LACOSAMIDE injection, solution
LACOSAMIDE by
Drug Labeling and Warnings
LACOSAMIDE by is a Prescription medication manufactured, distributed, or labeled by Virtus Pharmaceuticals, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LACOSAMIDE INJECTION safely and effectively. See full prescribing information for LACOSAMIDE INJECTION.
LACOSAMIDE injection, for intravenous use, CV
Initial U.S. Approval: 2008INDICATIONS AND USAGE
Lacosamide injection is indicated for:
- Treatment of partial-onset seizures in patients 17 years of age and older (1.1)
DOSAGE AND ADMINISTRATION
-
Adults (17 years and older):
- Initial dosage for monotherapy for the treatment of partial-onset seizures is 100 mg twice daily (2.1)
- Initial dosage for adjunctive therapy for the treatment of partial-onset seizures is 50 mg twice daily (2.1)
- Maximum recommended dosage for monotherapy and adjunctive therapy is 200 mg twice daily (2.1)
- Increase dosage based on clinical response and tolerability, no more frequently than once per week (2.1)
- Injection: for intravenous use only when oral administration is temporarily not feasible; the recommended dosage is administered two or three times daily over 15 to 60 minutes; obtaining ECG before initiation is recommended in certain patients (2.6, 5.3)
- Dose adjustment is recommended for severe renal impairment (2.3, 12.3)
- Dose adjustment is recommended for mild or moderate hepatic impairment; use in patients with severe hepatic impairment is not recommended (2.4, 12.3)
DOSAGE FORMS AND STRENGTHS
- 200 mg/20 mL single-dose vial for intravenous use (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Monitor patients for suicidal behavior and ideation (5.1)
- Lacosamide may cause dizziness and ataxia (5.2)
- Cardiac Rhythm and Conduction Abnormalities: Obtaining ECG before beginning and after titration to steady-state maintenance is recommended in patients with underlying proarrhythmic conditions or on concomitant medications that affect cardiac conduction; closely monitor these patients (5.3, 7.2)
- Lacosamide may cause syncope (5.4)
- Lacosamide should be gradually withdrawn to minimize the potential of increased seizure frequency (5.5)
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity: Discontinue if no alternate etiology (5.6)
ADVERSE REACTIONS
- Adjunctive therapy: Most common adverse reactions in adults (≥10% and greater than placebo) are diplopia, headache, dizziness, nausea, and somnolence (6.1)
- Monotherapy: Most common adverse reactions are similar to those seen in adjunctive therapy studies (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Virtus Pharmaceuticals, LLC at 1-888-848-3593 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
USE IN SPECIFIC POPULATIONS
- Pregnancy: Based on animal data, may cause fetal harm (8.1)
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Partial-Onset Seizures
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
2.2 Converting From a Single Antiepileptic (AED) to Lacosamide Monotherapy for the Treatment of Partial-Onset Seizures
2.3 Dosage Information for Patients with Renal Impairment
2.4 Dosage Information for Patients with Hepatic Impairment
2.6 Preparation and Administration Information for Lacosamide Injection
2.7 Discontinuation of Lacosamide
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Behavior and Ideation
5.2 Dizziness and Ataxia
5.3 Cardiac Rhythm and Conduction Abnormalities
5.4 Syncope
5.5 Withdrawal of Antiepileptic Drugs (AEDs)
5.6 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity
5.7 Risks in Patients with Phenylketonuria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 or CYP2C9 Inhibitors
7.2 Concomitant Medications that Affect Cardiac Conduction
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
11.2 Lacosamide Injection, USP
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Monotherapy in Patients with Partial-Onset Seizures
14.2 Adjunctive Therapy in Patients with Partial-Onset Seizures
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Partial-Onset Seizures
Lacosamide injection is indicated for the treatment of partial-onset seizures in patients 17 years of age and older.
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended dosage for monotherapy and adjunctive therapy for partial-onset seizures in 17 years of age and older is included in Table 1. Dosage should be increased based on clinical response and tolerability, no more frequently than once per week. Titration increments should not exceed those shown in Table 1.
Table 1: Recommended Dosages for Partial-Onset Seizures (Monotherapy or Adjunctive Therapy) in Patients 17 Years of Age and Older* Age and Body Weight Initial Dosage Titration Regimen Maintenance Dosage - * when not specified, the dosage is the same for monotherapy for partial-onset seizures and adjunctive therapy for partial-onset seizures
- † Monotherapy for partial-onset seizures only
Adults (17 years and older) Monotherapy†:
100 mg twice daily
(200 mg per day)
Adjunctive Therapy: 50 mg twice daily
(100 mg per day)Increase by 50 mg twice daily
(100 mg per day) every weekMonotherapy†:
150 mg to 200 mg twice daily (300 mg to 400 mg per day)
Adjunctive Therapy:
100 mg to 200 mg twice daily (200 mg to 400 mg per day)Alternate Initial Dosage: 200 mg single loading dose, followed 12 hours later by 100 mg twice daily In adjunctive clinical trials in adult patients with partial-onset seizures, a dosage higher than 200 mg twice daily (400 mg per day) was not more effective and was associated with a substantially higher rate of adverse reactions [see Adverse Reactions (6.1) and Clinical Studies (14.2)].
Lacosamide Injection Dosage
Lacosamide injection may be used when oral administration is temporarily not feasible [see Dosage and Administration (2.6) and Warnings and Precautions (5.3)]. Lacosamide injection can be administered intravenously to adult patients with the same dosing regimens described for oral dosing.
The clinical study experience of intravenous lacosamide is limited to 5 days of consecutive treatment.
Loading Dose in Adult Patients (17 Years and Older)
Lacosamide injection may be initiated in adult patients with a single loading dose of 200 mg, followed approximately 12 hours later by 100 mg twice daily (200 mg per day).
The maintenance dose regimen should be continued for one week. Lacosamide can then be titrated as recommended in Table 1. The adult loading dose should be administered with medical supervision because of the increased incidence of CNS adverse reactions [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
The use of a loading dose in pediatric patients has not been studied.
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
2.2 Converting From a Single Antiepileptic (AED) to Lacosamide Monotherapy for the Treatment of Partial-Onset Seizures
For patients who are already on a single AED and will convert to lacosamide monotherapy, withdrawal of the concomitant AED should not occur until the therapeutic dosage of lacosamide is achieved and has been administered for at least 3 days. A gradual withdrawal of the concomitant AED over at least 6 weeks is recommended.
2.3 Dosage Information for Patients with Renal Impairment
For patients with mild to moderate renal impairment, no dosage adjustment is necessary.
For patients with severe renal impairment [creatinine clearance (CLCR) less than 30 mL/min as estimated by the Cockcroft-Gault equation for adults; CLCR less than 30 mL/min/1.73m2 as estimated by the Schwartz equation for pediatric patients] or end-stage renal disease, a reduction of 25% of the maximum dosage is recommended.
In all patients with renal impairment, the dose titration should be performed with caution.
Hemodialysis
Lacosamide is effectively removed from plasma by hemodialysis. Following a 4-hour hemodialysis treatment, dosage supplementation of up to 50% should be considered.
Concomitant Strong CYP3A4 or CYP2C9 Inhibitors
Dose reduction may be necessary in patients with renal impairment who are taking strong inhibitors of CYP3A4 and CYP2C9 [see Drug Interactions (7.1), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
2.4 Dosage Information for Patients with Hepatic Impairment
For patients with mild or moderate hepatic impairment, a reduction of 25% of the maximum dosage is recommended. The dose titration should be performed with caution in patients with hepatic impairment. Lacosamide use is not recommended in patients with severe hepatic impairment.
Concomitant Strong CYP3A4 and CYP2C9 Inhibitors
Dose reduction may be necessary in patients with hepatic impairment who are taking strong inhibitors of CYP3A4 and CYP2C9 [see Drug Interactions (7.1), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
2.6 Preparation and Administration Information for Lacosamide Injection
Preparation
Lacosamide injection can be administered intravenously without further dilution or may be mixed with diluents listed below. The diluted solution should not be stored for more than 4 hours at room temperature.
Diluents:
Sodium Chloride Injection 0.9% (w/v)
Dextrose Injection 5% (w/v)
Lactated Ringer’s InjectionParenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Product with particulate matter or discoloration should not be used.
Lacosamide injection is for single-dose only. Any unused portion of lacosamide injection should be discarded.
Administration
The recommended infusion duration is 30 to 60 minutes; however, infusions as rapid as 15 minutes can be administered in adults if required [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)]. Infusion durations less than 30 minutes are generally not recommended in pediatric patients [see Adverse Reactions (6.1)].
Intravenous infusion of lacosamide may cause bradycardia, AV blocks, and ventricular tachyarrhythmia [see Warnings and Precautions (5.3)]. Obtaining an ECG before beginning lacosamide and after lacosamide is titrated to steady-state maintenance dose is recommended in patients with underlying proarrhythmic conditions or on concomitant medications that affect cardiac conduction [see Drug Interactions (7.2)].
Storage and Stability
The diluted solution should not be stored for more than 4 hours at room temperature. Any unused portion of lacosamide injection should be discarded.
2.7 Discontinuation of Lacosamide
When discontinuing lacosamide injection, a gradual withdrawal over at least 1 week is recommended [see Warnings and Precautions (5.5)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including lacosamide, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number of events is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 2 shows absolute and relative risk by indication for all evaluated AEDs.
Table 2: Risk by Indication for Antiepileptic Drugs in the Pooled Analysis Indication Placebo Patients
with Events
Per 1000 PatientsDrug Patients
with Events Per
1000 PatientsRelative Risk:
Incidence of Events in Drug
Patients/Incidence in Placebo PatientsRisk Difference:
Additional Drug
Patients with
Events Per 1000
PatientsEpilepsy 1.0 3.4 3.5 2.4 Psychiatric 5.7 8.5 1.5 2.9 Other 1.0 1.8 1.9 0.9 Total 2.4 4.3 1.8 1.9 The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar.
Anyone considering prescribing lacosamide or any other AED must balance this risk with the risk of untreated illness. Epilepsy and many other illnesses for which antiepileptics are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.2 Dizziness and Ataxia
Lacosamide may cause dizziness and ataxia in adult and pediatric patients. In adult patients with partial-onset seizures taking 1 to 3 concomitant AEDs, dizziness was experienced by 25% of patients randomized to the recommended doses (200 to 400 mg/day) of lacosamide (compared with 8% of placebo patients) and was the adverse event most frequently leading to discontinuation (3%). Ataxia was experienced by 6% of patients randomized to the recommended doses (200 to 400 mg/day) of lacosamide (compared to 2% of placebo patients). The onset of dizziness and ataxia was most commonly observed during titration. There was a substantial increase in these adverse events at doses higher than 400 mg/day [see Adverse Reactions (6.1)].
5.3 Cardiac Rhythm and Conduction Abnormalities
PR Interval Prolongation, Atrioventricular Block, and Ventricular Tachyarrhythmia
Dose-dependent prolongations in PR interval with lacosamide have been observed in clinical studies in adult patients and in healthy volunteers [see Clinical Pharmacology (12.2)]. In adjunctive clinical trials in adult patients with partial-onset seizures, asymptomatic first-degree atrioventricular (AV) block was observed as an adverse reaction in 0.4% (4/944) of patients randomized to receive lacosamide and 0% (0/364) of patients randomized to receive placebo. One case of profound bradycardia was observed in a patient during a 15-minute infusion of 150 mg lacosamide. When lacosamide is given with other drugs that prolong the PR interval, further PR prolongation is possible.
In the postmarketing setting, there have been reports of cardiac arrhythmias in patients treated with lacosamide, including bradycardia, AV block, and ventricular tachyarrhythmia, which have rarely resulted in asystole, cardiac arrest, and death. Most, although not all, cases have occurred in patients with underlying proarrhythmic conditions, or in those taking concomitant medications that affect cardiac conduction or prolong the PR interval. These events have occurred with both oral and intravenous routes of administration and at prescribed doses as well as in the setting of overdose [see Overdosage (10)].
Lacosamide should be used with caution in patients with underlying proarrhythmic conditions such as known cardiac conduction problems (e.g., marked first-degree AV block, second-degree or higher AV block and sick sinus syndrome without pacemaker), severe cardiac disease (such as myocardial ischemia or heart failure, or structural heart disease), and cardiac sodium channelopathies (e.g., Brugada Syndrome). Lacosamide should also be used with caution in patients on concomitant medications that affect cardiac conduction, including sodium channel blockers, beta-blockers, calcium channel blockers, potassium channel blockers, and medications that prolong the PR interval [see Drug Interactions (7.2)]. In such patients, obtaining an ECG before beginning lacosamide, and after lacosamide is titrated to steady-state maintenance dose, is recommended. In addition, these patients should be closely monitored if they are administered lacosamide through the intravenous route [see Adverse Reactions (6.1) and Drug Interactions (7.2)].
Atrial Fibrillation and Atrial Flutter
In the short-term investigational trials of lacosamide in adult patients with partial-onset seizures there were no cases of atrial fibrillation or flutter. Both atrial fibrillation and atrial flutter have been reported in open label partial-onset seizure trials and in postmarketing experience. In adult patients with diabetic neuropathy, for which lacosamide is not indicated, 0.5% of patients treated with lacosamide experienced an adverse reaction of atrial fibrillation or atrial flutter, compared to 0% of placebo-treated patients. Lacosamide administration may predispose to atrial arrhythmias (atrial fibrillation or flutter), especially in patients with diabetic neuropathy and/or cardiovascular disease.
5.4 Syncope
In the short-term controlled trials of lacosamide in adult patients with partial-onset seizures with no significant system illnesses, there was no increase in syncope compared to placebo. In the short-term controlled trials in adult patients with diabetic neuropathy, for which lacosamide is not indicated, 1.2% of patients who were treated with lacosamide reported an adverse reaction of syncope or loss of consciousness, compared with 0% of placebo-treated patients with diabetic neuropathy. Most of the cases of syncope were observed in patients receiving doses above 400 mg/day. The cause of syncope was not determined in most cases. However, several were associated with either changes in orthostatic blood pressure, atrial flutter/fibrillation (and associated tachycardia), or bradycardia. Cases of syncope have also been observed in open-label clinical partial-onset seizure studies in adult and pediatric patients. These cases were associated with a history of risk factors for cardiac disease and the use of drugs that slow AV conduction.
5.5 Withdrawal of Antiepileptic Drugs (AEDs)
As with all AEDs, lacosamide should be withdrawn gradually (over a minimum of 1 week) to minimize the potential of increased seizure frequency in patients with seizure disorders.
5.6 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multi-organ hypersensitivity, has been reported in patients taking antiepileptic drugs, including lacosamide. Some of these events have been fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematologic abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. This disorder is variable in its expression, and other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity (e.g., fever, lymphadenopathy) may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Lacosamide should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
5.7 Risks in Patients with Phenylketonuria
Phenylalanine can be harmful in patients with phenylketonuria (PKU). Lacosamide oral solution contains aspartame, a source of phenylalanine. A 200 mg dose of lacosamide oral solution (equivalent to 20 mL) contains 0.32 mg of phenylalanine. Before prescribing lacosamide oral solution to a patient with PKU, consider the combined daily amount of phenylalanine from all sources, including lacosamide oral solution.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.1)]
- Dizziness and Ataxia [see Warnings and Precautions (5.2)]
- Cardiac Rhythm and Conduction Abnormalities [see Warnings and Precautions (5.3)]
- Syncope [see Warnings and Precautions (5.4)]
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity Reactions [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Laboratory Abnormalities
Abnormalities in liver function tests have occurred in controlled trials with lacosamide in adult patients with partial-onset seizures who were taking 1 to 3 concomitant anti-epileptic drugs. Elevations of ALT to ≥3x ULN occurred in 0.7% (7/935) of lacosamide patients and 0% (0/356) of placebo patients. One case of hepatitis with transaminases >20x ULN occurred in one healthy subject 10 days after lacosamide treatment completion, along with nephritis (proteinuria and urine casts). Serologic studies were negative for viral hepatitis. Transaminases returned to normal within one month without specific treatment. At the time of this event, bilirubin was normal. The hepatitis/nephritis was interpreted as a delayed hypersensitivity reaction to lacosamide.
Other Adverse Reactions
The following is a list of adverse reactions reported by patients treated with lacosamide in all clinical trials in adult patients, including controlled trials and long-term open-label extension trials. Adverse reactions addressed in other tables or sections are not listed here.
Blood and lymphatic system disorders: neutropenia, anemia
Cardiac disorders: palpitations
Ear and labyrinth disorders: tinnitus
Gastrointestinal disorders: constipation, dyspepsia, dry mouth, oral hypoaesthesia
General disorders and administration site conditions: irritability, pyrexia, feeling drunk
Injury, poisoning, and procedural complications: fall
Musculoskeletal and connective tissue disorders: muscle spasms
Nervous system disorders: paresthesia, cognitive disorder, hypoaesthesia, dysarthria, disturbance in attention, cerebellar syndrome
Psychiatric disorders: confusional state, mood altered, depressed mood
Lacosamide Injection
Adult Patients (17 Years and Older)
Adverse reactions with intravenous administration to adult patients with partial-onset seizures generally were similar to those that occurred with the oral formulation, although intravenous administration was associated with local adverse reactions such as injection site pain or discomfort (2.5%), irritation (1%), and erythema (0.5%). One case of profound bradycardia (26 bpm: BP 100/60 mmHg) occurred in a patient during a 15-minute infusion of 150 mg lacosamide. This patient was on a beta-blocker. Infusion was discontinued and the patient experienced a rapid recovery.
The safety of a 15-minute loading dose administration of lacosamide injection 200 mg to 400 mg followed by oral administration of lacosamide given twice daily at the same total daily dose as the initial intravenous infusion was assessed in an open-label study in adult patients with partial-onset seizures. Patients had to have been maintained on a stable dose regimen of 1 to 2 marketed antiepileptics for at least 28 days prior to treatment assignment. Treatment groups were as follows:
- Single dose of intravenous lacosamide injection 200 mg followed by oral lacosamide 200 mg/day (100 mg every 12 hours)
- Single dose of intravenous lacosamide injection 300 mg followed by oral lacosamide 300 mg/day (150 mg every 12 hours)
- Single dose of intravenous lacosamide injection 400 mg followed by oral lacosamide 400 mg/day (200 mg every 12 hours).
Table 4 gives the incidence of adverse reactions that occurred in ≥5% of adult patients in any lacosamide dosing group.
Table 4: Adverse Reactions in a 15-minute Infusion Study in Adult Patients with Partial-Onset Seizures Adverse Reaction Lacosamide 200 mg N=25
%Lacosamide 300 mg N=50
%Lacosamide 400 mg N=25
%Lacosamide Total N=100
%Eye disorders Diplopia 4 6 20 9 Blurred Vision 0 4 12 5 Gastrointestinal disorders Nausea 0 16 24 14 Dry mouth 0 6 12 6 Vomiting 0 4 12 5 Oral Paresthesia 4 4 8 5 Oral Hypoesthesia 0 6 8 5 Diarrhea 0 8 0 4 General disorders/administration site conditions Fatigue 0 18 12 12 Gait disturbance 8 2 0 3 Chest pain 0 0 12 3 Nervous system disorders Dizziness 20 46 60 43 Somnolence 0 34 36 26 Headache 8 4 16 8 Paresthesia 8 6 4 6 Tremor 0 6 4 4 Abnormal Coordination 0 6 0 3 Skin & subcutaneous tissue disorders Pruritus 0 6 4 4 Hyperhidrosis 0 0 8 2 Adverse reactions that occurred with infusion of lacosamide 200 mg over 15-minutes followed by lacosamide 100 mg administered orally twice per day were similar in frequency to those that occurred in 3-month adjunctive therapy controlled trials. Considering the difference in period of observations (1 week vs. 3 months), the incidence of CNS adverse reactions, such as dizziness, somnolence, and paresthesia may be higher with 15-minute administration of lacosamide injection than with administration over a 30-to 60-minute period.
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of lacosamide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Agranulocytosis
Psychiatric disorders: Aggression, agitation, hallucination, insomnia, psychotic disorder
Skin and subcutaneous tissue disorders: Angioedema, rash, urticaria, Stevens-Johnson syndrome, toxic epidermal necrolysis.
Neurologic disorders: Dyskinesia, new or worsening seizures
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 or CYP2C9 Inhibitors
Patients with renal or hepatic impairment who are taking strong inhibitors of CYP3A4 and CYP2C9 may have a significant increase in exposure to lacosamide. Dose reduction may be necessary in these patients.
7.2 Concomitant Medications that Affect Cardiac Conduction
Lacosamide should be used with caution in patients on concomitant medications that affect cardiac conduction (sodium channel blockers, beta-blockers, calcium channel blockers, potassium channel blockers) including those that prolong PR interval (including sodium channel blocking AEDs), because of a risk of AV block, bradycardia, or ventricular tachyarrhythmia. In such patients, obtaining an ECG before beginning lacosamide, and after lacosamide is titrated to steady-state, is recommended. In addition, these patients should be closely monitored if they are administered lacosamide through the intravenous route [see Warnings and Precautions (5.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as lacosamide, during pregnancy. Encourage women who are taking lacosamide during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) pregnancy registry by calling 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org/.
Risk Summary
Available data from the North American Antiepileptic Drug (NAAED) pregnancy registry, a prospective cohort study, case reports, and a case series with lacosamide use in pregnant women are insufficient to identify a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Lacosamide produced developmental toxicity (increased embryofetal and perinatal mortality, growth deficit) in rats following administration during pregnancy. Developmental neurotoxicity was observed in rats following administration during a period of postnatal development corresponding to the third trimester of human pregnancy. These effects were observed at doses associated with clinically relevant plasma exposures (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
Oral administration of lacosamide to pregnant rats (20, 75, or 200 mg/kg/day) and rabbits (6.25, 12.5, or 25 mg/kg/day) during the period of organogenesis did not produce any effects on the incidences of fetal structural abnormalities. However, the maximum doses evaluated were limited by maternal toxicity in both species and embryofetal death in rats. These doses were associated with maternal plasma lacosamide exposures (AUC) approximately 2 and 1 times (rat and rabbit, respectively) that in humans at the maximum recommended human dose (MRHD) of 400 mg/day.
In two studies in which lacosamide (25, 70, or 200 mg/kg/day and 50, 100, or 200 mg/kg/day) was orally administered to rats throughout pregnancy and lactation, increased perinatal mortality and decreased body weights in the offspring were observed at the highest dose tested. The no-effect dose for pre- and postnatal developmental toxicity in rats (70 mg/kg/day) was associated with a maternal plasma lacosamide AUC similar to that in humans at the MRHD.
Oral administration of lacosamide (30, 90, or 180 mg/kg/day) to rats during the neonatal and juvenile periods of development resulted in decreased brain weights and long-term neurobehavioral changes (altered open field performance, deficits in learning and memory). The early postnatal period in rats is generally thought to correspond to late pregnancy in humans in terms of brain development. The no-effect dose for developmental neurotoxicity in rats was associated with a plasma lacosamide AUC less than that in humans at the MRHD.
In Vitro Data
Lacosamide has been shown in vitro to interfere with the activity of collapsin response mediator protein-2 (CRMP-2), a protein involved in neuronal differentiation and control of axonal outgrowth. Potential adverse effects on CNS development related to this activity cannot be ruled out.
8.2 Lactation
Risk Summary
Data from published literature indicate that lacosamide is present in human milk. There are reports of increased sleepiness in breastfed infants exposed to lacosamide (see Clinical Considerations). There is no information on the effects of lacosamide on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for lacosamide and any potential adverse effects on the breastfed infant from lacosamide or from the underlying maternal condition.
Monitor infants exposed to lacosamide through breastmilk for excess sedation.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients below 1 month of age have not been established.
Animal Data
Lacosamide has been shown in vitro to interfere with the activity of collapsin response mediator protein-2 (CRMP-2), a protein involved in neuronal differentiation and control of axonal outgrowth. Potential related adverse effects on CNS development cannot be ruled out. Administration of lacosamide to rats during the neonatal and juvenile periods of postnatal development (approximately equivalent to neonatal through adolescent development in humans) resulted in decreased brain weights and long-term neurobehavioral changes (altered open field performance, deficits in learning and memory). The no-effect dose for developmental neurotoxicity in rats was associated with a plasma lacosamide exposure (AUC) less than that in humans at the maximum recommended human dose of 400 mg/day.
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
8.5 Geriatric Use
There were insufficient numbers of elderly patients enrolled in partial-onset seizure trials (n=18) to adequately determine whether they respond differently from younger patients.
No lacosamide dose adjustment based on age is necessary. In elderly patients, dose titration should be performed with caution, usually starting at the lower end of the dosing range, reflecting the greater frequency of decreased hepatic function, decreased renal function, increased cardiac conduction abnormalities, and polypharmacy [see Dosage and Administration (2.1, 2.3, 2.4) and Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Based on data in adults, no dose adjustment is necessary in adult and pediatric patients with mild to moderate renal impairment (CLCR ≥30 mL/min). In adult and pediatric patients with severe renal impairment (CLCR <30 mL/min) and in those with end-stage renal disease, a reduction of 25% of the maximum dosage is recommended [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
In all patients with renal impairment, dose titration should be performed with caution.
Lacosamide is effectively removed from plasma by hemodialysis. Dosage supplementation of up to 50% following hemodialysis should be considered.
8.7 Hepatic Impairment
Based on data in adults, for adult and pediatric patients with mild to moderate hepatic impairment, a reduction of 25% of the maximum dosage is recommended. Patients with mild to moderate hepatic impairment should be observed closely during dose titration [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
The pharmacokinetics of lacosamide has not been evaluated in severe hepatic impairment. Lacosamide use is not recommended in patients with severe hepatic impairment.
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
In a human abuse potential study, single doses of 200 mg and 800 mg lacosamide produced euphoria-type subjective responses that differentiated statistically from placebo; at 800 mg, these euphoria-type responses were statistically indistinguishable from those produced by alprazolam, a Schedule IV drug. The duration of the euphoria-type responses following lacosamide was less than that following alprazolam. A high rate of euphoria was also reported as an adverse event in the human abuse potential study following single doses of 800 mg lacosamide (15% [5/34]) compared to placebo (0%) and in two pharmacokinetic studies following single and multiple doses of 300 to 800 mg lacosamide (ranging from 6% [2/33] to 25% [3/12]) compared to placebo (0%). However, the rate of euphoria reported as an adverse event in the lacosamide development program at therapeutic doses was less than 1%.
9.3 Dependence
Abrupt termination of lacosamide in clinical trials with diabetic neuropathic pain patients produced no signs or symptoms that are associated with a withdrawal syndrome indicative of physical dependence. However, psychological dependence cannot be excluded due to the ability of lacosamide to produce euphoria-type adverse events in humans.
-
10 OVERDOSAGE
Events reported after an intake of more than 800 mg (twice the maximum recommended daily dosage) of lacosamide include dizziness, nausea, and seizures (generalized tonic-clonic seizures, status epilepticus). Cardiac conduction disorders, confusion, decreased level of consciousness, cardiogenic shock, cardiac arrest, and coma have also been observed. Fatalities have occurred following lacosamide overdoses of several grams.
There is no specific antidote for overdose with lacosamide. Standard decontamination procedures should be followed. General supportive care of the patient is indicated including monitoring of vital signs and observation of the clinical status of patient. A Certified Poison Control Center should be contacted for up to date information on the management of overdose with lacosamide.
Standard hemodialysis procedures result in significant clearance of lacosamide (reduction of systemic exposure by 50% in 4 hours). Hemodialysis may be indicated based on the patient’s clinical state or in patients with significant renal impairment.
-
11 DESCRIPTION
The chemical name of lacosamide, the single (R)-enantiomer, is (R)-2-acetamido-N-benzyl-3- methoxypropionamide (IUPAC). Lacosamide is a functionalized amino acid. Its molecular formula is C13H18N2O3 and its molecular weight is 250.30. The chemical structure is:
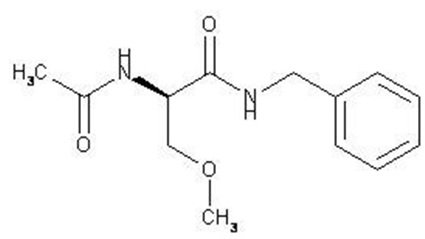
Lacosamide is a white to light yellow powder. It is sparingly soluble in water and slightly soluble in acetonitrile and ethanol.
11.2 Lacosamide Injection, USP
Lacosamide injection, USP is a clear, colorless, sterile solution containing 10 mg lacosamide per mL for intravenous infusion. One 20-mL vial contains 200 mg of lacosamide drug substance. The inactive ingredients are sodium chloride (7.5 mg/mL) and water for injection. Hydrochloric acid is used for pH adjustment. Lacosamide injection, USP has a pH of 3.8 to 5.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which lacosamide exerts its antiepileptic effects in humans remains to be fully elucidated. In vitro electrophysiological studies have shown that lacosamide selectively enhances slow inactivation of voltage-gated sodium channels, resulting in stabilization of hyperexcitable neuronal membranes and inhibition of repetitive neuronal firing.
12.2 Pharmacodynamics
A pharmacokinetic-pharmacodynamic (efficacy) analysis was performed based on the pooled data from the 3 efficacy trials for partial-onset seizures. Lacosamide exposure is correlated with the reduction in seizure frequency. However, doses above 400 mg/day do not appear to confer additional benefit in group analyses.
Cardiac Electrophysiology
Electrocardiographic effects of lacosamide were determined in a double-blind, randomized clinical pharmacology trial of 247 healthy subjects. Chronic oral doses of 400 and 800 mg/day were compared with placebo and a positive control (400 mg moxifloxacin). Lacosamide did not prolong QTc interval and did not have a dose-related or clinically important effect on QRS duration. Lacosamide produced a small, dose-related increase in mean PR interval. At steady-state, the time of the maximum observed mean PR interval corresponded with tmax. The placebo-subtracted maximum increase in PR interval (at tmax) was 7.3 ms for the 400 mg/day group and 11.9 ms for the 800 mg/day group. For patients who participated in the controlled trials, the placebo-subtracted mean maximum increase in PR interval for a 400 mg/day lacosamide dose was 3.1 ms in patients with partial-onset seizures and 9.4 ms for patients with diabetic neuropathy.12.3 Pharmacokinetics
The pharmacokinetics of lacosamide have been studied in healthy adult subjects (age range 18 to 87), adults with partial-onset seizures, adults with diabetic neuropathy, and subjects with renal and hepatic impairment. The pharmacokinetics of lacosamide are similar in healthy subjects, patients with partial-onset seizures, and patients with primary generalized tonic-clonic seizures.
Lacosamide is completely absorbed after oral administration with negligible first-pass effect with a high absolute bioavailability of approximately 100%. The maximum lacosamide plasma concentrations occur approximately 1 to 4 hour post-dose after oral dosing, and elimination half-life is approximately 13 hours. Steady state plasma concentrations are achieved after 3 days of twice daily repeated administration. Pharmacokinetics of lacosamide are dose proportional (100 to 800 mg) and time invariant, with low inter- and intra-subject variability. Compared to lacosamide the major metabolite, O-desmethyl metabolite, has a longer Tmax (0.5 to 12 hours) and elimination half-life (15 to 23 hours).
Absorption and Bioavailability
Lacosamide is completely absorbed after oral administration. The oral bioavailability of lacosamide tablets is approximately 100%. Food does not affect the rate and extent of absorption.After intravenous administration, Cmax is reached at the end of infusion. The 30- and 60-minute intravenous infusions are bioequivalent to the oral tablet. For the 15-minute intravenous infusion, bioequivalence was met for AUC(0-tz) but not for Cmax. The point estimate of Cmax was 20% higher than Cmax for oral tablet and the 90% CI for Cmax exceeded the upper boundary of the bioequivalence range.
In a trial comparing the oral tablet with an oral solution containing 10 mg/mL lacosamide, bioequivalence between both formulations was shown.
A single loading dose of 200 mg approximates steady-state concentrations comparable to the 100 mg twice daily oral administration.
Distribution
The volume of distribution is approximately 0.6 L/kg and thus close to the volume of total body water. Lacosamide is less than 15% bound to plasma proteins.Metabolism and Elimination
Lacosamide is primarily eliminated from the systemic circulation by renal excretion and biotransformation.After intravenous administration of 100 mg [14C]-lacosamide approximately 95% of radioactivity administered was recovered in the urine and less than 0.5% in the feces. The major compounds excreted were unchanged lacosamide (approximately 40% of the dose), its O-desmethyl metabolite (approximately 30%), and a structurally unknown polar fraction (~20%). The plasma exposure of the major human metabolite, O-desmethyl-lacosamide, is approximately 10% of that of lacosamide. This metabolite has no known pharmacological activity.
The CYP isoforms mainly responsible for the formation of the major metabolite (O-desmethyl) are CYP3A4, CYP2C9, and CYP2C19. The elimination half-life of the unchanged drug is approximately 13 hours and is not altered by different doses, multiple dosing or intravenous administration.
There is no enantiomeric interconversion of lacosamide.Specific Populations
Renal Impairment
Lacosamide and its major metabolite are eliminated from the systemic circulation primarily by renal excretion.The AUC of lacosamide was increased approximately 25% in mildly (CLCR 50 to 80 mL/min) and moderately (CLCR 30 to 50 mL/min) and 60% in severely (CLCR≤30 mL/min) renally impaired patients compared to subjects with normal renal function (CLCR>80 mL/min), whereas Cmax was unaffected. Lacosamide is effectively removed from plasma by hemodialysis. Following a 4-hour hemodialysis treatment, AUC of lacosamide is reduced by approximately 50% [see Dosage and Administration (2.3)].
Hepatic Impairment
Lacosamide undergoes metabolism. Subjects with moderate hepatic impairment (Child-Pugh B) showed higher plasma concentrations of lacosamide (approximately 50 to 60% higher AUC compared to healthy subjects). The pharmacokinetics of lacosamide have not been evaluated in severe hepatic impairment [see Dosage and Administration (2.4)].Pediatric Patients
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.Geriatric Patients
In the elderly (>65 years), dose and body-weight normalized AUC and Cmax is about 20% increased compared to young subjects (18 to 64 years). This may be related to body weight and decreased renal function in elderly subjects.Gender
Lacosamide clinical trials indicate that gender does not have a clinically relevant influence on the pharmacokinetics of lacosamide.Race
There are no clinically relevant differences in the pharmacokinetics of lacosamide between Asian, Black, and Caucasian subjects.CYP2C19 Polymorphism
There are no clinically relevant differences in the pharmacokinetics of lacosamide between CYP2C19 poor metabolizers and extensive metabolizers. Results from a trial in poor metabolizers (PM) (N=4) and extensive metabolizers (EM) (N=8) of cytochrome P450 (CYP) 2C19 showed that lacosamide plasma concentrations were similar in PMs and EMs, but plasma concentrations and the amount excreted into urine of the O-desmethyl metabolite were about 70% reduced in PMs compared to EMs.Drug Interactions
In Vitro Assessment of Drug Interactions
In vitro metabolism studies indicate that lacosamide does not induce the enzyme activity of drug metabolizing cytochrome P450 isoforms CYP1A2, 2B6, 2C9, 2C19 and 3A4. Lacosamide did not inhibit CYP 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 2E1, 3A4/5 at plasma concentrations observed in clinical studies.In vitro data suggest that lacosamide has the potential to inhibit CYP2C19 at therapeutic concentrations. However, an in vivo study with omeprazole did not show an inhibitory effect on omeprazole pharmacokinetics.
Lacosamide was not a substrate or inhibitor for P-glycoprotein.
Lacosamide is a substrate of CYP3A4, CYP2C9, and CYP2C19. Patients with renal or hepatic impairment who are taking strong inhibitors of CYP3A4 and CYP2C9 may have increased exposure to lacosamide.
Since <15% of lacosamide is bound to plasma proteins, a clinically relevant interaction with other drugs through competition for protein binding sites is unlikely.
In Vivo Assessment of Drug Interactions
- Drug interaction studies with AEDs
-
Effect of lacosamide on concomitant AEDs
Lacosamide 400 mg/day had no influence on the pharmacokinetics of 600 mg/day valproic acid and 400 mg/day carbamazepine in healthy subjects.
The placebo-controlled clinical studies in patients with partial-onset seizures showed that steady-state plasma concentrations of levetiracetam, carbamazepine, carbamazepine epoxide, lamotrigine, topiramate, oxcarbazepine monohydroxy derivative (MHD), phenytoin, valproic acid, phenobarbital, gabapentin, clonazepam, and zonisamide were not affected by concomitant intake of lacosamide at any dose. -
Effect of concomitant AEDs on lacosamide
Drug-drug interaction studies in healthy subjects showed that 600 mg/day valproic acid had no influence on the pharmacokinetics of 400 mg/day lacosamide. Likewise, 400 mg/day carbamazepine had no influence on the pharmacokinetics of lacosamide in a healthy subject study. Population pharmacokinetics results in patients with partial-onset seizures showed small reductions (15% to 20% lower) in lacosamide plasma concentrations when lacosamide was coadministered with carbamazepine, phenobarbital or phenytoin.
-
Effect of lacosamide on concomitant AEDs
- Drug-drug interaction studies with other drugs
-
Digoxin
There was no effect of lacosamide (400 mg/day) on the pharmacokinetics of digoxin (0.5 mg once daily) in a study in healthy subjects. -
Metformin
There were no clinically relevant changes in metformin levels following coadministration of lacosamide (400 mg/day).
Metformin (500 mg three times a day) had no effect on the pharmacokinetics of lacosamide (400 mg/day). -
Omeprazole
Omeprazole is a CYP2C19 substrate and inhibitor.
There was no effect of lacosamide (600 mg/day) on the pharmacokinetics of omeprazole (40 mg single dose) in healthy subjects. The data indicated that lacosamide had little in vivo inhibitory or inducing effect on CYP2C19.
Omeprazole at a dose of 40 mg once daily had no effect on the pharmacokinetics of lacosamide (300 mg single dose). However, plasma levels of the O-desmethyl metabolite were reduced about 60% in the presence of omeprazole. -
Midazolam
Midazolam is a 3A4 substrate.
There was no effect of lacosamide (200 mg single dose or repeat doses of 400 mg/day given as 200 mg BID) on the pharmacokinetics of midazolam (single dose, 7.5 mg), indicating no inhibitory or inducing effects on CYP3A4. -
Oral Contraceptives
There was no influence of lacosamide (400 mg/day) on the pharmacodynamics and pharmacokinetics of an oral contraceptive containing 0.03 mg ethinylestradiol and 0.15 mg levonorgestrel in healthy subjects, except that a 20% increase in ethinylestradiol Cmax was observed. -
Warfarin
Co-administration of lacosamide (400 mg/day) with warfarin (25 mg single dose) did not result in a clinically relevant change in the pharmacokinetic and pharmacodynamic effects of warfarin in a study in healthy male subjects.
-
Digoxin
- Drug interaction studies with AEDs
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
There was no evidence of drug related carcinogenicity in mice or rats. Mice and rats received lacosamide once daily by oral administration for 104 weeks at doses producing plasma exposures (AUC) up to approximately 1 and 3 times, respectively, the plasma AUC in humans at the maximum recommended human dose (MRHD) of 400 mg/day.Mutagenesis
Lacosamide was negative in an in vitro Ames test and an in vivo mouse micronucleus assay. Lacosamide induced a positive response in the in vitro mouse lymphoma assay.Fertility
No adverse effects on male or female fertility or reproduction were observed in rats at doses producing plasma exposures (AUC) up to approximately 2 times the plasma AUC in humans at the MRHD. -
14 CLINICAL STUDIES
14.1 Monotherapy in Patients with Partial-Onset Seizures
The efficacy of lacosamide in monotherapy was established in a historical-control, multicenter, randomized trial that included 425 patients, age 16 to 70 years, with partial-onset seizures (Study 1). To be included in Study 1, patients were required to be taking stable doses of 1 or 2 marketed antiepileptic drugs. This treatment continued into the 8 week baseline period. To remain in the study, patients were required to have at least 2 partial-onset seizures per 28 days during the 8 week baseline period. The baseline period was followed by a 3 week titration period, during which lacosamide was added to the ongoing antiepileptic regimen. This was followed by a 16-week maintenance period (i.e., a 6-week withdrawal period for background antiepileptic drugs, followed by a 10-week monotherapy period). Patients were randomized 3 to 1 to receive lacosamide 400 mg/day or lacosamide 300 mg/day. Treatment assignments were blinded. Response to treatment was based upon a comparison of the number of patients who met exit criteria during the maintenance phase, compared to historical controls. The historical control consisted of a pooled analysis of the control groups from 8 studies of similar design, which utilized a sub-therapeutic dose of an antiepileptic drug. Statistical superiority to the historical control was considered to be demonstrated if the upper limit from a 2-sided 95% confidence interval for the percentage of patients meeting exit criteria in patients receiving lacosamide remained below the lower 95% prediction limit of 65% derived from the historical control data.
The exit criteria were one or more of the following: (1) doubling of average monthly seizure frequency during any 28 consecutive days, (2) doubling of highest consecutive 2-day seizure frequency, (3) occurrence of a single generalized tonic-clonic seizure, (4) clinically significant prolongation or worsening of overall seizure duration, frequency, type or pattern considered by the investigator to require trial discontinuation, (5) status epilepticus or new onset of serial/cluster seizures. The study population profile appeared comparable to that of the historical control population.
For the lacosamide 400 mg/day group, the estimate of the percentage of patients meeting at least 1 exit criterion was 30% (95% CI: 25%, 36%). The upper limit of the 2-sided 95% CI (36%) was below the threshold of 65% derived from the historical control data, meeting the pre-specified criteria for efficacy. Lacosamide 300 mg/day also met the pre-specified criteria for efficacy.
14.2 Adjunctive Therapy in Patients with Partial-Onset Seizures
The efficacy of lacosamide as adjunctive therapy in partial-onset seizures was established in three 12-week, randomized, double-blind, placebo-controlled, multicenter trials in adult patients (Study 2, Study 3, and Study 4). Enrolled patients had partial-onset seizures with or without secondary generalization, and were not adequately controlled with 1 to 3 concomitant AEDs. During an 8-week baseline period, patients were required to have an average of ≥4 partial-onset seizures per 28 days with no seizure-free period exceeding 21 days. In these 3 trials, patients had a mean duration of epilepsy of 24 years and a median baseline seizure frequency ranging from 10 to 17 per 28 days. 84% of patients were taking 2 to 3 concomitant AEDs with or without concurrent vagal nerve stimulation.
Study 2 compared doses of lacosamide 200, 400, and 600 mg/day with placebo. Study 3 compared doses of lacosamide 400 and 600 mg/day with placebo. Study 4 compared doses of lacosamide 200 and 400 mg/day with placebo. In all three trials, following an 8-week baseline phase to establish baseline seizure frequency prior to randomization, patients were randomized and titrated to the randomized dose (a 1-step back-titration of lacosamide 100 mg/day or placebo was allowed in the case of intolerable adverse events at the end of the titration phase). During the titration phase, in all 3 adjunctive therapy trials, treatment was initiated at 100 mg/day (50 mg twice daily), and increased in weekly increments of 100 mg/day to the target dose. The titration phase lasted 6 weeks in Study 2 and Study 3, and 4 weeks in Study 4. In all three trials, the titration phase was followed by a maintenance phase that lasted 12 weeks, during which patients were to remain on a stable dose of lacosamide.
A reduction in 28 day seizure frequency (baseline to maintenance phase), as compared to the placebo group, was the primary variable in all three adjunctive therapy trials. A statistically significant effect was observed with lacosamide treatment (Figure 1) at doses of 200 mg/day (Study 4), 400 mg/day (Studies 2, 3, and 4), and 600 mg/day (Studies 2 and 3).
Subset evaluations of lacosamide demonstrate no important differences in seizure control as a function of gender or race, although data on race was limited (about 10% of patients were non-Caucasian).
Figure 1 - Median Percent Reduction in Seizure Frequency per 28 days from Baseline to the Maintenance Phase by Dose 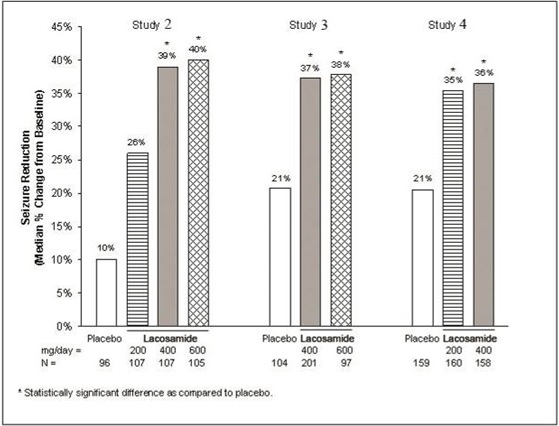
Figure 2 presents the percentage of patients (X-axis) with a percent reduction in partial seizure frequency (responder rate) from baseline to the maintenance phase at least as great as that represented on the Y-axis. A positive value on the Y-axis indicates an improvement from baseline (i.e., a decrease in seizure frequency), while a negative value indicates a worsening from baseline (i.e., an increase in seizure frequency). Thus, in a display of this type, a curve for an effective treatment is shifted to the left of the curve for placebo. The proportion of patients achieving any particular level of reduction in seizure frequency was consistently higher for the lacosamide groups, compared to the placebo group. For example, 40% of patients randomized to lacosamide (400 mg/day) experienced a 50% or greater reduction in seizure frequency, compared to 23% of patients randomized to placebo. Patients with an increase in seizure frequency >100% are represented on the Y- axis as equal to or greater than -100%.
Figure 2 - Proportion of Patients by Responder Rate for Lacosamide and Placebo Groups in Studies 2, 3, and 4 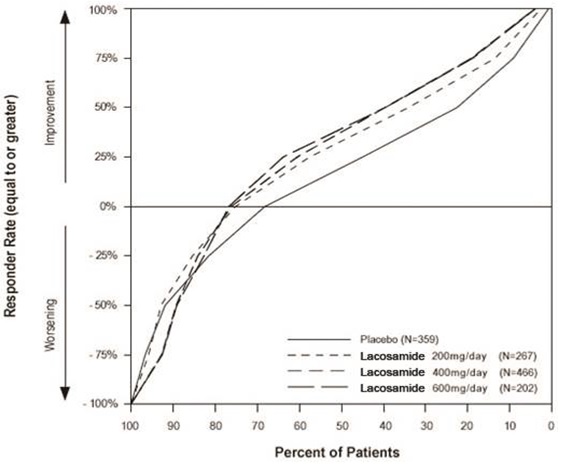
Pediatric use information is approved for UCB, Inc.’s VIMPAT® (lacosamide) injection. However, due to UCB, Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Lacosamide Injection, USP
- 200 mg/20 mL is a clear, colorless sterile solution supplied in 20 mL colorless single-dose glass vials.
200 mg/20 mL vial in cartons of 10 vials NDC: 69543-455-10
- 200 mg/20 mL is a clear, colorless sterile solution supplied in 20 mL colorless single-dose glass vials.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide). The Medication Guide accompanies the product.
Suicidal Thinking and Behavior
Patients, their caregivers, and families should be counseled that AEDs, including lacosamide, may increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers [see Warnings and Precautions (5.1)].Dizziness and Ataxia
Patients should be counseled that lacosamide use may cause dizziness, double vision, abnormal coordination and balance, and somnolence. Patients taking lacosamide should be advised not to drive, operate complex machinery, or engage in other hazardous activities until they have become accustomed to any such effects associated with lacosamide [see Warnings and Precautions (5.2)].Cardiac Rhythm and Conduction Abnormalities
Patients should be counseled that lacosamide is associated with electrocardiographic changes that may predispose to irregular heart beat and syncope. Cardiac arrest has been reported. This risk is increased in patients with underlying cardiovascular disease, with heart conduction problems, or who are taking other medications that affect the heart. Patients should be made aware of and report cardiac signs or symptoms to their healthcare provider right away. Patients who develop syncope should lay down with raised legs and contact their healthcare provider [see Warnings and Precautions (5.3)].Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity
Patients should be aware that lacosamide may cause serious hypersensitivity reactions affecting multiple organs such as the liver and kidney. Lacosamide should be discontinued if a serious hypersensitivity reaction is suspected. Patients should also be instructed to report promptly to their physicians any symptoms of liver toxicity (e.g., fatigue, jaundice, dark urine) [see Warnings and Precautions (5.6)].Pregnancy Registry
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during lacosamide therapy. Encourage patients to enroll in the North American Antiepileptic Drug (NAAED) pregnancy registry if they become pregnant. This registry is collecting information about the safety of AEDs during pregnancy [see Use in Specific Populations (8.1)].Lactation
Advise breastfeeding women using lacosamide to monitor infants for excess sleepiness and to seek medical care if they notice this sign [see Use in Specific Populations (8.2)].Manufactured by:
Gland Pharma Limited
Hyderabad-502307, India.Distributed by:
Virtus Pharmaceuticals, LLC
Langhorne, PA 19047Revised: January 2023
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration
- PRINCIPAL DISPLAY PANEL - 200mg per 20mL Label
- PRINCIPAL DISPLAY PANEL - 200mg per 20mL Carton
-
INGREDIENTS AND APPEARANCE
LACOSAMIDE
lacosamide injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 69543-455 Route of Administration INTRAVENOUS DEA Schedule CV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LACOSAMIDE (UNII: 563KS2PQY5) (LACOSAMIDE - UNII:563KS2PQY5) LACOSAMIDE 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 69543-455-10 10 in 1 CARTON 04/17/2023 09/30/2027 1 NDC: 69543-455-20 20 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA215628 04/17/2023 09/30/2027 Labeler - Virtus Pharmaceuticals, LLC (079659493)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.