthcare provider [see Drug Interactions (7)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: May 2023 Patient Information
Docetaxel (doe-se-TAKS-el) Injection
for intravenous useWhat is the most important information I should know about Docetaxel Injection?
Docetaxel Injection can cause serious side effects, including death.-
The chance of death in people who receive Docetaxel Injection is higher if you:
- o have liver problems
- o receive high doses of Docetaxel Injection
- o have non-small cell lung cancer and have been treated with chemotherapy medicines that contain platinum
- Docetaxel Injection can affect your blood cells. Your healthcare provider should do routine blood tests during treatment with Docetaxel Injection. This will include regular checks of your white blood cell counts. If your white blood cells are too low, your healthcare provider may not treat you with Docetaxel Injection until you have enough white blood cells. People with low white blood cell counts can develop life-threatening infections. The earliest sign of infection may be fever. Follow your healthcare provider's instructions for how often to take your temperature during treatment with Docetaxel Injection. Call your healthcare provider right away if you have a fever.
- Swelling (inflammation) of the small intestine and colon. This can happen at any time during treatment and could lead to death as early as the first day you get symptoms. Tell your healthcare provider right away if you develop new or worse symptoms of intestinal problems, including stomach (abdominal) pain or tenderness or diarrhea, with or without fever.
-
Severe allergic reactions are medical emergencies that can happen in people who receive Docetaxel Injection and can lead to death. You may be at higher risk of developing a severe allergic reaction to Docetaxel Injection if you are allergic to paclitaxel. Your healthcare provider will monitor you closely for allergic reactions during your Docetaxel Injection infusion.
Tell your healthcare provider right away if you have any of these signs of a severe allergic reaction:- o trouble breathing
- o sudden swelling of your face, lips, tongue, throat, or trouble swallowing
- o hives (raised bumps), rash, or redness all over your body
- Your body may hold too much fluid (severe fluid retention) during treatment with Docetaxel Injection. This can be life threatening. To decrease the chance of this happening, you must take another medicine, a corticosteroid, before each Docetaxel Injection treatment. You must take the corticosteroid exactly as your healthcare provider tells you. Tell your healthcare provider or nurse before your Docetaxel Injection treatment if you forgot to take your corticosteroid dose or do not take it as your healthcare provider tells you. Tell your healthcare provider right away if you have swelling in your legs or feet, weight gain or shortness of breath.
-
Risk of new cancers. An increase in new (second) cancers has happened in people treated with Docetaxel Injection together with certain other anticancer treatments. This includes certain blood cancers, such as acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), non-Hodgkin's Lymphoma (NHL), and kidney cancer.
- o Changes in blood counts due to leukemia and other blood disorders may occur years after treatment with Docetaxel Injection.
- Your healthcare provider will check you for new cancers during and after your treatment with Docetaxel Injection.
-
Severe skin problems.
Tell your healthcare provider right away if you have any of these signs of a severe skin reaction:- o redness and swelling of your arms and legs.
- o blistering, peeling, or bleeding on any part of your skin (including your lips, eyes, mouth, nose, genitals, hands or feet) with or without a rash. You may also have flu-like symptoms such as fever, chills, or muscle aches.
- o red, scaly rash all over your body with blisters, small red or white bumps under the skin that contain pus (pustules), and fever.
What is Docetaxel Injection?
Docetaxel Injection is a prescription anticancer medicine used to treat certain people with:- breast cancer
- non-small cell lung cancer
- prostate cancer
- stomach cancer
- head and neck cancer
It is not known if Docetaxel Injection is effective in children.
Do not receive Docetaxel Injection if you:
- have a low white blood cell count.
-
have had a severe allergic reaction to:
- o docetaxel, the active ingredient in Docetaxel Injection, or
- o any other medicines that contain polysorbate 80. Ask your healthcare provider or pharmacist if you are not sure.
See "What is the most important information I should know about Docetaxel Injection?" for the signs and symptoms of a severe allergic reaction.
See the end of this Patient Information for a complete list of the ingredients in Docetaxel Injection.Before you receive Docetaxel Injection, tell your healthcare provider about all of your medical conditions, including if you:
- are allergic to any medicines, including paclitaxel. See "Do not receive Docetaxel Injection if you".
- have liver problems
- have kidney problems
-
are pregnant or plan to become pregnant. Docetaxel Injection can harm your unborn baby. You should not become pregnant during treatment with Docetaxel Injection. Tell your healthcare provider if you become pregnant or you think you may be pregnant during treatment with Docetaxel Injection.
Females who are able to become pregnant:- o Your healthcare provider will check to see if you are pregnant before you start treatment with Docetaxel Injection.
- o You should use effective birth control (contraception) during treatment with Docetaxel Injection and for 2 months after the last dose.
- Males with female partners who are able to become pregnant should use effective birth control during treatment with Docetaxel Injection and for 4 months after the last dose.
- Talk to your healthcare provider if you have questions about birth control options that are right for you.
- are breastfeeding or plan to breastfeed. It is not known if Docetaxel Injection passes into your breast milk. Do not breastfeed during treatment with Docetaxel Injection and for 1 week after the last dose.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Docetaxel Injection may affect the way other medicines work, and other medicines may affect the way Docetaxel Injection works.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How will I receive Docetaxel Injection?
- Docetaxel Injection will be given to you as an intravenous (IV) injection into your vein, usually over 1 hour.
- Docetaxel Injection is usually given every 3 weeks.
- Your healthcare provider will decide how long you will receive treatment with Docetaxel Injection.
- Your healthcare provider will check your blood cell counts and other blood tests during your treatment with Docetaxel Injection to check for side effects of Docetaxel Injection.
- Your healthcare provider may stop your treatment, change the timing of your treatment, or change the dose of your treatment if you have certain side effects while receiving Docetaxel Injection.
What are the possible side effects of Docetaxel Injection?
Docetaxel Injection may cause serious side effects including death.- See "What is the most important information I should know about Docetaxel Injection?"
- Neurologic problems. Neurologic symptoms are common in people who receive Docetaxel Injection but can be severe. Tell your healthcare provider right away if you have numbness, tingling, or burning in your hands or feet (peripheral neuropathy) or weakness of your legs, feet, arms, or hands (motor weakness).
- Vision problems including blurred vision or loss of vision. Tell your healthcare provider right away if you have any vision changes.
- Docetaxel Injection contains alcohol. The alcohol content in Docetaxel Injection may impair your ability to drive or use machinery right after receiving Docetaxel Injection. Consider whether you should drive, operate machinery or do other dangerous activities right after you receive Docetaxel Injection treatment.
- Tumor lysis syndrome (TLS). TLS is caused by the fast breakdown of cancer cells. TLS can cause kidney failure, the need for dialysis treatment, or heart problems, and may lead to death. Your healthcare provider will do blood tests to check for TLS when you first start treatment and during treatment with Docetaxel Injection. Tell your healthcare provider right away if you have any symptoms of TLS during treatment with Docetaxel Injection, including:
- o nausea
- o vomiting
- o confusion
- o shortness of breath
- o irregular heartbeat
- o dark or cloudy urine
- o reduced amount of urine
- o unusual tiredness
- o muscle cramps
- You may experience side effects of Docetaxel Injection that may impair your ability to drive, use tools, or operate machines. If this happens, do not drive or use any tools or machines before discussing with your healthcare provider.
The most common side effects of Docetaxel Injection include:
- infections
- low white blood cells (help fight infections), low red blood cells (anemia), and low platelets (help blood to clot)
- allergic reactions (See "What is the most important information I should know about Docetaxel Injection?")
- changes in your sense of taste
- shortness of breath
- constipation
- decreased appetite
- changes in your fingernails or toenails
- swelling of your hands, face, or feet
- feeling weak or tired
- joint and muscle pain
- nausea and vomiting
- diarrhea
- mouth or lip sores
- hair loss: in some people, permanent hair loss has been reported
- redness of the eye, excess tearing
- skin reactions at the site of Docetaxel Injection administration such as increased skin pigmentation, redness, tenderness, swelling, warmth or dryness of the skin
- tissue damage if Docetaxel Injection leaks out of the vein into the tissues
Tell your healthcare provider if you have a fast or irregular heartbeat, severe shortness of breath, dizziness or fainting during your infusion. If any of these events occurs after your infusion, get medical help right away.
Docetaxel Injection may affect fertility in males. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of Docetaxel Injection. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of Docetaxel Injection.
Medicines are sometimes prescribed for purposes other than those listed in this Patient Information. You can ask your pharmacist or healthcare provider for information about Docetaxel Injection that is written for health professionals.What are the ingredients in Docetaxel Injection?
Active ingredient: docetaxel (anhydrous)
Inactive ingredients: (10 mg injection): polysorbate 80, anhydrous citric acid, dehydrated alcohol and polyethylene glycol.
Distributed by Hospira, Inc., Lake Forest, IL 60045 USA

Novaplus is a registered trademark of Vizient, Inc.
LAB-1496-2.0For more information, call 1-800-441-4100, or go to www.pfizer.com
Every three-week injection of Docetaxel Injection for breast, non-small cell lung and stomach, and head and neck cancers
Take your oral corticosteroid medicine as your healthcare provider tells you.
Oral corticosteroid dosing:
Day 1 Date: Time: AM PM
Day 2 Date: Time: AM PM
(Docetaxel Injection Treatment Day)
Day 3 Date: Time: AM PMEvery three-week injection of Docetaxel Injection for prostate cancer
Take your oral corticosteroid medicine as your healthcare provider tells you.
Oral corticosteroid dosing:
Date: Time:
Date: Time:
(Docetaxel Injection Treatment Day)
Time: -
The chance of death in people who receive Docetaxel Injection is higher if you:
- PRINCIPAL DISPLAY PANEL - 2 mL Vial Label
-
PRINCIPAL DISPLAY PANEL - 2 mL Vial Carton
20 mg/2 mL (10 mg/mL)
NDC: 0409-2026-01
1 x 2 mL
Single-dose vial
Discard unused portion.Docetaxel
Injection, USP20 mg/2 mL
(10 mg/mL)For Intravenous Use only
Ready to add to
infusion solutionCaution: Hazardous Drug
Check concentration
prior to preparation.
See package insert for
complete instructions. -
PRINCIPAL DISPLAY PANEL - 16 mL Vial Label
16 mL
Multiple-dose VialNDC: 0409-0016-01
Rx only
SterileDocetaxel
Injection, USP160 mg/16 mL
(10 mg/mL)For Intravenous Use only
Ready to add to infusion solution. Check
concentration prior to preparation. See
package insert for complete instructions. -
PRINCIPAL DISPLAY PANEL - 16 mL Vial Carton
160 mg/16 mL (10 mg/mL)
NDC: 0409-0016-01
1 x 16 mL
Multiple-dose vialDocetaxel
Injection, USP160 mg/16 mL
(10 mg/mL)For Intravenous Use only
Ready to add to
infusion solutionCaution: Hazardous Drug
Check concentration
prior to preparation.
See package insert for
complete instructions. -
INGREDIENTS AND APPEARANCE
DOCETAXEL
docetaxel anhydrous injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-2026 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOCETAXEL ANHYDROUS (UNII: 699121PHCA) (DOCETAXEL ANHYDROUS - UNII:699121PHCA) DOCETAXEL ANHYDROUS 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) 260 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 4 mg in 1 mL ALCOHOL (UNII: 3K9958V90M) 0.23 mL in 1 mL POLYETHYLENE GLYCOL 300 (UNII: 5655G9Y8AQ) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-2026-01 1 in 1 CARTON 01/10/2022 1 2 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022234 01/10/2022 DOCETAXEL
docetaxel anhydrous injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-0016 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOCETAXEL ANHYDROUS (UNII: 699121PHCA) (DOCETAXEL ANHYDROUS - UNII:699121PHCA) DOCETAXEL ANHYDROUS 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) 260 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 4 mg in 1 mL ALCOHOL (UNII: 3K9958V90M) 0.23 mL in 1 mL POLYETHYLENE GLYCOL 300 (UNII: 5655G9Y8AQ) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-0016-01 1 in 1 CARTON 01/10/2022 1 16 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022234 01/10/2022 Labeler - Hospira, Inc. (141588017) Establishment Name Address ID/FEI Business Operations Zydus Hospira Oncology Private Limited 676190889 ANALYSIS(0409-0016, 0409-2026) , MANUFACTURE(0409-0016, 0409-2026) , PACK(0409-0016, 0409-2026) , LABEL(0409-0016, 0409-2026)
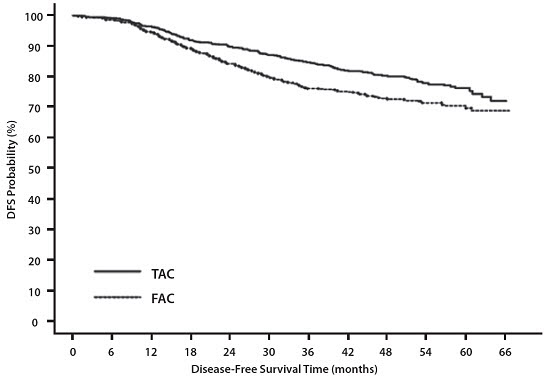
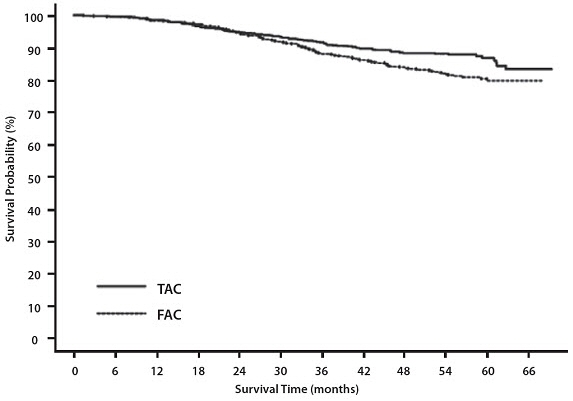
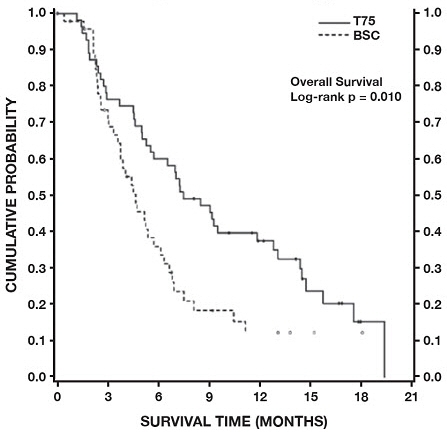
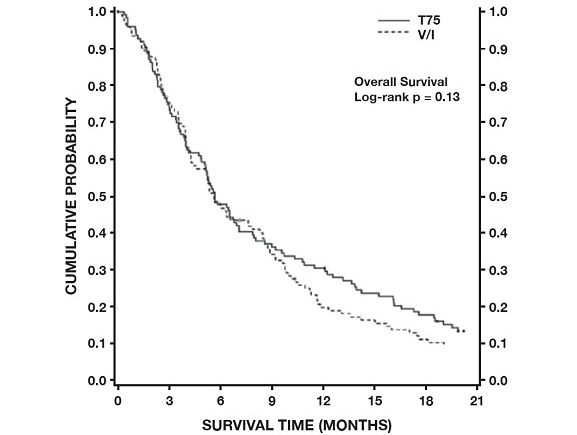
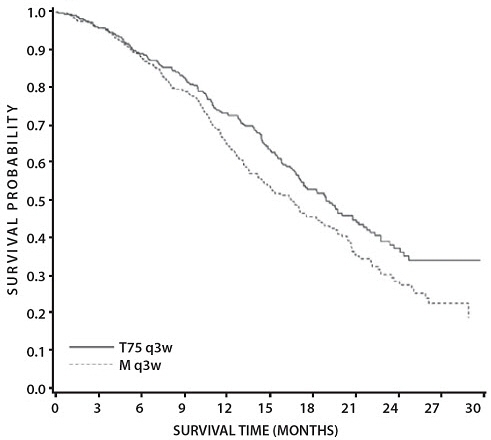
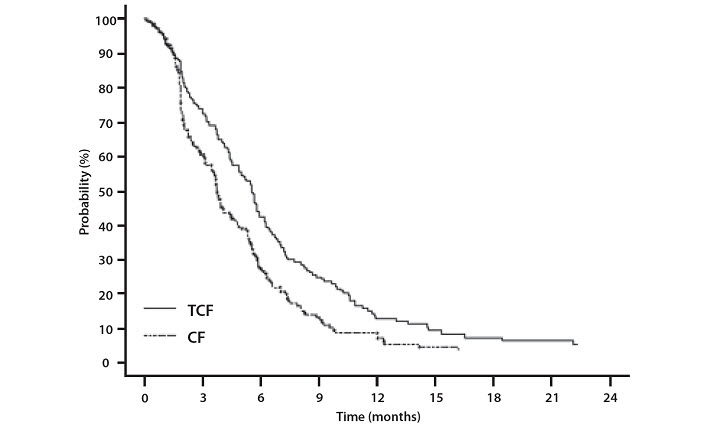
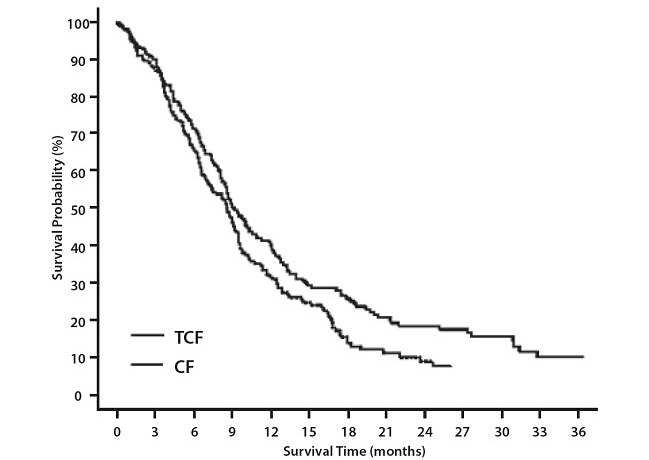
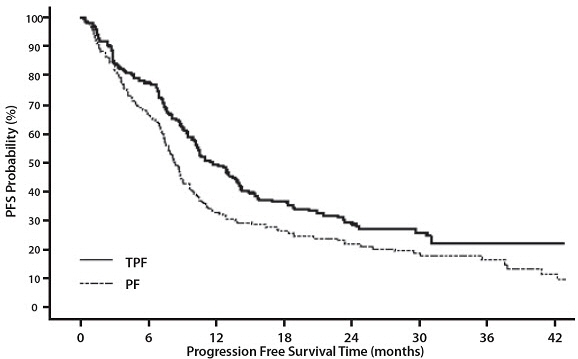
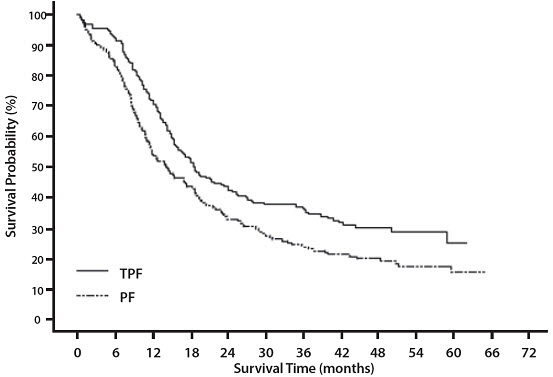
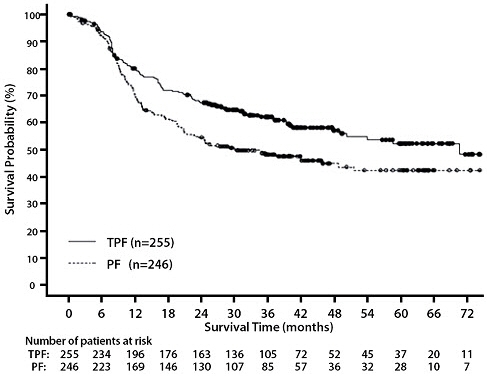




© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.