NINLARO- ixazomib capsule
NINLARO by
Drug Labeling and Warnings
NINLARO by is a Prescription medication manufactured, distributed, or labeled by Takeda Pharmaceuticals America, Inc., Ash Stevens LLC, Curia Indiana, LLC., Eurofins Lancaster Laboratories, Inc, Haupt Pharma Amareg GmbH, AndersonBrecon (UK) Limited, Takeda Ireland Ltd., Eurofins Biopharma Product Testing Ireland Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NINLARO safely and effectively. See full prescribing information for NINLARO.
NINLARO® (ixazomib) capsules, for oral use
Initial U.S. Approval: 2015RECENT MAJOR CHANGES
Warnings and Precautions
Thrombotic Microangiopathy (5.6)02/2020 INDICATIONS AND USAGE
NINLARO is a proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Capsules: 4 mg, 3 mg, and 2.3 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Thrombocytopenia: Monitor platelet counts at least monthly during treatment and adjust dosing, as needed. (2.2, 5.1)
- Gastrointestinal Toxicities: Adjust dosing for severe diarrhea, constipation, nausea, and vomiting, as needed. (2.2, 5.2)
- Peripheral Neuropathy: Monitor patients for symptoms of peripheral neuropathy and adjust dosing, as needed. (2.2, 5.3)
- Peripheral Edema: Monitor for fluid retention. Investigate for underlying causes, when appropriate. Adjust dosing, as needed. (2.2, 5.4)
- Cutaneous Reactions: Monitor patients for rash and adjust dosing, as needed. (2.2, 5.5)
- Thrombotic Microangiopathy: Monitor for signs and symptoms. Discontinue NINLARO if suspected. (5.6)
- Hepatotoxicity: Monitor hepatic enzymes during treatment. (5.7)
- Embryo-Fetal Toxicity: NINLARO can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.8, 8.1)
ADVERSE REACTIONS
The most common adverse reactions (≥ 20%) are diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting, and back pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals America, Inc. at 1-844-617-6468 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Hepatic Impairment: Reduce the NINLARO starting dose to 3 mg in patients with moderate or severe hepatic impairment. (2.3, 8.6)
- Renal Impairment: Reduce the NINLARO starting dose to 3 mg in patients with severe renal impairment or end-stage renal disease requiring dialysis. (2.4, 8.7)
- Lactation: Discontinue nursing. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing and Administration Guidelines
2.2 Dose Modification Guidelines
2.3 Dosage in Patients with Hepatic Impairment
2.4 Dosage in Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombocytopenia
5.2 Gastrointestinal Toxicities
5.3 Peripheral Neuropathy
5.4 Peripheral Edema
5.5 Cutaneous Reactions
5.6 Thrombotic Microangiopathy
5.7 Hepatotoxicity
5.8 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
16.3 Handling and Disposal
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing and Administration Guidelines
NINLARO in combination with lenalidomide and dexamethasone
The recommended starting dose of NINLARO is 4 mg administered orally once a week on Days 1, 8, and 15 of a 28-day treatment cycle.
The recommended starting dose of lenalidomide is 25 mg administered daily on Days 1 through 21 of a 28-day treatment cycle.
The recommended starting dose of dexamethasone is 40 mg administered on Days 1, 8, 15, and 22 of a 28-day treatment cycle.
Table 1: Dosing Schedule for NINLARO taken with Lenalidomide and Dexamethasone ✔ Take medicine 28-Day Cycle (a 4-week cycle) Week 1 Week 2 Week 3 Week 4 Day 1 Days 2-7 Day 8 Days 9-14 Day 15 Days 16-21 Day 22 Days 23-28 NINLARO ✔ ✔ ✔ Lenalidomide ✔ ✔ Daily ✔ ✔ Daily ✔ ✔ Daily Dexamethasone ✔ ✔ ✔ ✔ For additional information regarding lenalidomide and dexamethasone, refer to their prescribing information.
NINLARO should be taken once a week on the same day and at approximately the same time for the first three weeks of a four week cycle. NINLARO should be taken at least one hour before or at least two hours after food [see Clinical Pharmacology (12.3)]. The whole capsule should be swallowed with water. The capsule should not be crushed, chewed or opened [see How Supplied/Storage and Handling (16.3)].
If a NINLARO dose is delayed or missed, the dose should be taken only if the next scheduled dose is ≥ 72 hours away. A missed dose should not be taken within 72 hours of the next scheduled dose. A double dose should not be taken to make up for the missed dose.
If vomiting occurs after taking a dose, the patient should not repeat the dose. The patient should resume dosing at the time of the next scheduled dose.
Prior to initiating a new cycle of therapy:
- Absolute neutrophil count should be at least 1,000/mm3
- Platelet count should be at least 75,000/mm3
- Non-hematologic toxicities should, at the physician's discretion, generally be recovered to patient's baseline condition or Grade 1 or lower
Treatment should be continued until disease progression or unacceptable toxicity.
Concomitant Medications
Consider antiviral prophylaxis in patients being treated with NINLARO to decrease the risk of herpes zoster reactivation [see Adverse Reactions (6.1)].
2.2 Dose Modification Guidelines
The NINLARO dose reduction steps are presented in Table 2 and the dose modification guidelines are provided in Table 3.
Table 2: NINLARO Dose Reductions due to Adverse Reactions - * Recommended starting dose of 3 mg in patients with moderate or severe hepatic impairment, severe renal impairment or end-stage renal disease requiring dialysis [see Dosage and Administration (2.3, 2.4)].
Recommended starting dose* First reduction to Second reduction to Discontinue 4 mg 3 mg 2.3 mg An alternating dose modification approach is recommended for NINLARO and lenalidomide for thrombocytopenia, neutropenia, and rash as described in Table 3. Refer to the lenalidomide prescribing information if dose reduction is needed for lenalidomide.
Table 3: Dose Modifications Guidelines for NINLARO in Combination with Lenalidomide and Dexamethasone - * For additional occurrences, alternate dose modification of lenalidomide and NINLARO
- † Grading based on National Cancer Institute Common Terminology Criteria (CTCAE) Version 4.03
Hematological Toxicities Recommended Actions Thrombocytopenia (Platelet Count) Platelet count less than 30,000/mm3 - Withhold NINLARO and lenalidomide until platelet count is at least 30,000/mm3.
- Following recovery, resume lenalidomide at the next lower dose according to its prescribing information and resume NINLARO at its most recent dose.
- If platelet count falls to less than 30,000/mm3 again, withhold NINLARO and lenalidomide until platelet count is at least 30,000/mm3.
- Following recovery, resume NINLARO at the next lower dose and resume lenalidomide at its most recent dose.*
Neutropenia (Absolute Neutrophil Count) Absolute neutrophil count less than 500/mm3 - Withhold NINLARO and lenalidomide until absolute neutrophil count is at least 500/mm3. Consider adding G-CSF as per clinical guidelines.
- Following recovery, resume lenalidomide at the next lower dose according to its prescribing information and resume NINLARO at its most recent dose.
- If absolute neutrophil count falls to less than 500/mm3 again, withhold NINLARO and lenalidomide until absolute neutrophil count is at least 500/mm3.
- Following recovery, resume NINLARO at the next lower dose and resume lenalidomide at its most recent dose.*
Non-Hematological Toxicities Recommended Actions Rash Grade† 2 or 3 - Withhold lenalidomide until rash recovers to Grade 1 or lower.
- Following recovery, resume lenalidomide at the next lower dose according to its prescribing information.
- If Grade 2 or 3 rash occurs again, withhold NINLARO and lenalidomide until rash recovers to Grade 1 or lower.
- Following recovery, resume NINLARO at the next lower dose and resume lenalidomide at its most recent dose.*
Grade 4 Discontinue treatment regimen. Peripheral Neuropathy Grade 1 Peripheral Neuropathy with Pain or Grade 2 Peripheral Neuropathy - Withhold NINLARO until peripheral neuropathy recovers to Grade 1 or lower without pain or patient's baseline.
- Following recovery, resume NINLARO at its most recent dose.
Grade 2 Peripheral Neuropathy with Pain or Grade 3 Peripheral Neuropathy - Withhold NINLARO. Toxicities should, at the physician's discretion, generally recover to patient's baseline condition or Grade 1 or lower prior to resuming NINLARO.
- Following recovery, resume NINLARO at the next lower dose.
Grade 4 Peripheral Neuropathy Discontinue treatment regimen. Other Non-Hematological Toxicities Other Grade 3 or 4 Non-Hematological Toxicities - Withhold NINLARO. Toxicities should, at the physician's discretion, generally recover to patient's baseline condition or Grade 1 or lower prior to resuming NINLARO.
- If attributable to NINLARO, resume NINLARO at the next lower dose following recovery.
2.3 Dosage in Patients with Hepatic Impairment
Reduce the starting dose of NINLARO to 3 mg in patients with moderate (total bilirubin greater than 1.5-3 × ULN) or severe (total bilirubin greater than 3 × ULN) hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Dosage in Patients with Renal Impairment
Reduce the starting dose of NINLARO to 3 mg in patients with severe renal impairment (creatinine clearance less than 30 mL/min) or end-stage renal disease (ESRD) requiring dialysis. NINLARO is not dialyzable and therefore can be administered without regard to the timing of dialysis [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
Refer to the lenalidomide prescribing information for dosing recommendations in patients with renal impairment.
-
3 DOSAGE FORMS AND STRENGTHS
NINLARO is available in the following capsule strengths:
- 4 mg: Light orange gelatin capsule imprinted with "Takeda" on the cap and "4 mg" on the body in black ink. NINLARO 4 mg capsules contain 4 mg of ixazomib equivalent to 5.7 mg of ixazomib citrate.
- 3 mg: Light grey gelatin capsule imprinted with "Takeda" on the cap and "3 mg" on the body in black ink. NINLARO 3 mg capsules contain 3 mg of ixazomib equivalent to 4.3 mg of ixazomib citrate.
- 2.3 mg: Light pink gelatin capsule imprinted with "Takeda" on the cap and "2.3 mg" on the body in black ink. NINLARO 2.3 mg capsules contain 2.3 mg of ixazomib equivalent to 3.3 mg of ixazomib citrate.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombocytopenia
Thrombocytopenia has been reported with NINLARO with platelet nadirs typically occurring between Days 14-21 of each 28-day cycle and recovery to baseline by the start of the next cycle. Three percent of patients in the NINLARO regimen and 1% of patients in the placebo regimen had a platelet count ≤ 10,000/mm3 during treatment. Less than 1% of patients in both regimens had a platelet count ≤ 5000/mm3 during treatment. Discontinuations due to thrombocytopenia were similar in both regimens (< 1% of patients in the NINLARO regimen and 2% of patients in the placebo regimen discontinued one or more of the three drugs).The rate of platelet transfusions was 6% in the NINLARO regimen and 5% in the placebo regimen.
Monitor platelet counts at least monthly during treatment with NINLARO. Consider more frequent monitoring during the first three cycles. Manage thrombocytopenia with dose modifications [see Dosage and Administration (2.2)] and platelet transfusions as per standard medical guidelines.
5.2 Gastrointestinal Toxicities
Diarrhea, constipation, nausea, and vomiting, have been reported with NINLARO, occasionally requiring use of antidiarrheal and antiemetic medications, and supportive care. Diarrhea was reported in 42% of patients in the NINLARO regimen and 36% in the placebo regimen, constipation in 34% and 25%, respectively, nausea in 26% and 21%, respectively, and vomiting in 22% and 11%, respectively. Diarrhea resulted in discontinuation of one or more of the three drugs in 1% of patients in the NINLARO regimen and < 1% of patients in the placebo regimen. Adjust dosing for Grade 3 or 4 symptoms [see Dosage and Administration (2.2)].
5.3 Peripheral Neuropathy
The majority of peripheral neuropathy adverse reactions were Grade 1 (18% in the NINLARO regimen and 14% in the placebo regimen) and Grade 2 (8% in the NINLARO regimen and 5% in the placebo regimen). Grade 3 adverse reactions of peripheral neuropathy were reported at 2% in both regimens; there were no Grade 4 or serious adverse reactions.
The most commonly reported reaction was peripheral sensory neuropathy (19% and 14% in the NINLARO and placebo regimen, respectively). Peripheral motor neuropathy was not commonly reported in either regimen (< 1%). Peripheral neuropathy resulted in discontinuation of one or more of the three drugs in 1% of patients in both regimens. Patients should be monitored for symptoms of neuropathy. Patients experiencing new or worsening peripheral neuropathy may require dose modification [see Dosage and Administration (2.2)].
5.4 Peripheral Edema
Peripheral edema was reported in 25% and 18% of patients in the NINLARO and placebo regimens, respectively. The majority of peripheral edema adverse reactions were Grade 1 (16% in the NINLARO regimen and 13% in the placebo regimen) and Grade 2 (7% in the NINLARO regimen and 4% in the placebo regimen).
Grade 3 peripheral edema was reported in 2% and 1% of patients in the NINLARO and placebo regimens, respectively. There was no Grade 4 peripheral edema reported. There were no discontinuations reported due to peripheral edema. Evaluate for underlying causes and provide supportive care, as necessary. Adjust dosing of dexamethasone per its prescribing information or NINLARO for Grade 3 or 4 symptoms [see Dosage and Administration (2.2)].
5.5 Cutaneous Reactions
Rash was reported in 19% of patients in the NINLARO regimen and 11% of patients in the placebo regimen. The majority of the rash adverse reactions were Grade 1 (10% in the NINLARO regimen and 7% in the placebo regimen) or Grade 2 (6% in the NINLARO regimen and 3% in the placebo regimen). Grade 3 rash was reported in 3% of patients in the NINLARO regimen and 1% of patients in the placebo regimen. There were no Grade 4 or serious adverse reactions of rash reported. The most common type of rash reported in both regimens included maculo-papular and macular rash. Rash resulted in discontinuation of one or more of the three drugs in < 1% of patients in both regimens. Manage rash with supportive care or with dose modification if Grade 2 or higher [see Dosage and Administration (2.2)].
5.6 Thrombotic Microangiopathy
Cases, sometimes fatal, of thrombotic microangiopathy, including thrombotic thrombocytopenic purpura/hemolytic uremic syndrome (TTP/HUS), have been reported in patients who received NINLARO. Monitor for signs and symptoms of TTP/HUS. If the diagnosis is suspected, stop NINLARO and evaluate. If the diagnosis of TTP/HUS is excluded, consider restarting NINLARO. The safety of reinitiating NINLARO therapy in patients previously experiencing TTP/HUS is not known.
5.7 Hepatotoxicity
Drug-induced liver injury, hepatocellular injury, hepatic steatosis, hepatitis cholestatic and hepatotoxicity have each been reported in < 1% of patients treated with NINLARO. Events of liver impairment have been reported (6% in the NINLARO regimen and 5% in the placebo regimen). Monitor hepatic enzymes regularly and adjust dosing for Grade 3 or 4 symptoms [see Dosage and Administration (2.2)].
5.8 Embryo-Fetal Toxicity
NINLARO can cause fetal harm when administered to a pregnant woman based on the mechanism of action and findings in animals. There are no adequate and well-controlled studies in pregnant women using NINLARO. Ixazomib caused embryo-fetal toxicity in pregnant rats and rabbits at doses resulting in exposures that were slightly higher than those observed in patients receiving the recommended dose.
Females of reproductive potential should be advised to avoid becoming pregnant while being treated with NINLARO. If NINLARO is used during pregnancy or if the patient becomes pregnant while taking NINLARO, the patient should be apprised of the potential hazard to the fetus. Advise females of reproductive potential that they must use effective contraception during treatment with NINLARO and for 90 days following the final dose. Women using hormonal contraceptives should also use a barrier method of contraception [see Use in Specific Populations (8.1, 8.3) and Nonclinical Toxicology (13.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described in detail in other sections of the prescribing information:
- Thrombocytopenia [see Warnings and Precautions (5.1)]
- Gastrointestinal Toxicities [see Warnings and Precautions (5.2)]
- Peripheral Neuropathy [see Warnings and Precautions (5.3)]
- Peripheral Edema [see Warnings and Precautions (5.4)]
- Cutaneous Reactions [see Warnings and Precautions (5.5)]
- Thrombotic Microangiopathy [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety population from the randomized, double-blind, placebo-controlled clinical study included 720 patients with relapsed and/or refractory multiple myeloma, who received NINLARO in combination with lenalidomide and dexamethasone (NINLARO regimen; N=360) or placebo in combination with lenalidomide and dexamethasone (placebo regimen; N=360).
The most frequently reported adverse reactions (≥ 20%) in the NINLARO regimen and greater than the placebo regimen were diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting, and back pain. Serious adverse reactions reported in ≥ 2% of patients included thrombocytopenia (2%) and diarrhea (2%). For each adverse reaction, one or more of the three drugs was discontinued in ≤ 1% of patients in the NINLARO regimen.
Table 4 summarizes the adverse reactions occurring in at least 5% of patients with at least a 5% difference between the NINLARO regimen and the placebo regimen.
Table 4: Non-Hematologic Adverse Reactions Occurring in ≥ 5% of Patients with a ≥ 5% Difference Between the NINLARO Regimen and the Placebo Regimen (All Grades, Grade 3 and Grade 4) NINLARO + Lenalidomide and Dexamethasone
N=360Placebo + Lenalidomide and Dexamethasone
N=360System Organ Class /
Preferred TermN (%) N (%) All Grade 3 Grade 4 All Grade 3 Grade 4 Note: Adverse reactions included as preferred terms are based on MedDRA version 16.0. - * Represents a pooling of preferred terms
Infections and infestations Upper respiratory tract infection 69 (19) 1 (< 1) 0 52 (14) 2 (< 1) 0 Nervous system disorders Peripheral neuropathies* 100 (28) 7 (2) 0 77 (21) 7 (2) 0 Gastrointestinal disorders Diarrhea 151 (42) 22 (6) 0 130 (36) 8 (2) 0 Constipation 122 (34) 1 (< 1) 0 90 (25) 1 (< 1) 0 Nausea 92 (26) 6 (2) 0 74 (21) 0 0 Vomiting 79 (22) 4 (1) 0 38 (11) 2 (< 1) 0 Skin and subcutaneous tissue disorders Rash* 68 (19) 9 (3) 0 38 (11) 5 (1) 0 Musculoskeletal and connective tissue disorders Back pain 74 (21) 2 (< 1) 0 57 (16) 9 (3) 0 General disorders and administration site conditions Edema peripheral 91 (25) 8 (2) 0 66 (18) 4 (1) 0 Table 5 represents pooled information from adverse event and laboratory data.
Table 5: Thrombocytopenia and Neutropenia NINLARO + Lenalidomide and Dexamethasone
N=360Placebo + Lenalidomide and Dexamethasone
N=360N (%) N (%) Any Grade Grade 3-4 Any Grade Grade 3-4 Thrombocytopenia 281 (78) 93 (26) 196 (54) 39 (11) Neutropenia 240 (67) 93 (26) 239 (66) 107 (30) Herpes Zoster
Herpes zoster was reported in 4% of patients in the NINLARO regimen and 2% of patients in the placebo regimen. Antiviral prophylaxis was allowed at the physician's discretion. Patients treated in the NINLARO regimen who received antiviral prophylaxis had a lower incidence (< 1%) of herpes zoster infection compared to patients who did not receive prophylaxis (6%).
Eye Disorders
Eye disorders were reported with many different preferred terms but in aggregate, the frequency was 26% in patients in the NINLARO regimen and 16% of patients in the placebo regimen. The most common adverse reactions were blurred vision (6% in the NINLARO regimen and 3% in the placebo regimen), dry eye (5% in the NINLARO regimen and 1% in the placebo regimen), and conjunctivitis (6% in the NINLARO regimen and 1% in the placebo regimen). Grade 3 adverse reactions were reported in 2% of patients in the NINLARO regimen and 1% in the placebo regimen.
Adverse Reactions Reported Outside of the Randomized Controlled Trial
The following serious adverse reactions have each been reported at a frequency of < 1%: acute febrile neutrophilic dermatosis (Sweet's syndrome), Stevens-Johnson syndrome, transverse myelitis, posterior reversible encephalopathy syndrome, tumor lysis syndrome, and thrombotic thrombocytopenic purpura.
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A Inducers
Avoid concomitant administration of NINLARO with strong CYP3A inducers (such as rifampin, phenytoin, carbamazepine, and St. John's Wort) [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and data from animal reproduction studies, NINLARO can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no human data available regarding the potential effect of NINLARO on pregnancy or development of the embryo or fetus. Ixazomib caused embryo-fetal toxicity in pregnant rats and rabbits at doses resulting in exposures that were slightly higher then those observed in patients receiving the recommended dose (see Data). Advise women of the potential risk to a fetus and to avoid becoming pregnant while being treated with NINLARO.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rabbits there were increases in fetal skeletal variations/abnormalities (fused caudal vertebrae, number of lumbar vertebrae, and full supernumerary ribs) at doses that were also maternally toxic (≥ 0.3 mg/kg). Exposures in the rabbit at 0.3 mg/kg were 1.9 times the clinical time averaged exposures at the recommended dose of 4 mg. In a rat dose range-finding embryo-fetal development study, at doses that were maternally toxic, there were decreases in fetal weights, a trend towards decreased fetal viability, and increased post-implantation losses at 0.6 mg/kg. Exposures in rats at the dose of 0.6 mg/kg was 2.5 times the clinical time averaged exposures at the recommended dose of 4 mg.
8.2 Lactation
Risk Summary
No data are available regarding the presence of NINLARO or its metabolites in human milk, the effects of the drug on the breast fed infant, or the effects of the drug on milk production. Because the potential for serious adverse reactions from NINLARO in breastfed infants is unknown, advise nursing women not to breastfeed during treatment with NINLARO and for 90 days after the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Male and female patients of childbearing potential must use effective contraceptive measures during and for 90 days following treatment. Dexamethasone is known to be a weak to moderate inducer of CYP3A4 as well as other enzymes and transporters. Because NINLARO is administered with dexamethasone, the risk for reduced efficacy of contraceptives needs to be considered. Advise women using hormonal contraceptives to also use a barrier method of contraception.
8.5 Geriatric Use
Of the total number of subjects in clinical studies of NINLARO, 55% were 65 and over, while 17% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
In patients with moderate or severe hepatic impairment, the mean AUC increased by 20% when compared to patients with normal hepatic function. Reduce the starting dose of NINLARO in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
In patients with severe renal impairment or ESRD requiring dialysis, the mean AUC increased by 39% when compared to patients with normal renal function. Reduce the starting dose of NINLARO in patients with severe renal impairment or ESRD requiring dialysis. NINLARO is not dialyzable and therefore can be administered without regard to the timing of dialysis [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no known specific antidote for NINLARO overdose. In the event of an overdose, monitor the patient for adverse reactions [see Adverse Reactions (6.1)] and provide appropriate supportive care.
-
11 DESCRIPTION
Ixazomib is a proteasome inhibitor. Ixazomib citrate, a prodrug, rapidly hydrolyzes under physiological conditions to its biologically active form, ixazomib. The chemical name of ixazomib citrate is 1,3,2-dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- and the structural formula is:
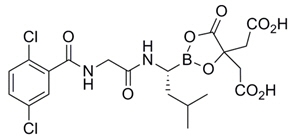
The molecular formula for ixazomib citrate is C20H23BCl2N2O9 and its molecular weight is 517.12. Ixazomib citrate has one chiral center and is the R-stereoisomer. The solubility of ixazomib citrate in 0.1N HCl (pH 1.2) at 37°C is 0.61 mg/mL (reported as ixazomib). The solubility increases as the pH increases.
NINLARO (ixazomib) capsules for oral use contain 4, 3 or 2.3 mg of ixazomib equivalent to 5.7, 4.3 or 3.3 mg of ixazomib citrate, respectively. Inactive ingredients include microcrystalline cellulose, magnesium stearate, and talc. Capsule shells contain gelatin and titanium dioxide. The 4 mg capsule shell contains red and yellow iron oxide, the 3 mg capsule shell contains black iron oxide and the 2.3 mg capsule shell contains red iron oxide. The printing ink contains shellac, propylene glycol, potassium hydroxide, and black iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ixazomib is a reversible proteasome inhibitor. Ixazomib preferentially binds and inhibits the chymotrypsin-like activity of the beta 5 subunit of the 20S proteasome.
Ixazomib induced apoptosis of multiple myeloma cell lines in vitro. Ixazomib demonstrated in vitro cytotoxicity against myeloma cells from patients who had relapsed after multiple prior therapies, including bortezomib, lenalidomide, and dexamethasone. The combination of ixazomib and lenalidomide demonstrated synergistic cytotoxic effects in multiple myeloma cell lines. In vivo, ixazomib demonstrated antitumor activity in a mouse multiple myeloma tumor xenograft model.
12.3 Pharmacokinetics
Absorption
After oral administration, the median time to achieve peak ixazomib plasma concentrations was one hour. The mean absolute oral bioavailability was 58%, based on population PK analysis. Ixazomib AUC increases in a dose proportional manner over a dose range of 0.2 to 10.6 mg.
A food effect study conducted in patients with a single 4 mg dose of ixazomib showed that a high-fat meal decreased ixazomib AUC by 28% and Cmax by 69% [see Dosage and Administration (2.1)].
Distribution
Ixazomib is 99% bound to plasma proteins and distributes into red blood cells with a blood-to-plasma ratio of 10. The steady-state volume of distribution is 543 L.
Elimination
Based on a population PK analysis, systemic clearance was approximately 1.9 L/hr with inter-individual variability of 44%. The terminal half-life (t1/2) of ixazomib was 9.5 days. Following weekly oral dosing, the accumulation ratio was determined to be 2-fold.
Metabolism
After oral administration of a radiolabeled dose, ixazomib represented 70% of total drug-related material in plasma. Metabolism by multiple CYP enzymes and non-CYP proteins is expected to be the major clearance mechanism for ixazomib. At clinically relevant ixazomib concentrations, in vitro studies using human cDNA-expressed cytochrome P450 isozymes showed that no specific CYP isozyme predominantly contributes to ixazomib metabolism. At higher than clinical concentrations, ixazomib was metabolized by multiple CYP isoforms with estimated relative contributions of 3A4 (42%), 1A2 (26%), 2B6 (16%), 2C8 (6%), 2D6 (5%), 2C19 (5%) and 2C9 (< 1%).
Specific Populations
There was no clinically meaningful effect of age (range 23-91 years), sex, body surface area (range 1.2-2.7 m2), or race on the clearance of ixazomib based on population PK analysis.
Patients with Hepatic Impairment
The PK of ixazomib was similar in patients with normal hepatic function and in patients with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin > 1-1.5 × ULN and any AST) based on population PK analysis.
The PK of ixazomib was characterized in patients with normal hepatic function at 4 mg (N=12), moderate hepatic impairment at 2.3 mg (total bilirubin > 1.5-3 × ULN, N=13) or severe hepatic impairment at 1.5 mg (total bilirubin > 3 × ULN, N=18). Dose-normalized mean AUC was 20% higher in patients with moderate or severe hepatic impairment as compared to patients with normal hepatic function [see Dosage and Administration (2.3)].
Patients with Renal Impairment
The PK of ixazomib was similar in patients with normal renal function and in patients with mild or moderate renal impairment (creatinine clearance ≥ 30 mL/min) based on population PK analysis.
The PK of ixazomib was characterized at a dose of 3 mg in patients with normal renal function (creatinine clearance ≥ 90 mL/min, N=18), severe renal impairment (creatinine clearance < 30 mL/min, N=14), or ESRD requiring dialysis (N=6). Mean AUC was 39% higher in patients with severe renal impairment or ESRD requiring dialysis as compared to patients with normal renal function. Pre- and post-dialyzer concentrations of ixazomib measured during the hemodialysis session were similar, suggesting that ixazomib is not dialyzable [see Dosage and Administration (2.4)].
Drug Interaction Studies
Effect of Other Drugs on NINLARO
Strong CYP3A Inducers
Co-administration of NINLARO with rifampin decreased ixazomib Cmax by 54% and AUC by 74% [see Drug Interactions (7.1)].
Effect of NINLARO on Other Drugs
Ixazomib is neither a reversible nor a time-dependent inhibitor of CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4/5. Ixazomib did not induce CYP1A2, CYP2B6, and CYP3A4/5 activity or corresponding immunoreactive protein levels. NINLARO is not expected to produce drug-drug interactions via CYP inhibition or induction.
Transporter-Based Interactions
Ixazomib is a low affinity substrate of P-gp. Ixazomib is not a substrate of BCRP, MRP2 or hepatic OATPs. Ixazomib is not an inhibitor of P-gp, BCRP, MRP2, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1, or MATE2-K. NINLARO is not expected to cause transporter-mediated drug-drug interactions.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Ixazomib was not mutagenic in a bacterial reverse mutation assay (Ames assay). Ixazomib was considered positive in an in vitro clastogenicity test in human peripheral blood lymphocytes. However, in vivo, ixazomib was not clastogenic in a bone marrow micronucleus assay in mice and was negative in an in vivo comet assay in mice, as assessed in the stomach and liver. No carcinogenicity studies have been performed with ixazomib.
Developmental toxicity studies in rats and rabbits did not show direct embryo-fetal toxicity below maternally toxic doses of ixazomib. Studies of fertility and early embryonic development and pre- and postnatal toxicology were not conducted with ixazomib, but evaluation of reproductive tissues was conducted in the general toxicity studies. There were no effects due to ixazomib treatment on male or female reproductive organs in studies up to 6-months duration in rats and up to 9-months duration in dogs.
-
14 CLINICAL STUDIES
The efficacy and safety of NINLARO in combination with lenalidomide and dexamethasone was evaluated in a randomized, double-blind, placebo-controlled, multicenter study in patients with relapsed and/or refractory multiple myeloma who had received at least one prior line of therapy. Patients who were refractory to lenalidomide or proteasome inhibitors were excluded from the study.
A total of 722 patients were randomized in a 1:1 ratio to receive either the combination of NINLARO, lenalidomide and dexamethasone (N=360; NINLARO regimen) or the combination of placebo, lenalidomide and dexamethasone (N=362; placebo regimen) until disease progression or unacceptable toxicity. Randomization was stratified according to number of prior lines of therapy (1 versus 2 or 3), myeloma International Staging System (ISS) (stage I or II versus III), and previous therapy with a proteasome inhibitor (exposed or naïve). Twenty three percent (N=166) of the patients had light chain disease and 12% (N=87) of patients had free light chain-measurable only disease.
Thromboprophylaxis was recommended for all patients in both treatment groups according to the lenalidomide prescribing information. Antiemetics were used in 19% of patients in the NINLARO regimen and 12% of patients in the placebo regimen; antivirals in 64% and 60%, respectively, and antihistamines in 27% and 19%, respectively. These medications were given to patients at the physician's discretion as prophylaxis and/or management of symptoms.
Patients received NINLARO 4 mg or placebo on Days 1, 8, and 15 plus lenalidomide (25 mg) on Days 1 through 21 and dexamethasone (40 mg) on Days 1, 8, 15, and 22 of a 28-day cycle. Patients with renal impairment received a starting dose of lenalidomide according to its prescribing information. Treatment continued until disease progression or unacceptable toxicities.
Table 6 summarizes the baseline patient and disease characteristics in the study. The baseline demographics and disease characteristics were balanced and comparable between the study regimens.
Table 6: Baseline Patient and Disease Characteristics NINLARO + Lenalidomide and Dexamethasone
(N = 360)Placebo + Lenalidomide and Dexamethasone
(N = 362)- * Primary refractory, defined as best response of stable disease or disease progression on all prior lines of therapy, was documented in 7% and 6% of patients in the NINLARO regimen and placebo regimens, respectively.
Patient Characteristics Median age in years (range) 66 (38, 91) 66 (30, 89) Gender (%) Male/ Female 58/42 56/44 Age Group (% [< 65/ ≥ 65 years]) 41/59 43/57 Race n (%) White 310 (86) 301 (83) Black 7 (2) 6 (2) Asian 30 (8) 34 (9) Other or Not Specified 13 (4) 21 (6) ECOG performance status, n (%) 0 or 1 336 (93) 334 (92) 2 18 (5) 24 (7) Missing 6 (2) 4 (1) Creatinine clearance, n (%) < 30 mL/min 5 (1) 5 (1) 30-59 mL/min 74 (21) 95 (26) ≥ 60 mL/min 281 (78) 261 (72) Disease Characteristics Myeloma ISS stage, n (%) Stage I or II 315 (87) 320 (88) Stage III 45 (13) 42 (12) Prior line therapies n (%) Median (range) 1 (1, 3) 1 (1,3) 1 224 (62) 217 (60) 2 or 3 136 (38) 145 (40) Status at Baseline n (%) Relapsed 276 (77) 280 (77) Refractory* 42 (12) 40 (11) Relapsed and Refractory 41 (11) 42 (12) Type of Prior Therapy n (%) Bortezomib containing 248 (69) 250 (69) Carfilzomib containing 1 (<1) 4 (1) Thalidomide containing 157 (44) 170 (47) Lenalidomide containing 44 (12) 44 (12) Melphalan containing 293 (81) 291 (80) Stem cell transplantation 212 (59) 199 (55) High risk (deletion (del) 17, t(4:14) and/or t(14:16) 75 (21) 62 (17) deletion del (17) 36 (10) 33 (9) The efficacy of NINLARO was evaluated by progression-free survival (PFS) according to the 2011 International Myeloma Working Group (IMWG) Consensus Uniform Response Criteria as assessed by a blinded independent review committee (IRC) based on central lab results. Response was assessed every four weeks until disease progression.
The approval of NINLARO was based upon a statistically significant improvement in PFS of the NINLARO regimen compared to the placebo regimen. PFS results are summarized in Table 7 and shown in Figure 1.
Table 7: Progression-Free Survival and Response Rate NINLARO + Lenalidomide and Dexamethasone
(N = 360)Placebo + Lenalidomide and Dexamethasone
(N = 362)NE: Not evaluable. - * Hazard ratio is based on a stratified Cox's proportional hazard regression model. A hazard ratio less than 1 indicates an advantage for the NINLARO regimen.
- † P-value is based on the stratified log-rank test.
Progression-free Survival PFS Events, n (%) 129 (36) 157 (43) Median (months)
(95% CI)20.6
(17.0, NE)14.7
(12.9, 17.6)Hazard Ratio*
(95% CI)0.74
(0.59, 0.94)p-value† 0.012 Response Rate Overall Response Rate, n (%) 282 (78) 259 (72) Complete Response 42 (12) 24 (7) Very Good Partial Response 131 (36) 117 (32) Partial Response 109 (30) 118 (33) The median time to response was 1.1 months in the NINLARO regimen and 1.9 months in the placebo regimen. The median duration of response was 20.5 months in the NINLARO regimen and 15 months in the placebo regimen for responders in the response evaluable population.
Figure 1: Kaplan-Meier Plot of Progression-Free Survival
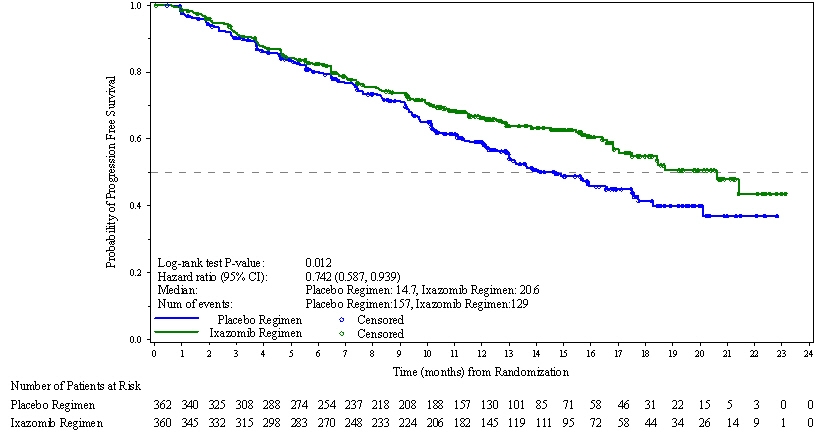
A non-inferential PFS analysis was conducted at a median follow up of 23 months with 372 PFS events. Hazard ratio of PFS was 0.82 (95% confidence interval [0.67, 1.0]) for NINLARO regimen versus placebo regimen, and estimated median PFS was 20 months in the NINLARO regimen and 15.9 months in the placebo regimen. At the same time, a planned interim OS analysis was conducted with 35% of the required number of deaths for final OS analysis; there were 81 deaths in the NINLARO regimen and 90 deaths in the placebo regimen. An OS benefit was not demonstrated.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
NINLARO is supplied as:
4 mg gelatin capsule: Light orange, size 3, imprinted with "Takeda" on the cap and "4 mg" on the body in black ink. NINLARO 4 mg capsules contain 4 mg of ixazomib equivalent to 5.7 mg of ixazomib citrate.
- One 4 mg capsule in a single blister pack (NDC: 63020-400-01)
- Three 4 mg single packs in a carton (NDC: 63020-400-02)
3 mg gelatin capsule: Light grey, size 4, imprinted with "Takeda" on the cap and "3 mg" on the body in black ink. NINLARO 3 mg capsules contain 3 mg of ixazomib equivalent to 4.3 mg of ixazomib citrate.
- One 3 mg capsule in a single blister pack (NDC: 63020-390-01)
- Three 3 mg single packs in a carton (NDC: 63020-390-02)
2.3 mg gelatin capsule: Light pink, size 4, imprinted with "Takeda" on the cap and "2.3 mg" on the body in black ink. NINLARO 2.3 mg capsules contain 2.3 mg of ixazomib equivalent to 3.3 mg of ixazomib citrate.
- One 2.3 mg capsule in a single blister pack (NDC: 63020-230-01)
- Three 2.3 mg single packs in a carton (NDC: 63020-230-02)
Capsules are individually packaged in a PVC-Aluminum/Aluminum blister.
16.2 Storage
NINLARO may be stored at room temperature. Do not store above 30°C (86°F). Do not freeze.
Store capsules in original packaging until immediately prior to use.
16.3 Handling and Disposal
NINLARO is a cytotoxic drug. Follow applicable special handling and disposal procedures1. Do not open or crush capsules. Avoid direct contact with the capsule contents. In case of capsule breakage, avoid direct contact of capsule contents with the skin or eyes. If contact occurs with the skin, wash thoroughly with soap and water. If contact occurs with the eyes, flush thoroughly with water.
Any unused medicinal product or waste material should be disposed in accordance with local requirements.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dosing Instructions
- Instruct patients to take NINLARO exactly as prescribed.
- Advise patients to take NINLARO once a week on the same day and at approximately the same time for the first three weeks of a four week cycle.
- Advise patients to take NINLARO at least one hour before or at least two hours after food.
- Advise patients that NINLARO and dexamethasone should not be taken at the same time, because dexamethasone should be taken with food and NINLARO should not be taken with food.
- Advise patients to swallow the capsule whole with water. The capsule should not be crushed, chewed or opened.
- Advise patients that direct contact with the capsule contents should be avoided. In case of capsule breakage, avoid direct contact of capsule contents with the skin or eyes. If contact occurs with the skin, wash thoroughly with soap and water. If contact occurs with the eyes, flush thoroughly with water.
- If a patient misses a dose, advise them to take the missed dose as long as the next scheduled dose is ≥ 72 hours away. Advise patients not to take a missed dose if it is within 72 hours of their next scheduled dose.
- If a patient vomits after taking a dose, advise them not to repeat the dose but resume dosing at the time of the next scheduled dose.
- Advise patients to store capsules in original packaging, and not to remove the capsule from the packaging until just prior to taking NINLARO.
Thrombocytopenia
Advise patients that they may experience low platelet counts (thrombocytopenia). Signs of thrombocytopenia may include bleeding and easy bruising. [see Warnings and Precautions (5.1)].
Gastrointestinal Toxicities
Advise patients they may experience diarrhea, constipation, nausea and vomiting and to contact their physician if these adverse reactions persist. [see Warnings and Precautions (5.2)].
Peripheral Neuropathy
Advise patients to contact their physicians if they experience new or worsening symptoms of peripheral neuropathy such as tingling, numbness, pain, a burning feeling in the feet or hands, or weakness in the arms or legs. [see Warnings and Precautions (5.3)].
Peripheral Edema
Advise patients to contact their physicians if they experience unusual swelling of their extremities or weight gain due to swelling [see Warnings and Precautions (5.4)].
Cutaneous Reactions
Advise patients to contact their physicians if they experience new or worsening rash [see Warnings and Precautions (5.5)].
Thrombotic Microangiopathy
Advise patients to seek immediate medical attention if any signs or symptoms of thrombotic microangiopathy occur [see Warnings and Precautions (5.6)].
Hepatotoxicity
Advise patients to contact their physicians if they experience jaundice or right upper quadrant abdominal pain [see Warnings and Precautions (5.7)].
Other Adverse Reactions
Advise patients to contact their physicians if they experience signs and symptoms of acute febrile neutrophilic dermatosis (Sweet's syndrome), Stevens-Johnson syndrome, transverse myelitis, posterior reversible encephalopathy syndrome, tumor lysis syndrome, and thrombotic thrombocytopenic purpura [see Adverse Reactions (6.1)].
Pregnancy
Advise women of the potential risk to a fetus and to avoid becoming pregnant while being treated with NINLARO and for 90 days following the final dose. Advise women using hormonal contraceptives to also use a barrier method of contraception. Advise patients to contact their physicians immediately if they or their female partner become pregnant during treatment or within 90 days of the final dose [see Warnings and Precautions (5.8)].
-
SPL UNCLASSIFIED SECTION
Distributed and Marketed by: Takeda Pharmaceutical Company Limited, Cambridge, MA 02139
NINLARO is a registered trademark of Millennium Pharmaceuticals, Inc. Millennium Pharmaceuticals, Inc. is a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
©2015-2020 Millennium Pharmaceuticals, Inc.
For more information, you may also go to www.NINLARO.com or call 1-844-617-6468.
Item Code: 101155/4
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: February 2020 PATIENT INFORMATION
NINLARO® (nin-LAR-oh)
(ixazomib)
capsulesNINLARO is used with two other prescription medicines called REVLIMID® (lenalidomide) and dexamethasone. Read the Medication Guide that comes with REVLIMID® (lenalidomide). You can ask your healthcare provider or pharmacist for information about dexamethasone.
What is NINLARO?
NINLARO is a prescription medicine used to treat multiple myeloma in combination with the medicines REVLIMID® (lenalidomide) and dexamethasone, in people who have received at least one prior treatment for their multiple myeloma.
It is not known if NINLARO is safe and effective in children.
Before taking NINLARO, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems
- have kidney problems or are on dialysis
- are pregnant or plan to become pregnant. NINLARO can harm your unborn baby.
- Avoid becoming pregnant during treatment with NINLARO.
- Females who are able to become pregnant must use effective birth control during treatment and for 90 days after your final dose of NINLARO. If using hormonal contraceptives (for example, the pill), an additional barrier method of contraception (for example, diaphragm or condom) must be used.
- Males with a female partner who is able to become pregnant must use effective birth control during treatment and for 90 days after your final dose of NINLARO.
- Talk to your healthcare provider about birth control methods that may be right for you.
- Tell your healthcare provider right away if you or your partner become pregnant while you are receiving NINLARO.
- are breastfeeding or plan to breastfeed. It is not known if NINLARO passes into breast milk, if it affects an infant who is breastfed, or breast milk production. Do not breastfeed during treatment with NINLARO and for 90 days after your final dose of NINLARO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Talk to your healthcare provider before starting any new medicines during treatment with NINLARO.
How should I take NINLARO?
- Take NINLARO exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking NINLARO without talking to your healthcare provider first.
- NINLARO is taken in "cycles." Each cycle lasts 4 weeks (28 days).
- The usual dose of NINLARO is 1 capsule taken 1 time each week, on the same day of the week for the first 3 weeks of each cycle.
- Take each dose of NINLARO at about the same time of day.
- Take REVLIMID (lenalidomide) and dexamethasone exactly as your healthcare provider tells you to.
- Your healthcare provider will do blood tests during treatment with NINLARO to check for side effects.
- Your healthcare provider may change your dose or stop NINLARO, REVLIMID (lenalidomide), or dexamethasone if you have side effects.
- Take NINLARO at least 1 hour before or at least 2 hours after food.
- On the days that you take both NINLARO and dexamethasone, do not take NINLARO and dexamethasone at the same time. Take dexamethasone with food.
- Swallow NINLARO capsules whole with water. Do not crush, chew or open the capsule.
- Avoid direct contact with the capsule contents. If you accidentally get powder from the NINLARO capsule on your skin, wash the area well with soap and water. If you accidentally get powder from the NINLARO capsule in your eyes, flush your eyes well with water.
- If you miss a dose of NINLARO, or if you are late taking a dose, take the dose as long as the next scheduled dose is more than 3 days (72 hours) away. Do not take a missed dose of NINLARO if it is within 3 days (72 hours) of your next scheduled dose.
- If you vomit after taking a dose of NINLARO, do not repeat the dose. Take your next dose of NINLARO on the next scheduled day and time.
- Your doctor may prescribe a medicine to take with NINLARO to decrease the risk of the chicken pox virus (herpes zoster) coming back (reactivation).
- If you take more NINLARO than your healthcare provider tells you to take, call your healthcare provider right away or go to the nearest hospital emergency room.
What are the possible side effects of NINLARO?
NINLARO may cause serious side effects, including:
- Low platelet counts (thrombocytopenia). Low platelet counts are common with NINLARO, and can sometimes be serious. You may need platelet transfusions if your counts are too low. Tell your healthcare provider if you have any signs of low platelet counts, including bleeding and easy bruising.
- Stomach and intestinal (gastrointestinal) problems. Diarrhea, constipation, nausea, and vomiting are common with NINLARO, and can sometimes be severe. Call your healthcare provider if you get any of these symptoms and they do not go away during treatment with NINLARO. Your healthcare provider may prescribe medicine to help treat your symptoms.
- Nerve problems. Nerve problems are common with NINLARO and may also be severe. Tell your healthcare provider if you get any new or worsening symptoms, including:
- tingling
- numbness
- pain
- a burning feeling in your feet or hands
- weakness in your arms or legs
- Swelling. Swelling is common with NINLARO and can sometimes be severe. Tell your healthcare provider if you develop swelling in your arms, hands, legs, ankles, or feet, or if you gain weight from swelling.
- Skin reactions. Tell your healthcare provider if you get a new or worsening rash.
- Thrombotic microangiopathy (TMA). This is a condition involving blood clots and injury to small blood vessels that may cause harm to your kidneys, brain, and other organs, and may lead to death. Get medical help right away if you get any of the following signs or symptoms during treatment with NINLARO:
- fever
- bruising
- nose bleeds
- tiredness
- decreased urination
-
Liver problems. Tell your healthcare provider if you get these signs of a liver problem:
- yellowing of your skin or the whites of your eyes
- pain in your right upper stomach-area
Other common side effects have occurred. Tell your healthcare provider if you get new or worsening signs or symptoms of the following:
- back pain
- skin rash and pain (shingles) as a result of reactivation of the chicken pox virus (herpes zoster)
- lowered white blood cells called neutrophils (neutropenia) that may increase the risk of infection
- vision conditions including blurred vision, dry eye and pink eye (conjunctivitis)
These are not all the possible side effects of NINLARO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NINLARO?
- Store NINLARO at room temperature. Do not store above 86°F (30°C).
- Do not freeze NINLARO.
- Store NINLARO capsules in the original packaging until just before each use.
- Ask your pharmacist or healthcare provider about how to dispose of (throw away) unused NINLARO.
Keep NINLARO and all medicines out of the reach of children.
General information about the safe and effective use of NINLARO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use NINLARO for a condition for which it was not prescribed. Do not give NINLARO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about NINLARO that is written for healthcare professionals.
What are the ingredients in NINLARO?
Active ingredient: ixazomib
Inactive ingredients: microcrystalline cellulose, magnesium stearate, and talc
Capsule shells: gelatin and titanium dioxide. The 4 mg capsule shell contains red and yellow iron oxide. The 3 mg capsule shell contains black iron oxide. The 2.3 mg capsule shell contains red iron oxide. The printing ink contains shellac, propylene glycol, potassium hydroxide, and black iron oxide.
Distributed and Marketed by: Takeda Pharmaceutical Company Limited, Cambridge, MA 02139
NINLARO is a registered trademark of Millennium Pharmaceuticals, Inc. Millennium Pharmaceuticals, Inc. is a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
All other trademarks are the property of their respective owners.
©2015-2020 Millennium Pharmaceuticals, Inc.
For more information, you may also go to www.NINLARO.com or call 1-844-617-6468. -
PRINCIPAL DISPLAY PANEL - 4 mg Capsule Blister Pack
RX only
NDC: 63020-400-011
PRESS
&
HOLD
HERENINLARO®
(ixazomib) capsules4mg
1
Press &
hold button2
Pull out
medication cardContains 1 Capsule
Please read Package Insert before use.Takeda
2
PULL OUT HERE

-
PRINCIPAL DISPLAY PANEL - 3 mg Capsule Blister Pack
RX only
NDC: 63020-390-011
PRESS
&
HOLD
HERENINLARO®
(ixazomib) capsules3mg
1
Press &
hold button2
Pull out
medication cardContains 1 Capsule
Please read Package Insert before use.Takeda
2
PULL OUT HERE

-
PRINCIPAL DISPLAY PANEL - 2.3 mg Capsule Blister Pack
RX only
NDC: 63020-230-011
PRESS
&
HOLD
HERENINLARO®
(ixazomib) capsules2.3mg
1
Press &
hold button2
Pull out
medication cardContains 1 Capsule
Please read Package Insert before use.Takeda
2
PULL OUT HERE

-
INGREDIENTS AND APPEARANCE
NINLARO
ixazomib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63020-400 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ixazomib citrate (UNII: 46CWK97Z3K) (ixazomib - UNII:71050168A2) ixazomib 4 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) talc (UNII: 7SEV7J4R1U) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color ORANGE (Light orange) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code 4;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63020-400-02 3 in 1 CARTON 11/20/2015 1 NDC: 63020-400-01 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208462 11/20/2015 NINLARO
ixazomib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63020-390 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ixazomib citrate (UNII: 46CWK97Z3K) (ixazomib - UNII:71050168A2) ixazomib 3 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) talc (UNII: 7SEV7J4R1U) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color GRAY (Light grey) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code 3;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63020-390-02 3 in 1 CARTON 11/20/2015 1 NDC: 63020-390-01 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208462 11/20/2015 NINLARO
ixazomib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 63020-230 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ixazomib citrate (UNII: 46CWK97Z3K) (ixazomib - UNII:71050168A2) ixazomib 2.3 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) talc (UNII: 7SEV7J4R1U) magnesium stearate (UNII: 70097M6I30) Product Characteristics Color PINK (Light pink) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code 2;3;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 63020-230-02 3 in 1 CARTON 11/20/2015 1 NDC: 63020-230-01 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208462 11/20/2015 Labeler - Millennium Pharmaceuticals, Inc. (804148757) Establishment Name Address ID/FEI Business Operations Ash Stevens Inc. 049265333 API MANUFACTURE(63020-400, 63020-390, 63020-230) , ANALYSIS(63020-400, 63020-390, 63020-230) Establishment Name Address ID/FEI Business Operations AMRI SSCI, LLC 020593403 ANALYSIS(63020-400, 63020-390, 63020-230) Establishment Name Address ID/FEI Business Operations Eurofins Lancaster Laboratories, Inc 069777290 ANALYSIS(63020-400, 63020-390, 63020-230) Establishment Name Address ID/FEI Business Operations Haupt Pharma Amareg GmbH 331334909 MANUFACTURE(63020-400, 63020-390, 63020-230) Establishment Name Address ID/FEI Business Operations AndersonBrecon (UK) Limited 762771269 PACK(63020-400, 63020-390, 63020-230) , LABEL(63020-400, 63020-390, 63020-230) Establishment Name Address ID/FEI Business Operations Takeda Ireland Ltd. 986017670 PACK(63020-400, 63020-390, 63020-230) , LABEL(63020-400, 63020-390, 63020-230)
Trademark Results [NINLARO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 NINLARO 85854620 4905454 Live/Registered |
Millennium Pharmaceuticals, Inc. 2013-02-20 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.