PRANDIN- repaglinide tablet
Prandin by
Drug Labeling and Warnings
Prandin by is a Prescription medication manufactured, distributed, or labeled by Physicians Total Care, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
DESCRIPTION
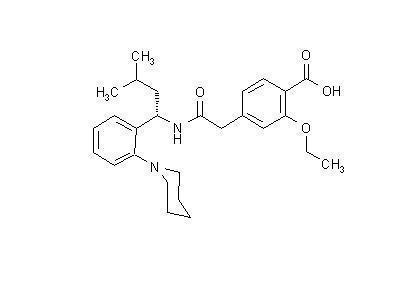
PRANDIN® (repaglinide) is an oral blood glucose-lowering drug of the meglitinide class used in the management of type 2 diabetes mellitus (also known as non-insulin dependent diabetes mellitus or NIDDM). Repaglinide, S(+)2-ethoxy-4(2((3-methyl-1-(2-(1-piperidinyl) phenyl)-butyl) amino)-2-oxoethyl) benzoic acid, is chemically unrelated to the oral sulfonylurea insulin secretagogues.
The structural formula is as shown below:
Repaglinide is a white to off-white powder with molecular formula C27 H36 N2 O4 and a molecular weight of 452.6. PRANDIN tablets contain 0.5 mg, 1 mg, or 2 mg of repaglinide. In addition each tablet contains the following inactive ingredients: calcium hydrogen phosphate (anhydrous), microcrystalline cellulose, maize starch, polacrilin potassium, povidone, glycerol (85%), magnesium stearate, meglumine, and poloxamer. The 1 mg and 2 mg tablets contain iron oxides (yellow and red, respectively) as coloring agents.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Repaglinide lowers blood glucose levels by stimulating the release of insulin from the pancreas. This action is dependent upon functioning beta (ß) cells in the pancreatic islets. Insulin release is glucose-dependent and diminishes at low glucose concentrations.
Repaglinide closes ATP-dependent potassium channels in the ß-cell membrane by binding at characterizable sites. This potassium channel blockade depolarizes the ß-cell, which leads to an opening of calcium channels. The resulting increased calcium influx induces insulin secretion. The ion channel mechanism is highly tissue selective with low affinity for heart and skeletal muscle.
Pharmacokinetics
Absorption: After oral administration, repaglinide is rapidly and completely absorbed from the gastrointestinal tract. After single and multiple oral doses in healthy subjects or in patients, peak plasma drug levels (Cmax) occur within 1 hour (Tmax). Repaglinide is rapidly eliminated from the blood stream with a half-life of approximately 1 hour. The mean absolute bioavailability is 56%. When repaglinide was given with food, the mean Tmax was not changed, but the mean Cmax and AUC (area under the time/plasma concentration curve) were decreased 20% and 12.4%, respectively.
Distribution: After intravenous (IV) dosing in healthy subjects, the volume of distribution at steady state (Vss) was 31 L, and the total body clearance (CL) was 38 L/h. Protein binding and binding to human serum albumin was greater than 98%.
Metabolism: Repaglinide is completely metabolized by oxidative biotransformation and direct conjugation with glucuronic acid after either an IV or oral dose. The major metabolites are an oxidized dicarboxylic acid (M2), the aromatic amine (M1), and the acyl glucuronide (M7). The cytochrome P-450 enzyme system, specifically 2C8 and 3A4, have been shown to be involved in the N-dealkylation of repaglinide to M2 and the further oxidation to M1. Metabolites do not contribute to the glucose-lowering effect of repaglinide.
Repaglinide appears to be a substrate for active hepatic uptake transporter (organic anion transporting protein OATP1B1).
Excretion: Within 96 hours after dosing with 14C-repaglinide as a single, oral dose, approximately 90% of the radiolabel was recovered in the feces and approximately 8% in the urine. Only 0.1% of the dose is cleared in the urine as parent compound. The major metabolite (M2) accounted for 60% of the administered dose. Less than 2% of parent drug was recovered in feces.
Pharmacokinetic Parameters: The pharmacokinetic parameters of repaglinide obtained from a single-dose, crossover study in healthy subjects and from a multiple-dose, parallel, dose-proportionality (0.5, 1, 2 and 4 mg) study in patients with type 2 diabetes are summarized in the following table:
- * dosed preprandially with three meals
Parameter
Patients with type 2 diabetes*
Dose
0.5 mg
1 mg
2 mg
4 mg
AUC0-24 hr Mean ±SD (ng/mL*hr):
68.9 ± 154.4
125.8 ± 129.8
152.4 ± 89.6
447.4 ± 211.3
Dose
0.5 mg
1 mg
2 mg
4 mg
Cmax0-5 hr Mean ±SD (ng/mL):
9.8 ± 10.2
18.3 ± 9.1
26.0 ± 13.0
65.8 ± 30.1
Dose
0.5 - 4 mg
Tmax0-5 hr Means (SD)
1.0 - 1.4 (0.3 - 0.5) hr
Dose
0.5 - 4 mg
T½ Means (Ind Range)
1.0 - 1.4 (0.4 - 8.0) hr
Parameter
Healthy Subjects
CL based on i.v.
38 ± 16 L/hr
Vss based on i.v.
31 ± 12 L
AbsBio
56 ± 9%
CL= total body clearance
Vss= volume of distribution at steady state
AbsBio = absolute bioavailability
These data indicate that repaglinide did not accumulate in serum. Clearance of oral repaglinide did not change over the 0.5 - 4 mg dose range, indicating a linear relationship between dose and plasma drug levels.
Variability of Exposure: Repaglinide AUC after multiple doses of 0.25 to 4 mg with each meal varies over a wide range. The intra-individual and inter-individual coefficients of variation were 36% and 69%, respectively. AUC over the therapeutic dose range included 69 to 1005 ng/mL*hr, but AUC exposure up to 5417 ng/mL*hr was reached in dose escalation studies without apparent adverse consequences.
Special Populations
Geriatric: Healthy volunteers were treated with a regimen of 2 mg taken before each of 3 meals. There were no significant differences in repaglinide pharmacokinetics between the group of patients <65 years of age and a comparably sized group of patients ≥65 years of age (See PRECAUTIONS, Geriatric Use).
Pediatric: No studies have been performed in pediatric patients.
Gender: A comparison of pharmacokinetics in males and females showed the AUC over the 0.5 mg to 4 mg dose range to be 15% to 70% higher in females with type 2 diabetes. This difference was not reflected in the frequency of hypoglycemic episodes (male: 16%; female: 17%) or other adverse events. With respect to gender, no change in general dosage recommendation is indicated since dosage for each patient should be individualized to achieve optimal clinical response.
Race: No pharmacokinetic studies to assess the effects of race have been performed, but in a U.S. 1-year study in patients with type 2 diabetes, the blood glucose-lowering effect was comparable between Caucasians (n=297) and African-Americans (n=33). In a U.S. dose-response study, there was no apparent difference in exposure (AUC) between Caucasians (n=74) and Hispanics (n=33).
Drug-Drug Interactions
Drug interaction studies performed in healthy volunteers show that PRANDIN had no clinically relevant effect on the pharmacokinetic properties of digoxin, theophylline, or warfarin. Co-administration of cimetidine with PRANDIN did not significantly alter the absorption and disposition of repaglinide.
Additionally, the following drugs were studied in healthy volunteers with co-administration of PRANDIN. Listed below are the results:
CYP2C8 and CYP3A4 Inhibitors/Inducer
Gemfibrozil and Itraconazole: Co-administration of gemfibrozil (600 mg) and a single dose of 0.25 mg PRANDIN (after 3 days of twice-daily 600 mg gemfibrozil) resulted in an 8.1-fold higher repaglinide AUC and prolonged repaglinide half-life from 1.3 to 3.7 hr. Co-administration with itraconazole and a single dose of 0.25 mg PRANDIN (on the third day of a regimen of 200 mg initial dose, twice-daily 100 mg itraconazole) resulted in a 1.4-fold higher repaglinide AUC. Co-administration of both gemfibrozil and itraconazole with PRANDIN resulted in a 19-fold higher repaglinide AUC and prolonged repaglinide half-life to 6.1 hr. Plasma repaglinide concentration at 7 h increased 28.6-fold with gemfibrozil co-administration and 70.4-fold with the gemfibrozil-itraconazole combination (see CONTRAINDICATIONS, PRECAUTIONS, Drug-Drug Interactions).
Fenofibrate: Co-administration of 200 mg fenofibrate with a single dose of 0.25 mg repaglinide (after 5 days of once daily fenofibrate 200 mg) resulted in unchanged AUC and Cmax values for both drugs.
Ketoconazole: Co-administration of 200 mg ketoconazole and a single dose of 2 mg PRANDIN (after 4 days of once daily ketoconazole 200 mg) resulted in a 15% and 16% increase in repaglinide AUC and Cmax, respectively. The increases were from 20.2 ng/mL to 23.5 ng/mL for Cmax and from 38.9 ng/mL*hr to 44.9 ng/mL*hr for AUC.
Trimethoprim: Co-administration of 160 mg trimethoprim and a single dose of 0.25 mg PRANDIN (after 2 days of twice daily and one dose on the third day of trimethoprim 160 mg) resulted in a 61% and 41% increase in repaglinide AUC and Cmax, respectively. The increase in AUC was from 5.9 ng/mL*hr to 9.6 ng/mL*hr and the increase in Cmax was from 4.7 ng/mL to 6.6 ng/mL.
Cyclosporine: Co-administration of 100 mg cyclosporine with a single dose of 0.25 mg repaglinide (after two 100 mg doses of cyclosporine twelve hours apart) increased the repaglinide (0.25 mg) Cmax 1.8-fold and the AUC 2.5-fold in an interaction study with healthy volunteers (see PRECAUTIONS, Drug-Drug Interactions).
Rifampin: Co-administration of 600 mg rifampin and a single dose of 4 mg PRANDIN (after 6 days of once daily rifampin 600 mg) resulted in a 32% and 26% decrease in repaglinide AUC and Cmax, respectively. The decreases were from 40.4 ng/mL to 29.7 ng/mL for Cmax and from 56.8 ng/mL*hr to 38.7 ng/mL*hr for AUC.
In another study, co-administration of 600 mg rifampin and a single dose of 4 mg PRANDIN (after 6 days of once daily rifampin 600 mg) resulted in a 48% and 17% decrease in repaglinide median AUC and median Cmax respectively. The median decreases were from 54 ng/mL*hr to 28 ng/mL*hr for AUC and from 35 ng/mL to 29 ng/mL for Cmax. PRANDIN administered by itself (after 7 days of once daily rifampin 600 mg) resulted in an 80% and 79% decrease in repaglinide median AUC and Cmax respectively. The decreases were from 54 ng/mL*hr to 11 ng/mL*hr for AUC and from 35 ng/mL to 7.5 ng/mL for Cmax.
Levonorgestrel & Ethinyl Estradiol: Co-administration of a combination tablet of 0.15 mg levonorgestrel and 0.03 mg ethinyl estradiol administered once daily for 21 days with 2 mg PRANDIN administered three times daily (days 1-4) and a single dose on Day 5 resulted in 20% increases in repaglinide, levonorgestrel, and ethinyl estradiol Cmax. The increase in repaglinide Cmax was from 40.5 ng/mL to 47.4 ng/mL. Ethinyl estradiol AUC parameters were increased by 20%, while repaglinide and levonorgestrel AUC values remained unchanged.
Simvastatin: Co-administration of 20 mg simvastatin and a single dose of 2 mg PRANDIN (after 4 days of once daily simvastatin 20 mg and three times daily PRANDIN 2 mg) resulted in a 26% increase in repaglinide Cmax from 23.6 ng/mL to 29.7 ng/mL. AUC was unchanged.
Nifedipine: Co-administration of 10 mg nifedipine with a single dose of 2 mg PRANDIN (after 4 days of three times daily nifedipine 10 mg and three times daily PRANDIN 2 mg) resulted in unchanged AUC and Cmax values for both drugs.
Clarithromycin: Co-administration of 250 mg clarithromycin and a single dose of 0.25 mg PRANDIN (after 4 days of twice daily clarithromycin 250 mg) resulted in a 40% and 67% increase in repaglinide AUC and Cmax, respectively. The increase in AUC was from 5.3 ng/mL*hr to 7.5 ng/mL*hr and the increase in Cmax was from 4.4 ng/mL to 7.3 ng/mL.
Deferasirox: Co-administration of deferasirox (30 mg/kg/day for 4 days) and repaglinide (single dose of 0.5 mg) resulted in an increase in repaglinide systemic exposure (AUC) to 2.3-fold of control and an increase in Cmax of 62% (see PRECAUTIONS, Drug-Drug Interactions).
Renal Insufficiency: Single-dose and steady-state pharmacokinetics of repaglinide were compared between patients with type 2 diabetes and normal renal function (CrCl > 80 mL/min), mild to moderate renal function impairment (CrCl = 40 – 80 mL/min), and severe renal function impairment (CrCl = 20 – 40 mL/min). Both AUC and Cmax of repaglinide were similar in patients with normal and mild to moderately impaired renal function (mean values 56.7 ng/mL*hr vs 57.2 ng/mL*hr and 37.5 ng/mL vs 37.7 ng/mL, respectively.) Patients with severely reduced renal function had elevated mean AUC and Cmax values (98.0 ng/mL*hr and 50.7 ng/mL, respectively), but this study showed only a weak correlation between repaglinide levels and creatinine clearance. Initial dose adjustment does not appear to be necessary for patients with mild to moderate renal dysfunction. However, patients with type 2 diabetes who have severe renal function impairment should initiate PRANDIN therapy with the 0.5 mg dose – subsequently, patients should be carefully titrated. Studies were not conducted in patients with creatinine clearances below 20 mL/min or patients with renal failure requiring hemodialysis.
Hepatic Insufficiency: A single-dose, open-label study was conducted in 12 healthy subjects and 12 patients with chronic liver disease (CLD) classified by Child-Pugh scale and caffeine clearance. Patients with moderate to severe impairment of liver function had higher and more prolonged serum concentrations of both total and unbound repaglinide than healthy subjects (AUChealthy: 91.6 ng/mL*hr; AUCCLD patients: 368.9 ng/mL*hr; Cmax, healthy: 46.7 ng/mL; Cmax, CLD patients: 105.4 ng/mL). AUC was statistically correlated with caffeine clearance. No difference in glucose profiles was observed across patient groups. Patients with impaired liver function may be exposed to higher concentrations of repaglinide and its associated metabolites than would patients with normal liver function receiving usual doses. Therefore, PRANDIN should be used cautiously in patients with impaired liver function. Longer intervals between dose adjustments should be utilized to allow full assessment of response.
Clinical Trials
Monotherapy Trials
A four-week, double-blind, placebo-controlled dose-response trial was conducted in 138 patients with type 2 diabetes using doses ranging from 0.25 to 4 mg taken with each of three meals. PRANDIN therapy resulted in dose-proportional glucose lowering over the full dose range. Plasma insulin levels increased after meals and reverted toward baseline before the next meal. Most of the fasting blood glucose-lowering effect was demonstrated within 1-2 weeks.
In a double-blind, placebo-controlled, 3-month dose titration study, PRANDIN or placebo doses for each patient were increased weekly from 0.25 mg through 0.5, 1, and 2 mg, to a maximum of 4 mg, until a fasting plasma glucose (FPG) level <160 mg/dL was achieved or the maximum dose reached. The dose that achieved the targeted control or the maximum dose was continued to end of study. FPG and 2-hour post-prandial glucose (PPG) increased in patients receiving placebo and decreased in patients treated with repaglinide. Differences between the repaglinide- and placebo-treated groups were -61 mg/dL (FPG) and -104 mg/dL (PPG). The between-group change in HbA1c, which reflects long-term glycemic control, was 1.7% units.
PRANDIN vs. Placebo Treatment: Mean FPG, PPG, and HbA1c Changes from baseline after 3 months of treatment: - * p < 0.05 for between group difference
FPG (mg/dL)
PPG (mg/dL)
HbA1c (%)
PL
R
PL
R
PL
R
Baseline
215.3
220.2
245.2
261.7
8.1
8.5
Change from Baseline
(at last visit)
30.3
-31.0*
56.5
-47.6*
1.1
-0.6*
FPG = fasting plasma glucose
PPG = post-prandial glucose
PL = placebo (N=33)
R = repaglinide (N=66)
Another double-blind, placebo-controlled trial was carried out in 362 patients treated for 24 weeks. The efficacy of 1 and 4 mg preprandial doses was demonstrated by lowering of fasting blood glucose and by HbA1c at the end of the study. HbA1c for the PRANDIN- treated groups (1 and 4 mg groups combined) at the end of the study was decreased compared to the placebo-treated group in previously naïve patients and in patients previously treated with oral hypoglycemic agents by 2.1% units and 1.7% units, respectively. In this fixed-dose trial, patients who were naïve to oral hypoglycemic agent therapy and patients in relatively good glycemic control at baseline (HbA1c below 8%) showed greater blood glucose-lowering including a higher frequency of hypoglycemia. Patients who were previously treated and who had baseline HbA1c ≥ 8% reported hypoglycemia at the same rate as patients randomized to placebo. There was no average gain in body weight when patients previously treated with oral hypoglycemic agents were switched to PRANDIN. The average weight gain in patients treated with PRANDIN and not previously treated with sulfonylurea drugs was 3.3%.
The dosing of PRANDIN relative to meal-related insulin release was studied in three trials including 58 patients. Glycemic control was maintained during a period in which the meal and dosing pattern was varied (2, 3 or 4 meals per day; before meals x 2, 3, or 4) compared with a period of 3 regular meals and 3 doses per day (before meals x 3). It was also shown that PRANDIN can be administered at the start of a meal, 15 minutes before, or 30 minutes before the meal with the same blood glucose-lowering effect.
PRANDIN was compared to other insulin secretagogues in 1-year controlled trials to demonstrate comparability of efficacy and safety. Hypoglycemia was reported in 16% of 1228 PRANDIN patients, 20% of 417 glyburide patients, and 19% of 81 glipizide patients. Of PRANDIN-treated patients with symptomatic hypoglycemia, none developed coma or required hospitalization.
Combination Trials
PRANDIN was studied in combination with metformin in 83 patients not satisfactorily controlled on exercise, diet, and metformin alone. PRANDIN dosage was titrated for 4 to 8 weeks, followed by a 3-month maintenance period. Combination therapy with PRANDIN and metformin resulted in significantly greater improvement in glycemic control as compared to repaglinide or metformin monotherapy. HbA1c was improved by 1% unit and FPG decreased by an additional 35 mg/dL. In this study where metformin dosage was kept constant, the combination therapy of PRANDIN and metformin showed dose-sparing effects with respect to PRANDIN. The greater efficacy response of the combination group was achieved at a lower daily repaglinide dosage than in the PRANDIN monotherapy group (see Table).
PRANDIN and Metformin Therapy: Mean Changes from Baseline in Glycemic Parameters and Weight after 4 to 5 Months of Treatment* - * based on intent-to-treat analysis
- † p < 0.05, for pairwise comparisons with PRANDIN and metformin.
- ‡ p < 0.05, for pairwise comparison with metformin.
PRANDIN
Combination
Metformin
N
28
27
27
Median Final Dose
(mg/day)
12
6 (PRANDIN)
1500 (metformin)
1500
HbA1c (% units)
-0.38
-1.41†
-0.33
FPG (mg/dL)
8.8
-39.2 †
-4.5
Weight (kg)
3.0
2.4 ‡
-0.90
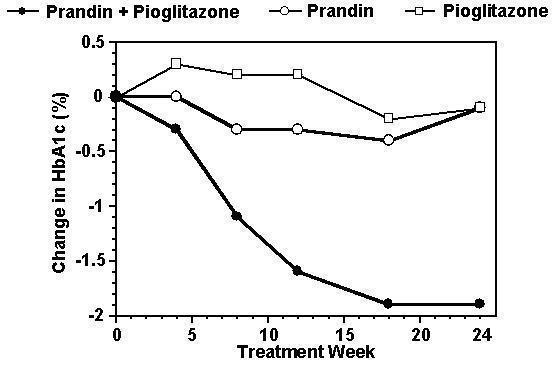
A combination therapy regimen of PRANDIN and pioglitazone was compared to monotherapy with either agent alone in a 24-week trial that enrolled 246 patients previously treated with sulfonylurea or metformin monotherapy (HbA1c > 7.0%). Numbers of patients treated were: PRANDIN (N = 61), pioglitazone (N = 62), combination (N = 123). PRANDIN dosage was titrated during the first 12 weeks, followed by a 12-week maintenance period. Combination therapy resulted in significantly greater improvement in glycemic control as compared to monotherapy (figure below). The changes from baseline for completers in FPG (mg/dL) and HbA1c (%), respectively were: -39.8 and -0.1 for PRANDIN, -35.3 and -0.1 for pioglitazone and -92.4 and -1.9 for the combination. In this study where pioglitazone dosage was kept constant, the combination therapy group showed dose-sparing effects with respect to PRANDIN (see figure legend). The greater efficacy response of the combination group was achieved at a lower daily repaglinide dosage than in the PRANDIN monotherapy group. Mean weight increases associated with combination, PRANDIN and pioglitazone therapy were 5.5 kg, 0.3 kg, and 2.0 kg respectively.
HbA1c Values from PRANDIN / Pioglitazone Combination Study
HbA1c values by study week for patients who completed study (combination, N = 101; PRANDIN, N = 35, pioglitazone, N = 26).
Subjects with FPG above 270 mg/dL were withdrawn from the study.
Pioglitazone dose: fixed at 30 mg/day; PRANDIN median final dose: 6 mg/day for combination and 10 mg/day for monotherapy.
A combination therapy regimen of PRANDIN and rosiglitazone was compared to monotherapy with either agent alone in a 24-week trial that enrolled 252 patients previously treated with sulfonylurea or metformin (HbA1c > 7.0%). Combination therapy resulted in significantly greater improvement in glycemic control as compared to monotherapy (table below). The glycemic effects of the combination therapy were dose-sparing with respect to both total daily PRANDIN dosage and total daily rosiglitazone dosage (see table legend). A greater efficacy response of the combination therapy group was achieved with half the median daily dose of PRANDIN and rosiglitazone, as compared to the respective monotherapy groups. Mean weight change associated with combination therapy was greater than that of PRANDIN monotherapy.
Mean Changes from Baseline in Glycemic Parameters and Weight in a 24-Week PRANDIN/Rosiglitazone Combination Study* - * based on intent-to-treat analysis
- † p-value ≤ 0.001 for comparison to either monotherapy
- ‡ p-value < 0.001 for comparison to PRANDIN
PRANDIN
Combination
Rosiglitazone
N
63
127
62
HbA1c (%)
Baseline
9.3
9.1
9.0
Change by 24 weeks
-0.17
-1.43 †
-0.56
FPG (mg/dL)
Baseline
269
257
252
Change by 24 weeks
-54
-94 †
-67
Change in Weight (kg)
+1.3
+4.5 ‡
+3.3
Final median doses: rosiglitazone - 4 mg/day for combination and 8 mg/day for monotherapy; PRANDIN - 6 mg/day for combination and 12 mg/day for monotherapy
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
PRECAUTIONS
General:
PRANDIN is not indicated for use in combination with NPH-insulin (See ADVERSE REACTIONS, Cardiovascular Events).
Macrovascular Outcomes:
There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with PRANDIN or any other anti-diabetic drug.
Hypoglycemia: All oral blood glucose-lowering drugs including repaglinide are capable of producing hypoglycemia. Proper patient selection, dosage, and instructions to the patients are important to avoid hypoglycemic episodes. Hepatic insufficiency may cause elevated repaglinide blood levels and may diminish gluconeogenic capacity, both of which increase the risk of serious hypoglycemia. Elderly, debilitated, or malnourished patients, and those with adrenal, pituitary, hepatic, or severe renal insufficiency may be particularly susceptible to the hypoglycemic action of glucose-lowering drugs.
Hypoglycemia may be difficult to recognize in the elderly and in people taking beta-adrenergic blocking drugs. Hypoglycemia is more likely to occur when caloric intake is deficient, after severe or prolonged exercise, when alcohol is ingested, or when more than one glucose-lowering drug is used.
The frequency of hypoglycemia is greater in patients with type 2 diabetes who have not been previously treated with oral blood glucose-lowering drugs (naïve) or whose HbA1c is less than 8%. PRANDIN should be administered with meals to lessen the risk of hypoglycemia.
Loss of Control of Blood Glucose: When a patient stabilized on any diabetic regimen is exposed to stress such as fever, trauma, infection, or surgery, a loss of glycemic control may occur. At such times, it may be necessary to discontinue PRANDIN and administer insulin. The effectiveness of any hypoglycemic drug in lowering blood glucose to a desired level decreases in many patients over a period of time, which may be due to progression of the severity of diabetes or to diminished responsiveness to the drug. This phenomenon is known as secondary failure, to distinguish it from primary failure in which the drug is ineffective in an individual patient when the drug is first given. Adequate adjustment of dose and adherence to diet should be assessed before classifying a patient as a secondary failure.
Information for Patients
Patients should be informed of the potential risks and advantages of PRANDIN and of alternative modes of therapy. They should also be informed about the importance of adherence to dietary instructions, of a regular exercise program, and of regular testing of blood glucose and HbA1c. The risks of hypoglycemia, its symptoms and treatment, and conditions that predispose to its development and concomitant administration of other glucose-lowering drugs should be explained to patients and responsible family members. Primary and secondary failure should also be explained.
Patients should be instructed to take PRANDIN before meals (2, 3, or 4 times a day preprandially). Doses are usually taken within 15 minutes of the meal but time may vary from immediately preceding the meal to as long as 30 minutes before the meal. Patients who skip a meal (or add an extra meal) should be instructed to skip (or add) a dose for that meal.
Laboratory Tests
Response to all diabetic therapies should be monitored by periodic measurements of fasting blood glucose and glycosylated hemoglobin levels with a goal of decreasing these levels towards the normal range. During dose adjustment, fasting glucose can be used to determine the therapeutic response. Thereafter, both glucose and glycosylated hemoglobin should be monitored. Glycosylated hemoglobin may be especially useful for evaluating long-term glycemic control. Postprandial glucose level testing may be clinically helpful in patients whose pre-meal blood glucose levels are satisfactory but whose overall glycemic control (HbA1c) is inadequate.
Drug-Drug Interactions
In vitro data indicate that PRANDIN is metabolized by cytochrome P450 enzymes 2C8 and 3A4. Consequently, repaglinide metabolism may be altered by drugs which influence these cytochrome P450 enzyme systems via induction and inhibition. Caution should therefore be used in patients who are on PRANDIN and taking inhibitors and/or inducers of CYP2C8 and CYP3A4. The effect may be very significant if both enzymes are inhibited at the same time resulting in a substantial increase in repaglinide plasma concentrations. Drugs that are known to inhibit CYP3A4 include antifungal agents like ketoconazole, itraconazole, and antibacterial agents like erythromycin. Drugs that are known to inhibit CYP2C8 include agents like trimethoprim, gemfibrozil and montelukast. Drugs that induce the CYP3A4 and/or 2C8 enzyme systems include rifampin, barbiturates, and carbamezapine. See CLINICAL PHARMACOLOGY section, Drug-Drug Interactions.
Repaglinide appears to be a substrate for active hepatic uptake transporter (organic anion transporting protein OATP1B1). Drugs that inhibit OATP1B1 (e.g. cyclosporine) may likewise have the potential to increase plasma concentrations of repaglinide. See CLINICAL PHARMACOLOGY section, Drug-Drug Interactions.
In vivo data from a study that evaluated the co-administration of a cytochrome P450 enzyme 3A4 inhibitor, clarithromycin, with PRANDIN resulted in a clinically significant increase in repaglinide plasma levels. In addition, an increase in repaglinide plasma levels was observed in studies that evaluated the co-administration of PRANDIN with trimethoprim and PRANDIN with deferasirox, both cytochrome P-450 enzyme 2C8 inhibitors. These increases in repaglinide plasma levels may necessitate a PRANDIN dose adjustment. See CLINICAL PHARMACOLOGY section, Drug-Drug Interactions.
Gemfibrozil significantly increased PRANDIN exposure. Therefore, patients should not take PRANDIN with gemfibrozil. See CLINICAL PHARMACOLOGY section, Drug-Drug Interactions, and CONTRAINDICATIONS.
The hypoglycemic action of oral blood glucose-lowering agents may be potentiated by certain drugs including nonsteroidal anti-inflammatory agents and other drugs that are highly protein bound, salicylates, sulfonamides, cyclosporine, chloramphenicol, coumarins, probenecid, monoamine oxidase inhibitors, and beta adrenergic blocking agents. When such drugs are administered to a patient receiving oral blood glucose-lowering agents, the patient should be observed closely for hypoglycemia. When such drugs are withdrawn from a patient receiving oral blood glucose-lowering agents, the patient should be observed closely for loss of glycemic control.
Certain drugs tend to produce hyperglycemia and may lead to loss of glycemic control. These drugs include the thiazides and other diuretics, corticosteroids, phenothiazines, thyroid products, estrogens, oral contraceptives, phenytoin, nicotinic acid, sympathomimetics, calcium channel blocking drugs, and isoniazid. When these drugs are administered to a patient receiving oral blood glucose-lowering agents, the patient should be observed for loss of glycemic control. When these drugs are withdrawn from a patient receiving oral blood glucose-lowering agents, the patient should be observed closely for hypoglycemia.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Long-term carcinogenicity studies were performed for 104 weeks at doses up to and including 120 mg/kg body weight/day (rats) and 500 mg/kg body weight/day (mice) or approximately 60 and 125 times clinical exposure, respectively, on a mg/m2 basis. No evidence of carcinogenicity was found in mice or female rats. In male rats, there was an increased incidence of benign adenomas of the thyroid and liver. The relevance of these findings to humans is unclear. The no-effect doses for these observations in male rats were 30 mg/kg body weight/day for thyroid tumors and 60 mg/kg body weight/day for liver tumors, which are over 15 and 30 times, respectively, clinical exposure on a mg/m2 basis.
Repaglinide was non-genotoxic in a battery of in vivo and in vitro studies: Bacterial mutagenesis (Ames test), in vitro forward cell mutation assay in V79 cells (HGPRT), in vitro chromosomal aberration assay in human lymphocytes, unscheduled and replicating DNA synthesis in rat liver, and in vivo mouse and rat micronucleus tests.
Fertility of male and female rats was unaffected by repaglinide administration at doses up to 80 mg/kg body weight/day (females) and 300 mg/kg body weight/day (males); over 40 times clinical exposure on a mg/m2 basis.
Pregnancy
Pregnancy category C
Teratogenic Effects: Safety in pregnant women has not been established. Repaglinide was not teratogenic in rats or rabbits at doses 40 times (rats) and approximately 0.8 times (rabbit) clinical exposure (on a mg/m2 basis) throughout pregnancy. Because animal reproduction studies are not always predictive of human response, PRANDIN should be used during pregnancy only if it is clearly needed.
Because recent information suggests that abnormal blood glucose levels during pregnancy are associated with a higher incidence of congenital abnormalities, many experts recommend that insulin be used during pregnancy to maintain blood glucose levels as close to normal as possible.
Nonteratogenic Effects: Offspring of rat dams exposed to repaglinide at 15 times clinical exposure on a mg/m2 basis during days 17 to 22 of gestation and during lactation developed nonteratogenic skeletal deformities consisting of shortening, thickening, and bending of the humerus during the postnatal period. This effect was not seen at doses up to 2.5 times clinical exposure (on a mg/m2 basis) on days 1 to 22 of pregnancy or at higher doses given during days 1 to 16 of pregnancy. Relevant human exposure has not occurred to date and therefore the safety of PRANDIN administration throughout pregnancy or lactation cannot be established.
Nursing Mothers
In rat reproduction studies, measurable levels of repaglinide were detected in the breast milk of the dams and lowered blood glucose levels were observed in the pups. Cross fostering studies indicated that skeletal changes (see Nonteratogenic Effects) could be induced in control pups nursed by treated dams, although this occurred to a lesser degree than those pups treated in utero. Although it is not known whether repaglinide is excreted in human milk some oral agents are known to be excreted by this route. Because the potential for hypoglycemia in nursing infants may exist, and because of the effects on nursing animals, a decision should be made as to whether PRANDIN should be discontinued in nursing mothers, or if mothers should discontinue nursing. If PRANDIN is discontinued and if diet alone is inadequate for controlling blood glucose, insulin therapy should be considered.
Geriatric Use
In repaglinide clinical studies of 24 weeks or greater duration, 415 patients were over 65 years of age. In one-year, active-controlled trials, no differences were seen in effectiveness or adverse events between these subjects and those less than 65 other than the expected age-related increase in cardiovascular events observed for PRANDIN and comparator drugs. There was no increase in frequency or severity of hypoglycemia in older subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals to PRANDIN therapy cannot be ruled out.
-
ADVERSE REACTIONS
Hypoglycemia: See PRECAUTIONS and OVERDOSAGE sections.
PRANDIN has been administered to 2931 individuals during clinical trials. Approximately 1500 of these individuals with type 2 diabetes have been treated for at least 3 months, 1000 for at least 6 months, and 800 for at least 1 year. The majority of these individuals (1228) received PRANDIN in one of five 1-year, active-controlled trials. The comparator drugs in these 1-year trials were oral sulfonylurea drugs (SU) including glyburide and glipizide. Over one year, 13% of PRANDIN patients were discontinued due to adverse events, as were 14% of SU patients. The most common adverse events leading to withdrawal were hyperglycemia, hypoglycemia, and related symptoms (see PRECAUTIONS). Mild or moderate hypoglycemia occurred in 16% of PRANDIN patients, 20% of glyburide patients, and 19% of glipizide patients.
The table below lists common adverse events for PRANDIN patients compared to both placebo (in trials 12 to 24 weeks duration) and to glyburide and glipizide in one year trials. The adverse event profile of PRANDIN was generally comparable to that for sulfonylurea drugs (SU).
Commonly Reported Adverse Events (% of Patients)* - * Events ≥ 2% for the PRANDIN group in the placebo-controlled studies and ≥ events in the placebo group
- † See trial description in CLINICAL PHARMACOLOGY, Clinical Trials
EVENT
PRANDIN
PLACEBO
PRANDIN
SU
N = 352
N = 108
N = 1228
N = 498
Placebo controlled studies
Active controlled studies
Metabolic
Hypoglycemia
31†
7
16
20
Respiratory
URI
16
8
10
10
Sinusitis
6
2
3
4
Rhinitis
3
3
7
8
Bronchitis
2
1
6
7
Gastrointestinal
Nausea
5
5
3
2
Diarrhea
5
2
4
6
Constipation
3
2
2
3
Vomiting
3
3
2
1
Dyspepsia
2
2
4
2
Musculoskeletal
Arthralgia
6
3
3
4
Back Pain
5
4
6
7
Other
Headache
11
10
9
8
Paresthesia
3
3
2
1
Chest pain
3
1
2
1
Urinary tract infection
2
1
3
3
Tooth disorder
2
0
<1
<1
Allergy
2
0
1
<1
Cardiovascular Events
In one-year trials comparing PRANDIN to sulfonylurea drugs, the incidence of angina was comparable (1.8%) for both treatments, with an incidence of chest pain of 1.8% for PRANDIN and 1.0% for sulfonylureas. The incidence of other selected cardiovascular events (hypertension, abnormal EKG, myocardial infarction, arrhythmias, and palpitations) was ≤ 1% and not different between PRANDIN and the comparator drugs.
The incidence of total serious cardiovascular adverse events, including ischemia, was higher for repaglinide (4%) than for sulfonylurea drugs (3%) in controlled comparator clinical trials. In 1-year controlled trials, PRANDIN treatment was not associated with excess mortality when compared to the rates observed with other oral hypoglycemic agent therapies.
Summary of Serious Cardiovascular Events (% of total patients with events) in Trials Comparing PRANDIN to Sulfonylureas - * glyburide and glipizide
PRANDIN
SU*
Total Exposed
1228
498
Serious CV Events
4%
3%
Cardiac Ischemic Events
2%
2%
Deaths due to CV Events
0.5%
0.4%
Seven controlled clinical trials included PRANDIN combination therapy with NPH-insulin (n=431), insulin formulations alone (n=388) or other combinations (sulfonylurea plus NPH-insulin or PRANDIN plus metformin) (n=120). There were six serious adverse events of myocardial ischemia in patients treated with PRANDIN plus NPH-insulin from two studies, and one event in patients using insulin formulations alone from another study.
Infrequent Adverse Events (<1% of Patients)
Less common adverse clinical or laboratory events observed in clinical trials included elevated liver enzymes, thrombocytopenia, leukopenia, and anaphylactoid reactions.
Although no causal relationship with repaglinide has been established, postmarketing experience includes reports of the following rare adverse events: alopecia, hemolytic anemia, pancreatitis, Stevens-Johnson Syndrome, and severe hepatic dysfunction including jaundice and hepatitis.
Combination Therapy with Thiazolidinediones
During 24-week treatment clinical trials of PRANDIN-rosiglitazone or PRANDIN-pioglitazone combination therapy (a total of 250 patients in combination therapy), hypoglycemia (blood glucose < 50 mg/dL) occurred in 7% of combination therapy patients in comparison to 7% for PRANDIN monotherapy, and 2% for thiazolidinedione monotherapy.
Peripheral edema was reported in 12 out of 250 PRANDIN-thiazolidinedione combination therapy patients and 3 out of 124 thiazolidinedione monotherapy patients, with no cases reported in these trials for PRANDIN monotherapy. When corrected for dropout rates of the treatment groups, the percentage of patients having events of peripheral edema per 24 weeks of treatment were 5% for PRANDIN-thiazolidinedione combination therapy, and 4% for thiazolidinedione monotherapy. There were reports in 2 of 250 patients (0.8%) treated with PRANDIN-thiazolidinedione therapy of episodes of edema with congestive heart failure. Both patients had a prior history of coronary artery disease and recovered after treatment with diuretic agents. No comparable cases in the monotherapy treatment groups were reported.
Mean change in weight from baseline was +4.9 kg for PRANDIN-thiazolidinedione therapy. There were no patients on PRANDIN-thiazolidinedione combination therapy who had elevations of liver transaminases (defined as 3 times the upper limit of normal levels).
-
OVERDOSAGE
In a clinical trial, patients received increasing doses of PRANDIN up to 80 mg a day for 14 days. There were few adverse effects other than those associated with the intended effect of lowering blood glucose. Hypoglycemia did not occur when meals were given with these high doses. Hypoglycemic symptoms without loss of consciousness or neurologic findings should be treated aggressively with oral glucose and adjustments in drug dosage and/or meal patterns. Close monitoring may continue until the physician is assured that the patient is out of danger. Patients should be closely monitored for a minimum of 24 to 48 hours, since hypoglycemia may recur after apparent clinical recovery. There is no evidence that repaglinide is dialyzable using hemodialysis.
Severe hypoglycemic reactions with coma, seizure, or other neurological impairment occur infrequently, but constitute medical emergencies requiring immediate hospitalization. If hypoglycemic coma is diagnosed or suspected, the patient should be given a rapid intravenous injection of concentrated (50%) glucose solution. This should be followed by a continuous infusion of more dilute (10%) glucose solution at a rate that will maintain the blood glucose at a level above 100 mg/dL.
-
DOSAGE AND ADMINISTRATION
There is no fixed dosage regimen for the management of type 2 diabetes with PRANDIN.
The patient's blood glucose should be monitored periodically to determine the minimum effective dose for the patient; to detect primary failure, i.e., inadequate lowering of blood glucose at the maximum recommended dose of medication; and to detect secondary failure, i.e., loss of an adequate blood glucose-lowering response after an initial period of effectiveness. Glycosylated hemoglobin levels are of value in monitoring the patient's longer term response to therapy.
Short-term administration of PRANDIN may be sufficient during periods of transient loss of control in patients usually well controlled on diet.
PRANDIN doses are usually taken within 15 minutes of the meal but time may vary from immediately preceding the meal to as long as 30 minutes before the meal.
Starting Dose
For patients not previously treated or whose HbA1c is < 8%, the starting dose should be 0.5 mg with each meal. For patients previously treated with blood glucose-lowering drugs and whose HbA1c is ≥ 8%, the initial dose is 1 or 2 mg with each meal preprandially (see previous paragraph).
Dose Adjustment
Dosing adjustments should be determined by blood glucose response, usually fasting blood glucose. Postprandial glucose levels testing may be clinically helpful in patients whose pre-meal blood glucose levels are satisfactory but whose overall glycemic control (HbA1c) is inadequate. The preprandial dose should be doubled up to 4 mg with each meal until satisfactory blood glucose response is achieved. At least one week should elapse to assess response after each dose adjustment.
The recommended dose range is 0.5 mg to 4 mg taken with meals. PRANDIN may be dosed preprandially 2, 3, or 4 times a day in response to changes in the patient’s meal pattern. The maximum recommended daily dose is 16 mg.
Patient Management
Long-term efficacy should be monitored by measurement of HbA1c levels approximately every 3 months. Failure to follow an appropriate dosage regimen may precipitate hypoglycemia or hyperglycemia. Patients who do not adhere to their prescribed dietary and drug regimen are more prone to exhibit unsatisfactory response to therapy including hypoglycemia. When hypoglycemia occurs in patients taking a combination of PRANDIN and a thiazolidinedione or PRANDIN and metformin, the dose of PRANDIN should be reduced.
Patients Receiving Other Oral Hypoglycemic Agents
When PRANDIN is used to replace therapy with other oral hypoglycemic agents, PRANDIN may be started on the day after the final dose is given. Patients should then be observed carefully for hypoglycemia due to potential overlapping of drug effects. When transferred from longer half-life sulfonylurea agents (e.g., chlorpropamide) to repaglinide, close monitoring may be indicated for up to one week or longer.
Combination Therapy
If PRANDIN monotherapy does not result in adequate glycemic control, metformin or a thiazolidinedione may be added. If metformin or thiazolidinedione monotherapy does not provide adequate control, PRANDIN may be added. The starting dose and dose adjustments for PRANDIN combination therapy is the same as for PRANDIN monotherapy. The dose of each drug should be carefully adjusted to determine the minimal dose required to achieve the desired pharmacologic effect. Failure to do so could result in an increase in the incidence of hypoglycemic episodes. Appropriate monitoring of FPG and HbA1c measurements should be used to ensure that the patient is not subjected to excessive drug exposure or increased probability of secondary drug failure.
-
HOW SUPPLIED
PRANDIN (repaglinide) tablets are supplied as unscored, biconvex tablets available in 0.5 mg (white), 1 mg (yellow) and 2 mg (peach) strengths. Tablets are embossed with the Novo Nordisk (Apis) bull symbol and colored to indicate strength.
1 mg tablets (yellow)
Bottles of 90 NDC: 54868-6439-0
2 mg tablets (peach)
Bottles of 10 NDC: 54868-5381-0
Bottles of 90 NDC: 54868-5381-1
Bottles of 60 NDC: 54868-5381-2
Do not store above 25° C (77° F).
Protect from moisture. Keep bottles tightly closed.
Dispense in tight containers with safety closures.
PRANDIN® is a registered trademark of Novo Nordisk A/S.
Manufactured in Germany for
Novo Nordisk Inc.
Plainsboro, NJ 08536
1-800-727-6500
www.novonordisk-us.com
© 2003-2014 Novo Nordisk
All rights reserved.
Date of Issue: October 2014
Version: 13
Distributed By:
Physicians Total Care, Inc.
Tulsa, OK 74146
- Principal Display Panel
- Principal Display Panel
-
INGREDIENTS AND APPEARANCE
PRANDIN
repaglinide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 54868-6439(NDC:0169-0082) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REPAGLINIDE (UNII: 668Z8C33LU) (REPAGLINIDE - UNII:668Z8C33LU) REPAGLINIDE 1 mg Product Characteristics Color yellow (YELLOW) Score no score Shape ROUND (ROUND) Size 5mm Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 54868-6439-0 90 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020741 01/03/2000 PRANDIN
repaglinide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 54868-5381(NDC:0169-0084) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REPAGLINIDE (UNII: 668Z8C33LU) (REPAGLINIDE - UNII:668Z8C33LU) REPAGLINIDE 2 mg Product Characteristics Color pink (PEACH) Score no score Shape ROUND (ROUND) Size 5mm Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 54868-5381-0 10 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC: 54868-5381-1 90 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 3 NDC: 54868-5381-2 60 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020741 01/03/2000 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel(54868-6439, 54868-5381) , repack(54868-6439, 54868-5381)


Trademark Results [Prandin]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 PRANDIN 75272035 2330531 Live/Registered |
NOVO NORDISK A/S 1997-04-09 |
 PRANDIN 72412854 0954329 Dead/Expired |
UPJOHN COMPANY, THE 1972-01-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.