LEUKINE- sargramostim liquid LEUKINE- sargramostim injection, powder, for solution
Leukine by
Drug Labeling and Warnings
Leukine by is a Prescription medication manufactured, distributed, or labeled by sanofi-aventis U.S. LLC, Hospira, Inc., Genzyme Corporation, Bayer HealthCare LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LEUKINE safely and effectively. See full prescribing information for LEUKINE.
LEUKINE® (sargramostim) for injection, for subcutaneous or intravenous use
LEUKINE® (sargramostim) injection, for subcutaneous or intravenous use
Initial U.S. Approval: 1991RECENT MAJOR CHANGES
INDICATIONS AND USAGE
LEUKINE is a leukocyte growth factor indicated:
- To shorten time to neutrophil recovery and to reduce the incidence of severe and life-threatening infections and infections resulting in death following induction chemotherapy in adult patients 55 years and older with acute myeloid leukemia (AML). (1.1)
- For the mobilization of hematopoietic progenitor cells into peripheral blood for collection by leukapheresis and autologous transplantation in adult patients. (1.2)
- For the acceleration of myeloid reconstitution following autologous bone marrow or peripheral blood progenitor cell transplantation in adult and pediatric patients 2 years of age and older. (1.3)
- For the acceleration of myeloid reconstitution following allogeneic bone marrow transplantation in adult and pediatric patients 2 years of age and older. (1.4)
- For treatment of delayed neutrophil recovery or graft failure after autologous or allogeneic bone marrow transplantation in adult and pediatric patients 2 years of age and older. (1.5)
- To increase survival in adult and pediatric patients from birth to 17 years of age acutely exposed to myelosuppressive doses of radiation (Hematopoietic Syndrome of Acute Radiation Syndrome [H-ARS]). (1.6)
DOSAGE AND ADMINISTRATION
See Full Prescribing Information for dosage adjustments and timing of administration (2.1–2.6).
- AML, Neutrophil recovery following chemotherapy:
- 250 mcg/m2/day administered intravenously over a 4-hour period. (2.1)
- Mobilization of peripheral blood progenitor cells:
- 250 mcg/m2/day administered intravenously over 24 hours or subcutaneous injection once daily. (2.2)
- Post peripheral blood progenitor cell transplantation:
- 250 mcg/m2/day administered intravenously over 24 hours or subcutaneous injection once. (2.3)
- Myeloid reconstitution after autologous or allogeneic BMT:
- 250 mcg/m2/day administered intravenously over a 2-hour period. (2.4)
- BMT failure or engraftment delayed:
- 250 mcg/m2/day for 14 days as a 2-hour intravenous infusion. (2.5)
- Patients acutely exposed to myelosuppressive doses of radiation, administer once daily as subcutaneous injection:
- Adults and pediatric patients weighing >40 kg: 7 mcg/kg
- Pediatric patients 15 kg to 40 kg: 10 mcg/kg
- Pediatric patients <15 kg: 12 mcg/kg (2.6)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- Do not administer LEUKINE to patients with a history of serious allergic reactions, including anaphylaxis, to human granulocyte-macrophage colony stimulating factor such as sargramostim, yeast-derived products, or any component of the product. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Permanently discontinue LEUKINE in patients with serious allergic reactions. (5.1)
- Infusion Related Reactions: Manage using infusion rate reductions or discontinuations. (5.2)
- Effusions and Capillary Leak Syndrome: Manage with dose-reduction, discontinuation, or diuretics. Monitor body weight and hydration status during therapy. (5.4)
- Supraventricular Arrhythmias: Risk may be increased in patients with history of cardiac arrhythmias. Manage medically and discontinue LEUKINE. (5.5)
ADVERSE REACTIONS
The most common adverse reactions (incidence >30%) were (6.1):
- In recipients of autologous BMT: fever, nausea, diarrhea, vomiting, mucous membrane disorder, alopecia, asthenia, malaise, anorexia, rash, gastrointestinal disorder and edema.
- In recipients of allogeneic BMT: diarrhea, fever, nausea, rash, vomiting, stomatitis, anorexia, high glucose, alopecia, abdominal pain, low albumin, headache and hypertension.
- In patients with AML: fever, liver toxicity, skin reactions, infections, metabolic laboratory abnormalities, nausea, diarrhea, genitourinary abnormalities, pulmonary toxicity, vomiting, neurotoxicity, stomatitis, alopecia and weight loss.
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme Corporation at 1-888-4RX-LEUKINE or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Use with caution in patients receiving drugs that may potentiate LEUKINE's myeloproliferative effects, such as lithium and corticosteroids. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Acute Myeloid Leukemia Following Induction Chemotherapy
1.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
1.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
1.4 Allogeneic Bone Marrow Transplantation
1.5 Allogeneic or Autologous Bone Marrow Transplantation: Treatment of Delayed Neutrophil Recovery or Graft Failure
1.6 Acute Exposure to Myelosuppressive Doses of Radiation (H-ARS)
2 DOSAGE AND ADMINISTRATION
2.1 Neutrophil Recovery Following Induction Chemotherapy for Acute Myeloid Leukemia
2.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
2.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
2.4 Allogeneic Bone Marrow Transplantation
2.5 Allogeneic or Autologous Bone Marrow Transplantation: Treatment of Delayed Neutrophil Recovery or Graft Failure
2.6 Acute Exposure to Myelosuppressive Doses of Radiation (H-ARS)
2.7 Preparation and Administration of LEUKINE
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Infusion Related Reactions
5.3 Risk of Severe Myelosuppression when LEUKINE Administered within 24 hours of Chemotherapy or Radiotherapy
5.4 Effusions and Capillary Leak Syndrome
5.5 Supraventricular Arrhythmias
5.6 Leukocytosis
5.7 Potential Effect on Malignant Cells
5.8 Immunogenicity
5.9 Risk of Serious Adverse Reactions in Infants Due to Benzyl Alcohol Preservative
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Use with Products that Induce Myeloproliferation
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Following Induction Chemotherapy for Acute Myelogenous Leukemia
14.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
14.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
14.4 Allogeneic Bone Marrow Transplantation
14.5 Treatment of Delayed Neutrophil Recovery or Graft Failure After Allogeneic or Autologous Bone Marrow Transplantation
14.6 Acute Exposure to Myelosuppressive Doses of Radiation (H-ARS)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Acute Myeloid Leukemia Following Induction Chemotherapy
LEUKINE is indicated to shorten time to neutrophil recovery and to reduce the incidence of severe, life-threatening, or fatal infections following induction chemotherapy in adult patients 55 years and older with acute myeloid leukemia (AML).
1.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
LEUKINE is indicated in adult patients with cancer undergoing autologous hematopoietic stem cell transplantation for the mobilization of hematopoietic progenitor cells into peripheral blood for collection by leukapheresis.
1.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
LEUKINE is indicated for the acceleration of myeloid reconstitution following autologous peripheral blood progenitor cell (PBPC) or bone marrow transplantation in adult and pediatric patients 2 years of age and older with non-Hodgkin's lymphoma (NHL), acute lymphoblastic leukemia (ALL) and Hodgkin's lymphoma (HL).
1.4 Allogeneic Bone Marrow Transplantation
LEUKINE is indicated for the acceleration of myeloid reconstitution in adult and pediatric patients 2 years of age and older undergoing allogeneic bone marrow transplantation from HLA-matched related donors.
1.5 Allogeneic or Autologous Bone Marrow Transplantation: Treatment of Delayed Neutrophil Recovery or Graft Failure
LEUKINE is indicated for the treatment of adult and pediatric patients 2 years and older who have undergone allogeneic or autologous bone marrow transplantation in whom neutrophil recovery is delayed or failed.
-
2 DOSAGE AND ADMINISTRATION
2.1 Neutrophil Recovery Following Induction Chemotherapy for Acute Myeloid Leukemia
The recommended dose is 250 mcg/m2/day administered intravenously over a 4-hour period starting approximately on day 11 or four days following the completion of induction chemotherapy, if the day 10 bone marrow is hypoplastic with less than 5% blasts. If a second cycle of induction chemotherapy is necessary, administer LEUKINE approximately four days after the completion of chemotherapy if the bone marrow is hypoplastic with less than 5% blasts. Continue LEUKINE until an absolute neutrophil count (ANC) greater than 1500 cells/mm3 for 3 consecutive days or a maximum of 42 days. Do not administer LEUKINE within 24 hours preceding or following receipt of chemotherapy or radiotherapy [see Warnings and Precautions (5.3)].
Dose Modifications
Obtain a CBC with differential twice per week during LEUKINE therapy and modify the dose for the following:
- Leukemic regrowth: Discontinue LEUKINE immediately
- Grade 3 or 4 adverse reactions: Reduce the dose of LEUKINE by 50% or interrupt dosing until the reaction abates
- ANC greater than 20,000 cells/mm3: Interrupt LEUKINE treatment or reduce the dose by 50%
2.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
The recommended dose is 250 mcg/m2/day administered intravenously over 24 hours or subcutaneously once daily. Continue at the same dose through the period of PBPC collection. The optimal schedule for PBPC collection has not been established. In clinical studies, collection of PBPC was usually begun after 5 days of LEUKINE and performed daily until protocol specified targets were achieved [see Clinical Studies (14)].
If WBC greater than 50,000 cells/mm3, reduce the LEUKINE dose by 50%. Consider other mobilization therapy if adequate numbers of progenitor cells are not collected.
2.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
Autologous Peripheral Blood Progenitor Cell Transplantation
The recommended dose is 250 mcg/m2/day administered intravenously over 24 hours or subcutaneously once daily beginning immediately following infusion of progenitor cells and continuing until an ANC greater than 1500 cells/mm3 for three consecutive days is attained. Do not administer LEUKINE within 24 hours preceding or following receipt of chemotherapy or radiotherapy.
Autologous Bone Marrow Transplantation
The recommended dose is 250 mcg/m2/day administered intravenously over a 2-hour period beginning two to four hours after bone marrow infusion, and not less than 24 hours after the last dose of chemotherapy or radiotherapy. Do not administer LEUKINE until the post marrow infusion ANC is less than 500 cells/mm3. Continue LEUKINE until an ANC greater than 1500 cells/mm3 for three consecutive days is attained. Do not administer LEUKINE within 24 hours preceding or following receipt of chemotherapy or radiotherapy [see Warnings and Precautions (5.3)].
2.4 Allogeneic Bone Marrow Transplantation
The recommended dose is 250 mcg/m2/day administered intravenously over a 2-hour period beginning two to four hours after bone marrow infusion, and not less than 24 hours after the last dose of chemotherapy or radiotherapy. Do not administer LEUKINE until the post marrow infusion ANC is less than 500 cells/mm3. Continue LEUKINE until an ANC greater than 1500 cells/mm3 for three consecutive days is attained. Do not administer LEUKINE within 24 hours preceding or following receipt of chemotherapy or radiotherapy [see Warnings and Precautions (5.3)].
Dose Modifications
Obtain a CBC with differential twice per week during LEUKINE therapy and modify the dose as for the following:
- Disease progression or blast cell appearance: Discontinue LEUKINE immediately
- Grade 3 or 4 adverse reactions: Reduce the dose of LEUKINE by 50% or temporarily discontinue until the reaction abates
- WBC greater than 50,000 cells/mm3 or ANC greater than 20,000 cells/mm3: Interrupt LEUKINE treatment or reduce the dose by 50%
2.5 Allogeneic or Autologous Bone Marrow Transplantation: Treatment of Delayed Neutrophil Recovery or Graft Failure
The recommended dose is 250 mcg/m2/day for 14 days as a 2-hour intravenous infusion. The dose can be repeated after 7 days off therapy if neutrophil recovery has not occurred. If neutrophil recovery still has not occurred, a third course of 500 mcg/m2/day for 14 days may be tried after another 7 days off therapy. If there is still no improvement, it is unlikely that further dose escalation will be beneficial.
Dose Modifications
Obtain a CBC with differential twice per week during LEUKINE therapy and modify the dose as for the following:
- Disease progression or blast cell appearance: Discontinue LEUKINE immediately
- Grade 3 or 4 adverse reactions: Reduce the dose of LEUKINE by 50% or temporarily discontinue until the reaction abates
- WBC greater than 50,000 cells/mm3 or ANC greater than 20,000 cells/mm3: Interrupt LEUKINE treatment or reduce the dose by 50%
2.6 Acute Exposure to Myelosuppressive Doses of Radiation (H-ARS)
For patients with H-ARS, the recommended dose of LEUKINE is a subcutaneous injection administered once daily as follows:
- 7 mcg/kg in adult and pediatric patients weighing greater than 40 kg
- 10 mcg/kg in pediatric patients weighing 15 kg to 40 kg
- 12 mcg/kg in pediatric patients weighing less than 15 kg
Administer LEUKINE as soon as possible after suspected or confirmed exposure to radiation doses greater than 2 gray (Gy).
Estimate a patient's absorbed radiation dose (i.e., level of radiation exposure) based on information from public health authorities, biodosimetry if available, or clinical findings such as time to onset of vomiting or lymphocyte depletion kinetics.
Obtain a baseline CBC with differential and then serial CBCs approximately every third day until the ANC remains greater than 1,000/mm3 for three consecutive CBCs. Do not delay administration of LEUKINE if a CBC is not readily available.
Continue administration of LEUKINE until the ANC remains greater than 1,000/mm3 for three consecutive CBCs or exceeds 10,000/mm3 after a radiation-induced nadir.
2.7 Preparation and Administration of LEUKINE
- Do not administer LEUKINE simultaneously with or within 24 hours preceding cytotoxic chemotherapy or radiotherapy or within 24 hours following chemotherapy [see Warnings and Precautions (5.3)].
- LEUKINE injection is formulated as a sterile solution preserved with 1.1% benzyl alcohol.
- LEUKINE for injection is a sterile, preservative-free lyophilized powder that requires reconstitution with 1 mL Sterile Water for Injection (without preservative), USP, to yield a clear, colorless single-dose solution or 1 mL Bacteriostatic Water for Injection, USP (with 0.9% benzyl alcohol as preservative) to yield a clear, colorless single-dose solution.
Use only LEUKINE for injection (lyophilized powder) reconstituted with Sterile Water for Injection without preservatives when administering LEUKINE to neonates or infants to avoid benzyl alcohol exposure [see Warnings and Precautions (5.9)].
Do NOT use an in-line membrane filter for intravenous infusion of LEUKINE.
- Store LEUKINE solution and reconstituted lyophilized LEUKINE solutions under refrigeration at 2°C–8°C (36°F–46°F); DO NOT FREEZE.
- In the absence of compatibility and stability information, do not add other medication to infusion solutions containing LEUKINE. Use only 0.9% Sodium Chloride Injection, USP to prepare intravenous infusion solutions.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. If particulate matter is present or the solution is discolored, the vial should not be used.
LEUKINE for Injection (lyophilized powder) Preparation
Reconstitute LEUKINE for Injection aseptically with 1 mL of diluent. Do not mix the contents of vials reconstituted with different diluents together. Reconstitute with either Sterile Water for Injection, USP (without preservative) or Bacteriostatic Water for Injection, USP (0.9% benzyl alcohol). Use reconstituted LEUKINE for injection (lyophilized powder) vials within 6 hours following constitution and/or dilution for intravenous infusion. Do not re-enter or reuse the vial. Discard any unused portions.
LEUKINE Injection (solution) Preparation
- For subcutaneous injection: Administer LEUKINE Injection without further dilution.
- For intravenous injection: Administer LEUKINE injection in 0.9% Sodium Chloride Injection, USP. Dilute LEUKINE for intravenous infusion in 0.9% Sodium Chloride Injection, USP. If the final concentration of LEUKINE is below 10 mcg/mL, add Albumin (Human) at a final concentration of 0.1% to the saline prior to addition of LEUKINE to prevent adsorption to the components of the drug delivery system. To obtain a final concentration of 0.1% Albumin (Human), add 1 mg Albumin (Human) per 1 mL 0.9% Sodium Chloride Injection, USP (e.g., use 1 mL 5% Albumin [Human] in 50 mL 0.9% Sodium Chloride Injection, USP).
- Store LEUKINE injection for up to 20 days at 2°C–8°C once the vial has been entered. Discard any remaining solution after 20 days.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Do not administer LEUKINE to patients with a history of serious allergic reactions, including anaphylaxis, to human granulocyte-macrophage colony stimulating factor such as sargramostim, yeast-derived products, or any component of the product. Anaphylactic reactions have been reported with LEUKINE [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic reactions, have been reported with LEUKINE. Parenteral administration of LEUKINE should be attended by appropriate precautions in case an allergic or untoward reaction occurs. If any serious allergic or anaphylactic reaction occurs, immediately discontinue LEUKINE therapy and institute medical management. Discontinue LEUKINE permanently for patients with serious allergic reactions.
5.2 Infusion Related Reactions
LEUKINE can cause infusion-related reactions. Infusion-related reactions may be characterized by respiratory distress, hypoxia, flushing, hypotension, syncope, and/or tachycardia following the first administration of LEUKINE in a particular cycle. These signs have resolved with symptomatic treatment and usually do not recur with subsequent doses in the same cycle of treatment.
Observe closely during infusion for symptoms, particularly in patients with pre-existing lung disease. If patients display dyspnea or other acute symptoms, reduce the rate of infusion by 50%. If symptoms persist or worsen despite rate reduction, discontinue the LEUKINE infusion. If patient experiences infusion-related reaction, subsequent intravenous infusions may be administered following the standard dose schedule with careful monitoring.
5.3 Risk of Severe Myelosuppression when LEUKINE Administered within 24 hours of Chemotherapy or Radiotherapy
Due to the potential sensitivity of rapidly dividing hematopoietic progenitor cells, LEUKINE should not be administered simultaneously with or within 24 hours preceding cytotoxic chemotherapy or radiotherapy or within 24 hours following chemotherapy. In one controlled study, patients with small cell lung cancer received LEUKINE and concurrent thoracic radiotherapy and chemotherapy or the identical radiotherapy and chemotherapy without LEUKINE. The patients randomized to LEUKINE had significantly higher incidence of adverse reactions, including higher mortality and a higher incidence of grade 3 and 4 infections and grade 3 and 4 thrombocytopenia.
5.4 Effusions and Capillary Leak Syndrome
Edema, capillary leak syndrome, and pleural and/or pericardial effusion, have been reported in patients after LEUKINE administration. In 156 patients enrolled in placebo-controlled studies using LEUKINE at a dose of 250 mcg/m2/day by 2-hour IV infusion, the reported incidences of fluid retention (LEUKINE vs. placebo) were as follows: peripheral edema, 11% vs. 7%; pleural effusion, 1% vs. 0%; and pericardial effusion, 4% vs. 1%. Capillary leak syndrome was not observed in this limited number of studies; based on other uncontrolled studies and reports from users of marketed LEUKINE, the incidence is estimated to be less than 1%. In patients with preexisting pleural and pericardial effusions, administration of LEUKINE may aggravate fluid retention; however, fluid retention associated with or worsened by LEUKINE has been reversible after interruption or dose reduction of LEUKINE with or without diuretic therapy. LEUKINE should be used with caution in patients with preexisting fluid retention, pulmonary infiltrates, or congestive heart failure. Body weight and hydration status should be carefully monitored during LEUKINE administration.
5.5 Supraventricular Arrhythmias
Supraventricular arrhythmia has been reported in uncontrolled studies during LEUKINE administration, particularly in patients with a previous history of cardiac arrhythmia. These arrhythmias have been reversible after discontinuation of LEUKINE. Use LEUKINE with caution in patients with preexisting cardiac disease.
5.6 Leukocytosis
White blood cell counts of ≥ 50,000/mm3 were observed in patients receiving LEUKINE. Monitor complete blood counts (CBC) with differential twice per week. Base the decision whether to reduce the dose or interrupt treatment on the clinical condition of the patient [see Dosage and Administration (2.1, 2.4, 2.5, 2.6)]. Following cessation of LEUKINE therapy, excessive blood counts have returned to normal or baseline levels within 3 to 7 days.
5.7 Potential Effect on Malignant Cells
LEUKINE is a growth factor that primarily stimulates normal myeloid precursors. However, the possibility that LEUKINE can act as a growth factor for any tumor type, particularly myeloid malignancies, cannot be excluded. Because of the possibility of tumor growth potentiation, precaution should be exercised when using this drug in any malignancy with myeloid characteristics.
Discontinue LEUKINE therapy if disease progression is detected during LEUKINE treatment.
5.8 Immunogenicity
Treatment with LEUKINE may induce neutralizing anti-drug antibodies. The incidence of anti-sargramostim neutralizing antibodies may be related to duration of exposure to LEUKINE. In a study of patients with normal neutrophil count and a solid tumor in complete response (an unapproved use) treated with LEUKINE for up to 12 months, 82.9% of 41 evaluable patients developed anti-sargramostim neutralizing antibodies and the myelostimulatory effect of LEUKINE was not sustained by day 155 as assessed by WBC count. Use LEUKINE for the shortest duration required [see Adverse Reactions (6.2)].
5.9 Risk of Serious Adverse Reactions in Infants Due to Benzyl Alcohol Preservative
Serious and fatal adverse reactions including "gasping syndrome" can occur in neonates and low birth weight infants treated with benzyl alcohol-preserved drugs, including LEUKINE injection (solution) or LEUKINE for injection (lyophilized powder) reconstituted with Bacteriostatic Water. The "gasping syndrome" is characterized by central nervous system depression, metabolic acidosis and gasping respirations.
Avoid administration of solutions containing benzyl alcohol (including LEUKINE injection [solution] or LEUKINE for injection [lyophilized powder] reconstituted with Bacteriostatic Water for Injection, USP [0.9% benzyl alcohol]) to neonates and low birth weight infants. Instead, administer lyophilized LEUKINE reconstituted with Sterile Water for Injection, USP [see Dosage and Administration (2.7)].
If LEUKINE injection (solution) or LEUKINE for injection (lyophilized powder) reconstituted with Bacteriostatic Water for Injection, USP (0.9% benzyl alcohol) must be used in neonates and low birth weight infants, consider the combined daily metabolic load of benzyl alcohol from all sources including LEUKINE (LEUKINE injection and LEUKINE for injection reconstituted with Bacteriostatic Water contain 11 mg and 9 mg of benzyl alcohol per mL, respectively). The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Use in Specific Populations (8.4) and Dosage and Administration (2.7)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Infusion Related Reactions [see Warnings and Precautions (5.2)]
- Risk of Severe Myelosuppression when LEUKINE Administered within 24 Hours of Chemotherapy or Radiotherapy [see Warnings and Precautions (5.3)]
- Effusions and Capillary Leak Syndrome [see Warnings and Precautions (5.4)]
- Supraventricular Arrhythmias [see Warnings and Precautions (5.5)]
- Leukocytosis [see Warnings and Precautions (5.6)]
- Potential Effect on Malignant Cells [see Warnings and Precautions (5.7)]
- Immunogenicity [see Warnings and Precautions (5.8)]
- Risk of Serious Adverse Reactions in Infants Due to Benzyl Alcohol Preservative [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Autologous Peripheral Blood Progenitor Cell (PBPC) and Bone Marrow Transplantation
Studies 301, 302 and 303 enrolled a total of 156 patients after autologous or allogeneic marrow or PBPC transplantation. In these placebo-controlled studies, pediatric and adult patients received once-daily intravenous infusions of LEUKINE 250 mcg/m2 or placebo for 21 days.
In Studies 301, 302, and 303, there was no difference in relapse rate between the LEUKINE and placebo-treated patients. Adverse reactions reported in at least 10% of patients who received intravenous LEUKINE or at a rate that was at least 5% higher than the placebo arm are shown in Table 1.
Table 1: Adverse Reactions after Autologous Marrow or PBPC Transplantation in at Least 10% of Patients Receiving Intravenous LEUKINE or at Least 5% Higher than the Placebo Arm Adverse Reactions by Body System LEUKINE
(n=79)
%Placebo
(n=77)
%Adverse Reactions by Body System LEUKINE
(n=79)
%Placebo
(n=77)
%Body, General Metabolic, Nutritional Disorder Fever 95 96 Edema 34 35 Mucous membrane disorder 75 78 Peripheral edema 11 7 Asthenia 66 51 Respiratory System Malaise 57 51 Dyspnea 28 31 Sepsis 11 14 Lung disorder 20 23 Digestive System Blood and Lymphatic System Nausea 90 96 Blood dyscrasia 25 27 Diarrhea 89 82 Cardiovascular Vascular System Vomiting 85 90 Hemorrhage 23 30 Anorexia 54 58 Urogenital System GI disorder 37 47 Urinary tract disorder 14 13 GI hemorrhage 27 33 Nervous System Stomatitis 24 29 CNS disorder 11 16 Liver damage 13 14 Skin and Appendages Alopecia 73 74 Rash 44 38 Allogeneic Bone Marrow Transplantation
In the placebo-controlled trial of 109 patients after allogeneic BMT (Study 9002), acute graft-vs-host disease occurred in 55% on the LEUKINE arm and in 59% on the placebo arm. Adverse reactions reported in at least 10% of patients who received IV LEUKINE or at a rate at least 5% higher than the placebo arm are shown in Table 2.
Table 2: Adverse Reactions after Allogeneic Marrow Transplantation in at Least 10% of Patients Receiving Intravenous LEUKINE or at Least 5% Higher than the Placebo Arm Adverse Reactions by Body System LEUKINE
(n=53)
%Placebo
(n=56)
%Adverse Reactions by Body System LEUKINE
(n=53)
%Placebo
(n=56)
%- * Grade 3 and 4 laboratory abnormalities only. Denominators may vary due to missing laboratory measures.
Body, General Eye hemorrhage 11 0 Fever 77 80 Cardiovascular System Abdominal pain 38 23 Hypertension 34 32 Headache 36 36 Tachycardia 11 9 Chills 25 20 Metabolic / Nutritional Disorders Pain 17 36 Bilirubinemia 30 27 Asthenia 17 20 Hyperglycemia 25 23 Chest pain 15 9 Peripheral edema 15 21 Digestive System Increased creatinine 15 14 Diarrhea 81 66 Hypomagnesemia 15 9 Nausea 70 66 Increased SGPT 13 16 Vomiting 70 57 Edema 13 11 Stomatitis 62 63 Respiratory System Anorexia 51 57 Pharyngitis 23 13 Dyspepsia 17 20 Epistaxis 17 16 Hematemesis 13 7 Dyspnea 15 14 Dysphagia 11 7 Rhinitis 11 14 GI hemorrhage 11 5 Blood and Lymphatic System Skin and Appendages Thrombocytopenia 19 34 Rash 70 73 Leukopenia 17 29 Alopecia 45 45 Nervous System Pruritus 23 13 Paresthesia 11 13 Musculoskeletal System Insomnia 11 9 Bone pain 21 5 Anxiety 11 2 Arthralgia 11 4 Laboratory Abnormalities* Special Senses High glucose 49 41 Low albumin 36 27 High BUN 17 23 Acute Myeloid Leukemia Following Induction Chemotherapy
Nearly all patients in both arms developed leukopenia, thrombocytopenia, and anemia. Adverse reactions reported in at least 10% of patients who received LEUKINE or at least 5% higher than the placebo arm are reported in Table 3.
Table 3: Adverse Reactions after Treatment of AML in at Least 10% of Patients Receiving Intravenous LEUKINE or at Least 5% Higher than the Placebo Arm Adverse Reactions by Body System LEUKINE
(n=52)
%Placebo
(n=47)
%Adverse Reactions by Body System LEUKINE
(n=52)
%Placebo
(n=47)
%Body, General Metabolic / Nutritional Disorder Fever (no infection) 81 74 Metabolic Laboratory Abnormalities 58 49 Infection 65 68 Edema 25 23 Weight loss 37 28 Respiratory System Chills 19 26 Pulmonary toxicity 48 64 Allergy 12 15 Blood and Lymphatic System Digestive System Coagulation 19 21 Nausea 58 55 Cardiovascular System Liver toxicity 77 83 Hemorrhage 29 43 Diarrhea 52 53 Hypertension 25 32 Vomiting 46 34 Cardiac 23 32 Stomatitis 42 43 Hypotension 13 26 Anorexia 13 11 Urogenital System Skin and Appendages GU abnormalities 50 57 Skin Reactions 77 45 Nervous System Alopecia 37 51 Neuro-clinical 42 53 Neuro-motor 25 26 Neuro-psych 15 26 There was no significant difference between the arms in the proportion of patients achieving complete remission (CR; 69% in the LEUKINE group and 55% in the placebo group). There was also no significant difference in relapse rates; 12 of 36 patients who received LEUKINE and five of 26 patients who received placebo relapsed within 180 days of documented CR (p=0.26). The study was not sized to assess the impact of LEUKINE treatment on response.
Graft Failure
In a historically controlled study of 86 patients with AML, the LEUKINE treated group exhibited an increased incidence of weight gain (p=0.007), low serum proteins, and prolonged prothrombin time (p=0.02) when compared to the control group. Two LEUKINE treated patients had progressive increase in circulating monocytes and promonocytes and blasts in the marrow, which reversed when LEUKINE was discontinued. The historical control group exhibited an increased incidence of cardiac events (p=0.018), liver function abnormalities (p=0.008), and neurocortical hemorrhagic events (p=0.025). Headache (26%), pericardial effusion (25%), arthralgia (21%), and myalgia (18%) were also reported in patients treated with LEUKINE in the graft failure study.
6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity with LEUKINE. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, duration of treatment, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to sargramostim in the studies described below with the incidence of antibodies in other studies or other products may be misleading.
In 214 patients with a variety of underlying diseases, neutralizing anti-sargramostim antibodies were detected in 5 patients (2.3%) after receiving LEUKINE by continuous IV infusion (3 patients) or SC injection (2 patients) for 28 to 84 days in multiple courses (as assessed by GM-CSF dependent human cell-line proliferation assay). All 5 patients had impaired hematopoiesis before the administration of LEUKINE, and consequently the effect of the development of anti-sargramostim antibodies on normal hematopoiesis could not be assessed.
Antibody studies of 75 patients with Crohn's disease (a disease for which LEUKINE is not indicated), with normal hematopoiesis and no other immunosuppressive drugs, receiving LEUKINE daily for 8 weeks by SC injection, showed 1 patient (1.3%) with detectable neutralizing anti-sargramostim antibodies (as assessed by GM-CSF dependent human cell-line proliferation assay).
In an experimental use trial where LEUKINE was given for an extended period, 53 patients with melanoma in complete remission (a disease for which LEUKINE is not indicated) received adjuvant therapy with LEUKINE 125 mcg/m2 once daily (maximum dose 250 mcg) from day 1 to 14 every 28 days for 1 year. Serum samples from patients assessed at day 0, 2 weeks, 1 month, and 5 and/or 12 months were tested retrospectively for the presence of anti-sargramostim antibodies. Of 43 evaluable patients (having at least 3 timepoint samples post treatment), 42 (97.7%) developed anti-sargramostim binding antibody as assessed by ELISA and confirmed using an immunoprecipitation assay. Of these 42 patients, 41 had sufficient sample and were further tested: 34 patients (82.9%) developed anti-sargramostim neutralizing antibodies (as determined by a cell based luciferase reporter gene neutralizing antibody assay); 17 (50%) of these patients did not have a sustained pharmacodynamic effect of LEUKINE by day 155 as assessed by WBC counts. This study provided limited assessment of the impact of antibody formation on the safety and efficacy of LEUKINE.
Serious allergic and anaphylactoid reactions have been reported with LEUKINE, but the rate of occurrence of antibodies in such patients has not been assessed.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of LEUKINE in clinical trials and/or postmarketing surveillance. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Infusion related reactions including dyspnea, hypoxia, flushing, hypotension, syncope and/or tachycardia [see Warnings and Precautions (5.1)]
- Serious allergic reactions/hypersensitivity, including anaphylaxis, skin rash, urticaria, generalized erythema, and flushing [see Warnings and Precautions (5.2)]
- Effusions and capillary leak syndrome [see Warnings and Precautions (5.3)]
- Supraventricular arrhythmias [see Warnings and Precautions (5.4)]
- Leukocytosis including eosinophilia [see Warnings and Precautions (5.6)]
- Thromboembolic events
- Pain, including chest, abdominal, back, and joint pain
- Injection site reactions
-
7 DRUG INTERACTIONS
7.1 Concomitant Use with Products that Induce Myeloproliferation
Avoid the concomitant use of LEUKINE and products that induce myeloproliferation (such as lithium and corticosteroids). Such products may increase the myeloproliferative effects of LEUKINE. Monitor patients receiving both LEUKINE and products that induce myeloproliferation frequently for clinical and laboratory signs of excess myeloproliferative effects.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Both LEUKINE injection (solution) and LEUKINE for injection (lyophilized powder) reconstituted with Bacteriostatic Water for Injection, USP contain benzyl alcohol, which has been associated with gasping syndrome in neonates and infants. The preservative benzyl alcohol can cause serious adverse reactions and death when administered intravenously to neonates and infants. If LEUKINE is needed during pregnancy, use only LEUKINE for injection (lyophilized powder) reconstituted with Sterile Water for injection without preservatives [see Dosage and Administration (2.7) and Use in Specific Populations (8.4)].
The limited available data on LEUKINE use in pregnant women are insufficient to inform the drug-associated risk of adverse developmental outcomes. Based on animal studies LEUKINE may cause embryofetal harm. In animal reproduction studies, administration of LEUKINE to pregnant rabbits during organogenesis resulted in adverse developmental outcomes including increased spontaneous abortion at systemic exposures ≥1.3 times the human exposure expected at the recommended human dose [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2%–4% and 15%–20%, respectively.
Data
Animal data
In an embryofetal developmental study and a prenatal and postnatal study, pregnant rabbits were administered SC doses of LEUKINE during the period of gestation day (GD) 6 to GD19, GD19 to GD28, or GD19 to parturition at 25, 70, and 200 mcg/kg/day. An increase in spontaneous abortions, late resorptions, and postimplantation loss, and a reduction in viable fetuses, mean live litter size, and offspring body weight were evident in rabbits treated with LEUKINE at 200 mcg/kg/day. No adverse effects were observed at ≤70 mcg/kg/day.
After the first administration in rabbits, the dose of 200 mcg/kg/day corresponds to a systemic exposure (AUC) of approximately 11–25.3 times the exposures observed in patients treated with the clinical LEUKINE dose of 250 mcg/m2; however, due to the production of anti-LEUKINE antibodies with repeat administration, the AUC in rabbits was reduced to 1.3–5.5 times the clinical exposure by the end of the dosing periods.
Similarly, after the first administration in rabbits, the dose of 70 mcg/kg/day corresponds to a systemic exposure (AUC) of approximately 7 to 11 times the exposures observed in patients treated with the clinical LEUKINE dose of 250 mcg/m2; however, due to the production of anti-LEUKINE antibodies with repeat administration, the AUC in rabbits was reduced to 1.0–1.2 times the clinical exposure by the end of the dosing periods.
8.2 Lactation
Risk Summary
There is no information regarding the presence of LEUKINE in human milk, the effects on the breastfed child, or the effects on milk production. Administration of LEUKINE to rabbits during lactation resulted in reduction in postnatal offspring survival [see Data]. Because of the potential for serious adverse reactions advise a lactating woman not to breastfeed during treatment and for at least 2 weeks after the last dose.
Data
There are no data regarding the presence of LEUKINE in rabbit milk. However, in the prenatal and postnatal study, lactating rabbits were administered SC doses of LEUKINE during the period of lactation day (LD) 1 to LD14 at 25, 70, and 200 mcg/kg/day. At doses ≥25 mcg/kg/day a reduction in postnatal offspring survival was observed. Maternal toxicity was also observed at LEUKINE doses ≥25 mcg/kg/day.
After the first administration in rabbits, the dose of 25 mcg/kg/day corresponds to a systemic AUC of approximately 2.6 times the exposure observed in patients treated with the clinical LEUKINE dose of 250 mcg/m2 however, due to the production of anti-LEUKINE antibodies with repeat administration, the exposure in rabbits decreased to 0.2 times the clinical exposure by the end of the dosing period.
8.4 Pediatric Use
The safety and effectiveness of LEUKINE have been established in pediatric patients 2 years of age and older for autologous peripheral blood progenitor cells and bone marrow transplantation, allogeneic bone marrow transplantation, and treatment of delayed neutrophil recovery or graft failure. Use of LEUKINE for these indications in this age group is based on adequate and well-controlled studies of LEUKINE in adults, in addition to clinical data in 12, 23, and 37 pediatric patients, respectively [See Clinical Studies (14.3, 14.4 and 14.5)]. The pediatric adverse reactions were consistent with those reported in the adult population.
The safety and effectiveness of LEUKINE for pediatric patients less than 2 years of age for autologous peripheral blood progenitor cells and bone marrow transplantation, allogeneic bone marrow transplantation, and treatment of delayed neutrophil recovery or graft failure have not been established.
The use of LEUKINE to increase survival in pediatric patients acutely exposed to myelosuppressive doses of radiation (H-ARS) is based on efficacy studies conducted in animals and clinical data supporting the use of LEUKINE in patients undergoing autologous or allogeneic BMT following myelosuppressive chemotherapy with or without total body irradiation. Efficacy studies of LEUKINE could not be conducted in humans with acute radiation syndrome for ethical and feasibility reasons. Modeling and simulation was used to derive dosing regimens that are predicted to provide pediatric patients with exposure comparable to the observed exposure in adults receiving 7 mcg/kg [see Clinical Pharmacology (12.3)]. The dose for pediatric patients is based on weight [see Dosage and Administration (2.2)].
Safety and effectiveness in pediatric patients have not been established in:
- Acute Myeloid Leukemia: Neutrophil Recovery Following Induction Chemotherapy
- Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
Avoid administration of solutions containing benzyl alcohol [including LEUKINE injection (solution) or LEUKINE for injection (lyophilized powder) reconstituted with Bacteriostatic Water for Injection, USP (0.9% benzyl alcohol)] to neonates and low birth weight infants. Instead, administer lyophilized LEUKINE reconstituted with Sterile Water for Injection, USP [see Dosage and Administration (2.7)].
Serious adverse reactions including fatal reactions and the "gasping syndrome" occurred in premature infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.38 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol.
If LEUKINE injection (solution) or LEUKINE for injection (lyophilized powder) reconstituted with Bacteriostatic Water for Injection, USP (0.9% benzyl alcohol) must be used in neonates and low birth weight infants, consider the combined daily metabolic load of benzyl alcohol from all sources including LEUKINE (LEUKINE injection [solution] and LEUKINE for injection [lyophilized powder] reconstituted with Bacteriostatic Water for Injection, USP [0.9% benzyl alcohol] contain 11 mg and 9 mg of benzyl alcohol per mL, respectively). The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Dosage and Administration (2.7)].
-
10 OVERDOSAGE
Doses up to 100 mcg/kg/day (4,000 mcg/m2/day or 16 times the recommended dose) were administered to four patients in a Phase 1 uncontrolled clinical study by continuous IV infusion for 7 to 18 days. Increases in WBC up to 200,000 cells/mm3 were observed. Adverse events reported were dyspnea, malaise, nausea, fever, rash, sinus tachycardia, headache, and chills. All these events were reversible after discontinuation of LEUKINE.
In case of overdosage, discontinue LEUKINE therapy and monitor the patient for WBC increase and respiratory symptoms.
-
11 DESCRIPTION
LEUKINE (sargramostim) injection and LEUKINE (sargramostim) for injection for subcutaneous or intravenous use are recombinant human granulocyte-macrophage colony stimulating factor (rhu GM-CSF) produced by recombinant DNA technology in a yeast (S. cerevisiae) expression system. LEUKINE is a glycoprotein of 127 amino acids characterized by three primary molecular species having molecular masses of 19,500, 16,800 and 15,500 Daltons.
The amino acid sequence of LEUKINE differs from the natural human GM-CSF by a substitution of leucine at position 23, and the carbohydrate moiety may be different from the native protein. LEUKINE differs from human GM-CSF by one amino acid at position 23, where leucine is substituted for arginine.
LEUKINE (sargramostim) injection is a sterile, clear, colorless solution preserved with 1.1% benzyl alcohol in a multiple-dose vial. Each 1 mL vial contains 500 mcg sargramostim and has a pH range of 6.7 – 7.7. LEUKINE (sargramostim) for injection is supplied as a sterile, preservative-free, white lyophilized powder in a single-dose vial. Each single-dose vial delivers 250 mcg sargramostim. Inactive ingredients are mannitol (40 mg), sucrose (10 mg), and tromethamine (1.2 mg). Reconstitution with 1 mL of the appropriate diluent (sterile water for injection or bacteriostatic water for injection) yields a solution containing 250 mcg/mL sargramostim at a pH range of 7.1 – 7.7 with a deliverable volume of 1 mL (250 mcg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sargramostim (GM-CSF) belongs to a group of growth factors termed colony stimulating factors which support survival, clonal expansion, and differentiation of hematopoietic progenitor cells. GM-CSF induces partially committed progenitor cells to divide and differentiate in the granulocyte-macrophage pathways which include neutrophils, monocytes/macrophages and myeloid-derived dendritic cells.
GM-CSF is also capable of activating mature granulocytes and macrophages. GM-CSF is a multilineage factor and, in addition to dose-dependent effects on the myelomonocytic lineage, can promote the proliferation of megakaryocytic and erythroid progenitors. However, other factors are required to induce complete maturation in these two lineages. The various cellular responses (i.e., division, maturation, activation) are induced through GM-CSF binding to specific receptors expressed on the cell surface of target cells.
The biological activity of GM-CSF is species-specific. Consequently, in vitro studies have been performed on human cells to characterize the pharmacological activity of GM-CSF. In vitro exposure of human bone marrow cells to GM-CSF at concentrations ranging from 1–100 ng/mL results in the proliferation of hematopoietic progenitors and in the formation of pure granulocyte, pure macrophage and mixed granulocyte macrophage colonies. Chemotactic, anti-fungal, and anti-parasitic activities of granulocytes and monocytes are increased by exposure to GM-CSF in vitro. GM-CSF increases the cytotoxicity of monocytes toward certain neoplastic cell lines and activates polymorphonuclear neutrophils to inhibit the growth of tumor cells.
12.2 Pharmacodynamics
LEUKINE stimulates hematopoietic precursor cells and increases neutrophil, eosinophil, megakaryocyte, macrophage, and dendritic cell production. In AML adult patients undergoing induction chemotherapy [see Clinical Studies (14.1)], LEUKINE at daily doses of 250 mcg/m2 significantly shortened the median duration of ANC <500/mm3 by 4 days and <1000/mm3 by 7 days following induction; 75% of patients receiving sargramostim achieved ANC greater than 500/mm3 by day 16 compared to day 25 for patients receiving placebo. Animal data and clinical data in humans suggest a correlation between sargramostim exposure and the duration of severe neutropenia as a predictor of efficacy. At doses of 250 mcg/m2 (approximately 7 mcg/kg in a 70 kg human with a body surface area of 1.96), daily LEUKINE treatment reduced the duration of severe neutropenia.
12.3 Pharmacokinetics
Intravenous Administration (IV)
Peak concentrations of sargramostim were observed in blood samples obtained during or immediately after completion of LEUKINE infusion. For LEUKINE injection, the mean maximum concentration (Cmax) was 16.7 ng/mL and the mean area under the time-concentration curve (AUC)0–inf was 32.9 ng∙h /mL. There is no accumulation of GM-CSF after repeat dosing and steady state conditions are met after a single dose.
Subcutaneous Administration (SC)
LEUKINE injection and LEUKINE for injection, at a dose of 6.5 mcg/kg (approximately 250 mcg/m2) given by the SC route, have been determined to be bioequivalent. Based on a population pharmacokinetics analysis of lyophilized LEUKINE data, the mean Cmax after a 7 mcg/kg SC dose (equivalent to a 250 mcg/m2 dose in a 70 kg human with a body surface area of 1.96) was 3.03 ng/mL and mean AUC0–24 was 21.3 ng∙h/mL (Table 4). There is no accumulation of GM-CSF after repeat SC dosing and steady state conditions are met after a single SC dose.
Table 4: Sargramostim serum Cmax and AUC Exposure (CV%) in Humans after Subcutaneous Administration Data type Sargramostim dose Formulation Number of healthy subjects AUC (CV%)
(ng∙h/mL )Cmax (CV%)
(ng/mL)- * 250 mcg/m2 is approximately 7 mcg/kg in 70 kg patient with a body surface area of 1.96 M2
Observed 250 mcg/m2 * LEUKINE
injection29
2921.9
(28.1%)
20.3 (32.4%)3.75 (45.5%)
3.24 (50.5%)Observed 6.5 mcg/kg Lyophilized LEUKINE 39 20.4 (28.7%) 3.15 (35.2%) Population PK model simulation 7 mcg/kg Lyophilized LEUKINE 500 21.3 (32.6) 3.03 (31.0) Absorption
After SC administration GM-CSF was detected in the serum early (15 min) and reached maximum serum concentrations between 2.5 and 4 h. The absolute bioavailability with the SC route, when compared to the IV route, was 75%.
Elimination
When a 500 mcg dose of LEUKINE (injection) was administered IV over two hours to healthy volunteers, the mean terminal elimination half-life was approximately 3.84 h and the mean clearance rate was approximately 17.2 L/h. When LEUKINE (lyophilized product) was administered SC to healthy adult volunteers, GM-CSF and had a terminal elimination half-life of 1.4 h. The observed total body clearance/subcutaneous bioavailability (CL/F) was 23 L/h. Specific metabolism studies were not conducted, because LEUKINE is a protein and is expected to degrade to small peptides and individual amino acids.
Special Populations
Adult patients acutely exposed to myelosuppressive doses of radiation (H-ARS)
The pharmacokinetics of sargramostim are not available in adult patients acutely exposed to myelosuppressive doses of radiation. Pharmacokinetic data in irradiated and non-irradiated non-human primates and in healthy human adults were used to derive human doses for patients acutely exposed to myelosuppressive doses of radiation. Modeling and simulation of the healthy human adult pharmacokinetic data indicate that sargramostim Cmax and AUC exposures at a LEUKINE dose of 7 mcg/kg in patients acutely exposed to myelosuppressive doses of radiation are expected to exceed sargramostim Cmax (97.6% of patients) and AUC (100% of patients) exposures at a LEUKINE dose of 7 mcg/kg in non-human primates.
Pediatric patients acutely exposed to myelosuppressive doses of radiation (H-ARS)
The pharmacokinetics of sargramostim was not available in pediatric patients acutely exposed to myelosuppressive doses of radiation. The pharmacokinetics of sargramostim in pediatric patients after being exposed to myelosuppressive doses of radiation were estimated by scaling the adult population pharmacokinetic model to the pediatric population. The model-predicted mean AUC0–24 values at 7, 10, and 12 mcg/kg doses of LEUKINE in pediatric patients weighing greater than 40 kg (~adolescents), 15 to 40 kg (~young children), and 0 to less than 15 kg (~newborns to toddlers), respectively, were similar to AUC values in adults after a 7 mcg/kg dose.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Carcinogenicity and genetic toxicology studies have not been conducted with LEUKINE.
Impairment of Fertility
LEUKINE had no effect on fertility of female rabbits up to a dose of 200 mcg/kg/day.
The toxicology studies with up to 6 weeks of exposure to LEUKINE in sexually mature female and male cynomolgus monkeys did not reveal findings in male or female reproductive organs that would suggest impairment of fertility up to a dose of 200 mcg/kg/day. At 200 mcg/kg, the AUC exposure of LEUKINE was 8.8 to 11.4 times (monkeys) and 2.0 to 25.3 times (rabbits) the exposure in humans at the recommended clinical dose of 250 mcg/m2.
After the first administration, a dose of 200 mcg/kg/day corresponds to an AUC of approximately 11.4 (monkeys) and 25.3 (rabbits) times the exposures observed in patients treated with the clinical LEUKINE dose of 250 mcg/m2; however, due to the production of anti-LEUKINE antibodies with repeat administration, the AUC decreased to 8.8 (monkeys) and 2.0 (rabbits) times the clinical exposure by the end of the dosing periods.
-
14 CLINICAL STUDIES
14.1 Following Induction Chemotherapy for Acute Myelogenous Leukemia
The efficacy of LEUKINE in the treatment of AML was evaluated in a multicenter, randomized, double-blind placebo-controlled trial (study 305) of 99 newly-diagnosed adult patients, 55–70 years of age, receiving induction with or without consolidation. A combination of standard doses of daunorubicin (days 1–3) and ara-C (days 1–7) was administered during induction and high dose ara-C was administered days 1–6 as a single course of consolidation, if given. Bone marrow evaluation was performed on day 10 following induction chemotherapy. If hypoplasia with <5% blasts was not achieved, patients immediately received a second cycle of induction chemotherapy. If the bone marrow was hypoplastic with <5% blasts on day 10 or four days following the second cycle of induction chemotherapy, LEUKINE (250 mcg/m2/day) or placebo was given intravenously over four hours each day, starting four days after the completion of chemotherapy. Study drug was continued until an ANC ≥1500 cells/mm3 for three consecutive days was attained or a maximum of 42 days. LEUKINE or placebo was also administered after the single course of consolidation chemotherapy if delivered (ara-C 3–6 weeks after induction following neutrophil recovery). Study drug was discontinued immediately if leukemic regrowth occurred.
LEUKINE significantly shortened the median duration of ANC <500 cells/mm3 by 4 days and <1000 cells/mm3 by 7 days following induction (see Table 5). Of patients receiving LEUKINE, 75% achieved ANC >500 cells/mm3 by day 16, compared to day 25 for patients receiving placebo. The proportion of patients receiving one cycle (70%) or two cycles (30%) of induction was similar in both treatment groups. LEUKINE significantly shortened the median times to neutrophil recovery whether one cycle (12 vs. 15 days) or two cycles (14 vs. 23 days) of induction chemotherapy was administered. Median times to platelet (>20,000 cells/mm3) and RBC transfusion independence were not significantly different between treatment groups.
Table 5: Hematological Recovery (in Days) in Patients with AML: Induction Dataset LEUKINE
n=52*
Median (25%, 75%)Placebo
n=47
Median (25%, 75%)p-value† - * Patients with missing data censored
- † p = Generalized Wilcoxon
- ‡ 2 patients on LEUKINE and 4 patients on placebo had missing values
- § 2 patients on LEUKINE and 3 patients on placebo had missing values
- ¶ 4 patients on placebo had missing values
- # 3 patients on LEUKINE and 4 patients on placebo had missing values
ANC >500/mm3 ‡ 13 (11, 16) 17 (13, 25) 0.009 ANC >1000/mm3 § 14 (12, 18) 21 (13, 34) 0.003 PLT >20,000/mm3 ¶ 11 (7, 14) 12 (9, >42) 0.10 RBC# 12 (9, 24) 14 (9, 42) 0.53 During the consolidation phase of treatment, LEUKINE did not shorten the median time to recovery of ANC to 500 cells/mm3 (13 days) or 1000 cells/mm3 (14.5 days) compared to placebo. There were no significant differences in time to platelet and RBC transfusion independence.
The incidence of severe infections and deaths associated with infections was significantly reduced in patients who received LEUKINE. During induction or consolidation, 27 of 52 patients receiving LEUKINE and 35 of 47 patients receiving placebo had at least one grade 3, 4 or 5 infection (p=0.02). Twenty-five patients receiving LEUKINE and 30 patients receiving placebo experienced severe and fatal infections during induction only. There were significantly fewer deaths from infectious causes in the LEUKINE arm (3 vs. 11, p=0.02). The majority of deaths in the placebo group were associated with fungal infections with pneumonia as the primary infection.
14.2 Autologous Peripheral Blood Progenitor Cell Mobilization and Collection
A retrospective review was conducted of data from adult patients with cancer undergoing collection of peripheral blood progenitor cells (PBPC) at a single transplant center. Mobilization of PBPC and myeloid reconstitution post transplant were compared between four groups of patients (n=196) receiving LEUKINE for mobilization and a historical control group who did not receive any mobilization treatment [progenitor cells collected by leukapheresis without mobilization (n=100)]. Sequential cohorts received LEUKINE. The cohorts differed by dose (125 or 250 mcg/m2/day), route (IV over 24 hours or SC) and use of LEUKINE post transplant. Leukaphereses were initiated for all mobilization groups after the WBC reached 10,000 cells/mm3. Leukaphereses continued until both a minimum number of mononucleated cells (MNC) were collected (6.5 or 8.0 × 108/kg body weight) and a minimum number of aphereses (5–8) were performed. Both minimum requirements varied by treatment cohort and planned conditioning regimen. If subjects failed to reach a WBC of 10,000 cells/mm3 by day 5, another cytokine was substituted for LEUKINE.
Marked mobilization effects were seen in patients administered the higher dose of LEUKINE (250 mcg/m2) either IV (n=63) or SC (n=41). PBPCs from patients treated at the 250 mcg/m2/day dose had a significantly higher number of granulocyte-macrophage colony-forming units (CFU-GM) than those collected without mobilization. The mean value after thawing was 11.41 × 104 CFU-GM/kg for all LEUKINE-mobilized patients, compared to 0.96 × 104/kg for the non-mobilized group. A similar difference was observed in the mean number of erythrocyte burst-forming units (BFU-E) collected (23.96 × 104/kg for patients mobilized with 250 mcg/m2 doses of LEUKINE administered SC vs. 1.63 × 104/kg for non-mobilized patients).
A second retrospective review of data from patients undergoing PBPC at another single transplant center was also conducted. LEUKINE was given SC at 250 mcg/m2/day once a day (n=10) or twice a day (n=21) until completion of apheresis. Apheresis was begun on day 5 of LEUKINE administration and continued until the targeted MNC count of 9 × 108/kg or CD34+ cell count of 1 × 106/kg was reached. There was no difference in CD34+ cell count in patients receiving LEUKINE once or twice a day.
14.3 Autologous Peripheral Blood Progenitor Cell and Bone Marrow Transplantation
The efficacy of LEUKINE to accelerate myeloid reconstitution following autologous PBPC was established in the retrospective review above. After transplantation, mobilized subjects had shorter times to neutrophil recovery and fewer days between transplantation and the last platelet transfusion compared to non-mobilized subjects. Neutrophil recovery (ANC >500 cells/mm3) was more rapid in patients administered LEUKINE following PBPC transplantation with LEUKINE-mobilized cells (see Table 6). Mobilized patients also had fewer days to the last platelet transfusion and last RBC transfusion, and a shorter duration of hospitalization than did non-mobilized subjects.
Table 6: ANC and Platelet Recovery after PBPC Transplantation LEUKINE
Route for MobilizationPost transplant LEUKINE Median Day ANC
>500 cells/mm3Median Day of Last platelet transfusion No Mobilization – No 29 28 LEUKINE
250 mcg/m2IV No 21 24 IV Yes 12 19 SC Yes 12 17 The efficacy of LEUKINE on time to myeloid reconstitution following autologous BMT was established by three single-center, randomized, placebo-controlled and double-blinded studies (studies 301, 302, and 303) in adult and pediatric patients undergoing autologous BMT for lymphoid malignancies. A total of 128 patients (65 LEUKINE, 63 placebo) were enrolled in these three studies. The median age was 38 years (range 3–62 years), and 12 patients were younger than 18 years of age. The majority of the patients had lymphoid malignancy (87 NHL, 17 ALL), 23 patients had Hodgkin lymphoma, and one patient had AML. In 72 patients with NHL or ALL, the bone marrow harvest was purged with one of several monoclonal antibodies prior to storage. No chemical agent was used for in vitro treatment of the bone marrow. Preparative regimens in the three studies included cyclophosphamide (total dose 120–150 mg/kg) and total body irradiation (total dose 1,200–1,575 rads). Other regimens used in patients with Hodgkin's disease and NHL without radiotherapy consisted of three or more of the following in combination (expressed as total dose): cytosine arabinoside (400 mg/m2) and carmustine (300 mg/m2), cyclophosphamide (140–150 mg/kg), hydroxyurea (4.5 grams/m2), and etoposide (375–450 mg/m2).
Compared to placebo, administration of LEUKINE in two studies (study 301: 44 patients, 23 patients treated with LEUKINE, and study 303: 47 patients, 24 treated with LEUKINE) significantly improved the following hematologic and clinical endpoints: time to neutrophil recovery, duration of hospitalization and infection experience or antibacterial usage. In the third study (study 302: 37 patients who underwent autologous BMT, 18 treated with LEUKINE) there was a positive trend toward earlier myeloid engraftment in favor of LEUKINE. This latter study differed from the other two in having enrolled a large number of patients with Hodgkin lymphoma who had also received extensive radiation and chemotherapy prior to harvest of autologous bone marrow. In the following combined analysis of the three studies, these two subgroups (NHL and ALL vs. Hodgkin lymphoma) are presented separately.
Patients with Lymphoid Malignancy (Non-Hodgkin's Lymphoma and Acute Lymphoblastic Leukemia)
Neutrophil recovery (ANC ≥500 cells/mm3) in 54 patients with NHL or ALL receiving LEUKINE on Studies 301, 302 and 303 was observed on day 18, and on day 24 in 50 patients treated with placebo (see Table 7). The median duration of hospitalization was six days shorter for the LEUKINE group than for the placebo group. Median duration of infectious episodes (defined as fever and neutropenia; or two positive cultures of the same organism; or fever >38°C and one positive blood culture; or clinical evidence of infection) was three days less in the group treated with LEUKINE. The median duration of antibacterial administration in the post transplantation period was four days shorter for the patients treated with LEUKINE than for placebo-treated patients.
Table 7: Autologous BMT: Combined Analysis from Placebo-Controlled Clinical Trials of Responses in Patients with NHL and ALL Median Values (days) ANC ≥500 cells /mm3 ANC ≥1000 cells/mm3 Duration of Hospitalization Duration of Infection Duration of Antibacterial Therapy - * p <0.05 Wilcoxon or Cochran-Mantel-Haenszel RIDIT chi-squared
- † p <0.05 Log rank
LEUKINE
n=5418*,† 24 *, † 25 * 1 * 21 * Placebo
n=5024 32 31 4 25 14.4 Allogeneic Bone Marrow Transplantation
A multicenter, randomized, placebo-controlled, and double-blinded study (study 9002) was conducted to evaluate the safety and efficacy of LEUKINE for promoting hematopoietic reconstitution following allogeneic BMT. A total of 109 adult and pediatric patients (53 LEUKINE, 56 placebo) were enrolled in the study. The median age was 34.7 years (range 2.2–65.1 years). Twenty-three patients (11 LEUKINE, 12 placebo) were 18 years old or younger. Sixty-seven patients had myeloid malignancies (33 AML, 34 CML), 17 had lymphoid malignancies (12 ALL, 5 NHL), three patients had Hodgkin's disease, six had multiple myeloma, nine had myelodysplastic disease, and seven patients had aplastic anemia. In 22 patients at one of the seven study sites, bone marrow harvests were depleted of T cells. Preparative regimens included cyclophosphamide, busulfan, cytosine arabinoside, etoposide, methotrexate, corticosteroids, and asparaginase. Some patients also received total body, splenic, or testicular irradiation. Primary GVHD prophylaxis was cyclosporine and a corticosteroid.
Accelerated myeloid engraftment was associated with significant laboratory and clinical benefits. Compared to placebo, administration of LEUKINE significantly improved the following: time to neutrophil engraftment, duration of hospitalization, number of patients with bacteremia, and overall incidence of infection (see Table 8).
Table 8: Allogeneic BMT: Analysis of Data from Placebo-Controlled Clinical Trial Median Values (days or number of patients) ANC ≥500/mm3 ANC ≥1000/mm3 Number of Patients with Infections Number of Patients with Bacteremia Days of Hospitalization - * p <0.05 generalized Wilcoxon test
- † p <0.05 simple chi-square test
LEUKINE
n=5313* 14 * 30 * 9† 25 * Placebo
n=5617 19 42 19 26 Median time to myeloid recovery (ANC ≥500 cells/mm3) in 53 patients receiving LEUKINE was 4 four days less than in 56 patients treated with placebo (see Table 8). The numbers of patients with bacteremia and infection were significantly lower in the LEUKINE group compared to the placebo group (9/53 versus 19/56 and 30/53 versus 42/56, respectively). There were a number of secondary laboratory and clinical endpoints. Of these, only the incidence of severe (grade 3/4) mucositis was significantly improved in the LEUKINE group (4/53) compared to the placebo group (16/56) at p<0.05. LEUKINE-treated patients also had a shorter median duration of post transplant IV antibiotic infusions, and a shorter median number of days to last platelet and RBC transfusions compared to placebo patients, but none of these differences reached statistical significance.
14.5 Treatment of Delayed Neutrophil Recovery or Graft Failure After Allogeneic or Autologous Bone Marrow Transplantation
A historically-controlled study (study 501) was conducted in patients experiencing graft failure following allogeneic or autologous BMT to determine whether LEUKINE improved survival after BMT failure.
Three categories of patients were eligible for this study:
- patients displaying a delay in neutrophil recovery (ANC ≤100 cells/mm3 by day 28 post transplantation);
- patients displaying a delay in neutrophil recovery (ANC ≤100 cells/mm3 by day 21 post transplantation) and who had evidence of an active infection; and
- patients who lost their marrow graft after a transient neutrophil recovery (manifested by an average of ANC ≥500 cells/mm3 for at least one week followed by loss of engraftment with ANC <500 cells/mm3 for at least one week beyond day 21 post transplantation).
A total of 140 eligible adult and pediatric patients from 35 institutions were treated with LEUKINE and evaluated in comparison to 103 historical control patients from a single institution. One hundred sixty-three patients had lymphoid or myeloid leukemia, 24 patients had NHL, 19 patients had Hodgkin's disease and 37 patients had other diseases, such as aplastic anemia, myelodysplasia or non-hematologic malignancy. The majority of patients (223 out of 243) had received prior chemotherapy with or without radiotherapy and/or immunotherapy prior to preparation for transplantation. The median age of enrolled patients was 27 years (range 1–66 years). Thirty-seven patients were younger than 18 years of age.
One hundred-day survival was improved in favor of the patients treated with LEUKINE for graft failure following either autologous or allogeneic BMT. In addition, the median survival was improved by greater than two-fold. The median survival of patients treated with LEUKINE after autologous failure was 474 days versus 161 days for the historical patients. Similarly, after allogeneic failure, the median survival was 97 days with LEUKINE treatment and 35 days for the historical controls. Improvement in survival was better in patients with fewer impaired organs. The Multiple Organ Failure (MOF) score is a clinical and laboratory assessment of seven major organ systems: cardiovascular, respiratory, gastrointestinal, hematologic, renal, hepatic and neurologic. Median survival by MOF category is presented in Table 9.
Table 9: Median Survival by Multiple Organ Failure (MOF) Category Median Survival (days) MOF ≤2 Organs MOF >2 Organs MOF (Composite of both groups) Autologous BMT LEUKINE 474 (n=58) 78.5 (n=10) 474 (n=68) Historical 165 (n=14) 39 (n=3) 161 (n=17) Allogeneic BMT LEUKINE 174 (n=50) 27 (n=22) 97 (n=72) Historical 52.5 (n=60) 15.5 (n=26) 35 (n=86) 14.6 Acute Exposure to Myelosuppressive Doses of Radiation (H-ARS)
Efficacy studies of LEUKINE could not be conducted in humans with acute radiation syndrome for ethical and feasibility reasons. The use of LEUKINE in the H-ARS indication was based on efficacy studies conducted in animals and data supporting LEUKINE's effect on severe neutropenia in patients undergoing autologous or allogeneic BMT following myelosuppressive chemotherapy with or without total body irradiation, and in patients with acute myelogenous leukemia following myelosuppressive chemotherapy [see Dosage and Administration (2.1 to 2.6)].
The recommended dose of LEUKINE for adults exposed to myelosuppressive doses of radiation is 7 mcg/kg as a single daily SC injection [see Dosage and Administration (2.6)]. The 7 mcg/kg dosing regimen is based on population modeling and simulation analyses. The sargramostim exposure associated with the 7 mcg/kg adult dose is expected to be higher than sargramostim exposure in the nonclinical efficacy study and therefore are expected to provide sufficient pharmacodynamic activity to treat humans exposed to myelosuppressive doses of radiation [see Clinical Pharmacology (12.3)]. The safety of LEUKINE at a dose of 250 mcg/m2/day (approximately 7 mcg/kg) has been assessed on the basis of clinical experience in myeloid reconstitution in patients after autologous or allogeneic BMT, and in patients with AML [see Adverse Reactions (6.1)].
The efficacy of LEUKINE was studied in a randomized, blinded, placebo-controlled study in a nonhuman primate model of radiation injury. Rhesus macaques (50% male) were randomized to a control (n = 36) or treated (n = 36) group. Animals were exposed to total body irradiation at a dose that would be lethal in 50% to 60% of animals (655 cGy) by day 60 post irradiation (lethal dose [LD]50–60/60). Starting 48 ± 1 hour after irradiation, animals received daily SC injections of placebo (sterile water for injection, USP) or LEUKINE (7 mcg/kg/day). Blinded treatment was stopped when one of the following criteria was met: ANC ≥1,000 cells/mm3 for 3 consecutive days or if the ANC ≥10,000 cells/mm3. Animals received minimal supportive care that included a prophylactic antibiotic, antiemetic, analgesics and parenteral fluids. No whole blood, blood products or individualized antibiotics were provided.
LEUKINE significantly (p=0.0018) increased survival at day 60 in irradiated nonhuman primates: 78% survival (28/36) in the LEUKINE group compared to 42% survival (15/36) in the control group.
In the same study, an exploratory cohort of 36 rhesus macaques randomized to control (n=18) or treated (n=18) was exposed to total body irradiation at a dose that would be lethal in 70–80% of animals (713 cGY) by day 60 post irradiation. LEUKINE increased survival at day 60 in irradiated nonhuman primates: 61% survival (11/18) in the LEUKINE group compared to 17% survival (3/18) in the control group.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
LEUKINE (sargramostim) for injection is a sterile, preservative-free, white lyophilized powder supplied in a carton containing five 250 mcg single-dose vials. (NDC: 0024-5843-05).
LEUKINE (sargramostim) injection is a sterile, clear, colorless solution preserved with 1.1% benzyl alcohol supplied in a carton containing one 500 mcg/mL multiple-dose vial (NDC: 0024-5844-01) and a carton containing five 500 mcg/mL multiple-dose vials (NDC: 0024-5844-05).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
LEUKINE should be used under the guidance and supervision of a health care professional. However, if the physician determines that LEUKINE may be used outside of the hospital or office setting, persons who will be administering LEUKINE should be instructed as to the proper dose, and the method of reconstituting and administering LEUKINE [see Dosage and Administration (2.7)]. If home use is prescribed, patients should be instructed in the importance of proper disposal and cautioned against the reuse of needles, syringes, drug product, and diluent. A puncture resistant container should be used by the patient for the disposal of used needles.
Advise patients of the following risks and potential risks with LEUKINE:
- Serious allergic reactions [see Warnings and Precautions (5.1)]
- Infusion related reactions [see Warnings and Precautions (5.2)]
- Risk of severe myelosuppression when LEUKINE administered within 24 hours of chemotherapy or radiotherapy [see Warnings and Precautions (5.3)]
- Effusions and capillary leak syndrome [see Warnings and Precautions (5.4)]
- Supraventricular arrhythmias [see Warnings and Precautions (5.5)]
- Leukocytosis including eosinophilia [see Warnings and Precautions (5.6)]
- Potential effect on malignant cells [see Warnings and Precautions (5.7)]
- Pain including chest, abdominal, back, and joint pain [see Adverse Reactions (6.1)]
- Thromboembolic events [see Adverse Reactions (6.3)]
- Embryofetal Toxicity: Advise females of reproductive potential that LEUKINE may cause fetal harm and to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)]
- Lactation: Advise lactating woman not to breastfeed during treatment and for at least 2 weeks after the last dose [see Use in Specific Populations (8.2)]
- Advise patients acutely exposed to myelosuppressive doses of radiation (H-ARS) that efficacy studies of LEUKINE for this indication could not be conducted in humans for ethical and feasibility reasons and that, therefore, approval of this use was based on efficacy studies conducted in animals [see Clinical Studies (14.6)]
Instruct patients who self-administer LEUKINE:
- Follow the Instructions for Use
- Do not reuse needles, syringes, or unused portions of vials
- Follow local requirements for proper disposal of used syringes, needles, and unused vials
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration
Revised: 03/2018Patient Information LEUKINE® (loo-kine)
(sargramostim)
injectionLEUKINE® (loo-kine)
(sargramostim)
for injectionWhat is LEUKINE?
LEUKINE is a man-made form of granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF is a substance produced by the body. It stimulates the growth of certain white blood cells that are important in the body's fight against infection.
Acute Radiation Syndrome: The effectiveness of LEUKINE for this use was only studied in animals because it could not be studied in people.Do not take LEUKINE if you have had a serious allergic reaction to human GM-CSF, such as sargramostim, products that are made from yeast, or any of the ingredients in LEUKINE. See the end of this leaflet for a complete list of ingredients in LEUKINE. Before receiving LEUKINE, tell your healthcare provider about all your medical conditions, including if you: - have heart or lung disease
- are allergic to benzyl alcohol
- are pregnant or plan to become pregnant. It is not known if LEUKINE will harm your unborn baby. See "What are the possible side effects of LEUKINE?" Talk to your healthcare provider about the type of LEUKINE that is right for you.
- are breastfeeding or plan to breastfeed. It is not known if LEUKINE passes into your breast milk. Do not breast feed during treatment and for at least 2 weeks after your last dose of LEUKINE.
How will I receive LEUKINE? - LEUKINE comes in two ways: as a solution or as a powder to be mixed for injection.
- LEUKINE is given as an injection under your skin (subcutaneous injection) by a healthcare provider.
- If your healthcare provider decides that the subcutaneous injections can be given at home by you or your caregiver, see the detailed "Instructions for Use" that comes with your LEUKINE for information on how to draw up and inject a dose of LEUKINE.
- If your healthcare provider gives you LEUKINE for injection (powder) to be mixed, carefully follow your healthcare provider's instructions on how to store, mix, draw up and inject the medicine.
- You and your caregiver should be shown how to prepare and inject LEUKINE before you use it, by your healthcare provider.
- Your healthcare provider will tell you how much LEUKINE to inject and when to inject it. Do not change your dose or stop LEUKINE unless your healthcare provider tells you to.
- If you are also receiving chemotherapy or radiation therapy, your dose of LEUKINE should be injected at least 24 hours before or 24 hours after your dose of chemotherapy and at least 24 hours before your dose of radiation therapy. Your healthcare provider will do blood tests to monitor your white blood cell count and, if necessary, adjust your LEUKINE dose.
- If you are receiving LEUKINE because you have been suddenly (acutely) exposed to an amount of radiation that can affect your bone marrow (Acute Radiation Syndrome), you will need to have blood tests about every 3 days during treatment with LEUKINE to check your white blood cell count until it returns to normal.
- If your child needs to receive LEUKINE for Acute Radiation Syndrome, follow your healthcare provider's instructions. LEUKINE injection contains the preservative benzyl alcohol.
- If you miss a dose of LEUKINE, talk to your healthcare provider about when you should give your next dose.
What are the possible side effects of LEUKINE?
LEUKINE may cause serious side effects, including:- Serious allergic reactions. LEUKINE can cause serious allergic reactions that can be severe. Get medical help right away if you get any of the following signs or symptoms of a serious allergic reaction with LEUKINE, including: skin rash over your entire body, hives, trouble breathing, wheezing, swelling around your mouth or eyes, a fast heartbeat, sweating, and dizziness or feeling faint.
- Infusion related reactions. When LEUKINE is given as an infusion, it can cause reactions during or soon after an infusion. Tell your healthcare provider or nurse right away if you develop trouble breathing, skin flushing, a fast pulse, dizziness or you feel faint during or soon after your infusion.
- Too much body fluid (fluid retention) and Capillary Leak Syndrome. LEUKINE can cause you to have too much fluid in your body, and fluid may leak from blood vessels into your body's tissues. This can happen anywhere in your body, including around your heart and lungs. This condition is called "Capillary Leak Syndrome" (CLS). CLS can quickly cause you to have symptoms that may become life-threatening. Get emergency medical help right away if you develop any of the following symptoms:
- swelling or puffiness of your feet or ankles, and are urinating less than usual
- swelling of your stomach area (abdomen) and feeling of fullness
- sudden weight gain
- swollen legs or feet
- trouble breathing
- dizziness or feeling faint
- a general feeling of tiredness
- Abnormal heart rhythm. An abnormal heart rhythm may happen during treatment with LEUKINE, especially in people with a history of an abnormal heart rhythm.
- Increased white blood cell count . LEUKINE can cause your white blood cell count to increase too high (leukocytosis). Your healthcare provider will check your blood during treatment with LEUKINE.
- Risks of using LEUKINE in newborns, babies born with a low birth weight, and in pregnant women. Some forms of LEUKINE may contain the preservative benzyl alcohol. Medicines that contain benzyl alcohol, including LEUKINE, can cause serious side effects that can lead to death in newborns and babies born with a low birth weight. Avoid using LEUKINE containing benzyl alcohol in newborns, babies born with a low birth weight, and in pregnant women. If LEUKINE needs to be given to your newborn or baby with a low birth weight, or in pregnant women, talk to your healthcare provider about this risk and which form of LEUKINE to use.
The most common side effects of LEUKINE in people who receive an allogeneic bone marrow transplant include: diarrhea, fever, nausea, rash, vomiting, mouth sores, decreased appetite, hair loss, stomach-area (abdomen) pain, headache and high blood pressure.
The most common side effects of LEUKINE in people with AML include: fever, liver problems, skin reactions, infections, abnormal amount of salts and other minerals in the blood, nausea, diarrhea, urinary tract problems, lung problems, vomiting, nervous system problems, mouth sores, hair loss and weight loss.
These are not all the possible side effects of LEUKINE.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store LEUKINE injection (solution)? - Store LEUKINE in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Do not freeze LEUKINE.
- Do not shake.
- Store LEUKINE vials in the original carton to protect from light.
- Do not use LEUKINE beyond the expiration date printed on the vial label.
- Used vials with remaining LEUKINE should be stored in the refrigerator and used within 20 days (be sure to mark down the date you first used the vial).
- Throw away (dispose of) any remaining LEUKINE after 20 days.
General information about the safe and effective use of LEUKINE.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use LEUKINE for a condition for which it was not prescribed. Do not give LEUKINE to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LEUKINE that is written for health professionals.What are the ingredients in LEUKINE?
Active ingredient: sargramostim
Inactive ingredients:
LEUKINE injection: benzyl alcohol
LEUKINE for injection: mannitol, sucrose, tromethamine
Manufactured by: sanofi-aventis U.S. LLC Bridgewater, NJ 08807 A SANOFI COMPANY
US License No. 1752 ©2018 sanofi-aventis U.S. LLC
LEUKINE® is a registered trademark licensed to Genzyme Corporation.
For more information go to www.leukine.com, or call 1-888-4RX-LEUKINE -
INSTRUCTIONS FOR USE
Instructions for Use
LEUKINE® (loo'-kine)
(sargramostim)
Injection, for subcutaneous use
Multiple-dose vial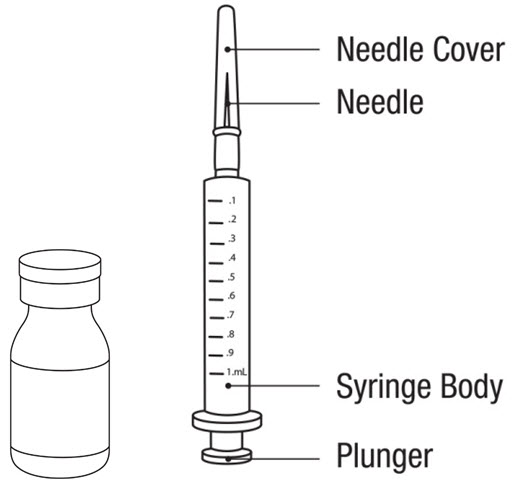
Important:
Read the Patient Information for important information you need to know about LEUKINE before using these Instructions for Use.
Storing the LEUKINE vial
- Store LEUKINE in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Do not freeze LEUKINE
- Do not shake LEUKINE.
- Keep the LEUKINE vial in the original carton to protect from light.
- Do not use LEUKINE beyond the expiration date printed on the vial label.
- Once the vial has been used, any remaining LEUKINE should be stored in the refrigerator and used within 20 days (be sure to mark down the date you first used the vial).
- Throw away (dispose of) any remaining LEUKINE in a used vial after 20 days. Throw away empty LEUKINE vials in the trash, unless otherwise instructed.
Keep LEUKINE, needles and syringes out of the reach of children.
Using LEUKINE vials
- It is important that you do not try to give the injection unless you or your caregiver have received training from your healthcare provider about how to draw up and inject LEUKINE, and how often to inject it. LEUKINE is given as an injection under the skin (subcutaneous injection). If you have any questions about your dose of LEUKINE, or how to draw up and inject a dose, talk to your healthcare provider.
- Use only the syringe and needle that your healthcare provider tells you to use to inject LEUKINE.
- Generally, a 1 mL syringe with a 25 to 30 gauge 5/8-inch needle is used. Your healthcare provider will either supply you with the correct syringes and needles, or will write you a prescription so you can get the correct syringes and needles from your pharmacy. The dose for LEUKINE will usually be measured in milliliters (mL). (For example: 0.8 mL). It is important that you use a syringe that is marked in tenths of a milliliter so that you are able to measure the correct dose prescribed by your healthcare provider. See the figure at the beginning of this leaflet.
- Make sure the name LEUKINE appears on the carton and vial label.
- Check the carton and vial label to make sure the dose strength is 500 mcg/mL.
- Look at the LEUKINE vial. LEUKINE should be clear and colorless. Do not use the vial of LEUKINE if the medicine is cloudy or discolored or contain flakes or particles. Contact your healthcare provider or pharmacist.
- Do not shake the vial.
- Do not use the vial if the carton is open or damaged.
- A skin reaction may occur at the injection site. Your skin may become red, painful, or swollen. If you develop a skin reaction, call your healthcare provider.
- Do not change your dose of LEUKINE unless your healthcare provider tells you to.
- Call your healthcare provider if you have any questions.
Step 1: Draw up the dose
- A.
Remove a LEUKINE vial from the refrigerator.
- Put original carton with any unused vials back in the refrigerator.
- Place the vial on a clean, flat, well-lit surface.
- B.
Inspect the LEUKINE vial.
Make sure the medicine in the vial is clear and colorless.-
Do not use the vial if:
- The medicine is cloudy, discolored or contains lumps, flakes, or particles.
- The expiration date printed on the label has passed.
-
Do not use the vial if:
- C.
Gather supplies needed for an injection.
Wash your hands well with soap and water.
Place the following supplies on your clean, flat, well-lit surface:- 1 vial of LEUKINE
- 1 sterile disposable syringe and needle
- 2 sterile alcohol swabs
- 1 cotton ball or gauze pad
- 1 adhesive bandage
- sharps disposal container. See Step 5.
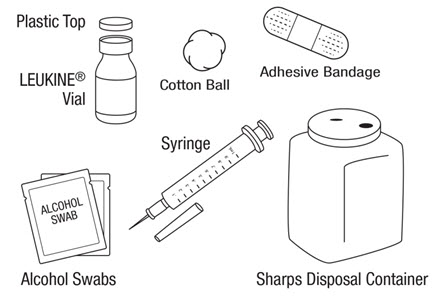
- Only use the disposable syringes and needles that your healthcare provider prescribes.
- Only use the syringes and needles 1 time. Discard (throw away) any used syringes and needles. See Step 5.
Step 2: Get ready
- D.
Take the cap off of the LEUKINE vial. Do not remove the gray rubber stopper.
- Wipe the top of the rubber stopper with a new alcohol swab.
-
Do not touch the rubber stopper with your hands or fingers. If you do touch the stopper, clean it again with a new alcohol swab.
- E.
Check the syringe packaging. If the packaging is opened or damaged, do not use that syringe. Dispose of (throw away) that syringe in the sharps disposal container. See Step 5.
- Remove the syringe and needle from its packaging.
- F.
Hold the syringe by the barrel with the needle cover pointing up. With the needle cover still on the needle, draw air into the syringe by pulling back on the plunger. The amount of air (mLs) you draw into the syringe should be equal to your prescribed LEUKINE dose.
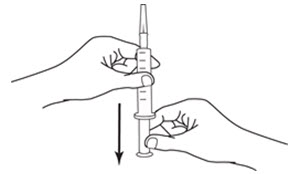
- Carefully remove the needle cover by pulling straight off and away from your body. Throw the needle cover into the sharps disposal container.
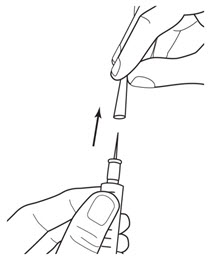
- Do not lay down the syringe or allow the needle to touch anything. If the needle touches any surface, including your hands, throw away the needle and syringe in your sharps disposal container and start over with a new syringe and needle.
- Carefully remove the needle cover by pulling straight off and away from your body. Throw the needle cover into the sharps disposal container.
- G. Keep the vial on your flat work surface and insert the needle downward, through the center of the rubber stopper.
- H.
Push the plunger down all the way to inject the air into the vial. Make sure the needle is above the LEUKINE liquid. Leave the needle in the rubber stopper.
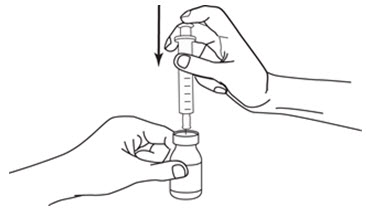
- I. Keep the needle in the vial and turn the vial upside down. Make sure that the LEUKINE liquid is covering the needle tip.
- J.
Keep the vial upside down and slowly pull back on the plunger to fill the syringe barrel with LEUKINE until the plunger lines up with the amount (mL) that matches your prescribed dose.
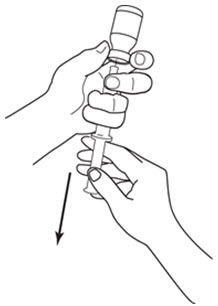
- K.
Keep the needle in the vial and check for air bubbles in the syringe. If there are air bubbles, gently tap the syringe barrel with your finger until the air bubbles rise to the top of the syringe. Slowly push the plunger up to push the air bubbles out of the syringe.
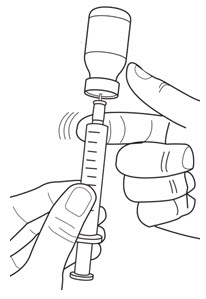
- L. Keep the tip of the needle in the LEUKINE liquid and again pull the plunger back to the number on the syringe barrel that matches your prescribed dose. Check again for bubbles. The air in the syringe will not hurt you, but too large of an air bubble can reduce the amount of LEUKINE you inject. If there are still air bubbles, repeat the above steps to remove them.
- M.
Check again that you have the correct dose of LEUKINE in the syringe. It is important that you use the correct dose as prescribed by your healthcare provider.
- Do not remove the needle from the vial. Place the vial down on its side on your flat work surface with the needle still in the vial.
Step 3: Choose and prepare the injection site
You can use
- Thigh
- Stomach-area (abdomen), except for a 2-inch area right around your navel area (belly button) or waistline.
- Outer area of upper arm (only if someone else is giving you the injection).
- Choose a different site with each injection. Injection sites should be at least one inch apart.
-
Do not inject into an area where the skin is tender, bruised, red, or hard. Avoid injecting into areas with scars or stretch marks.
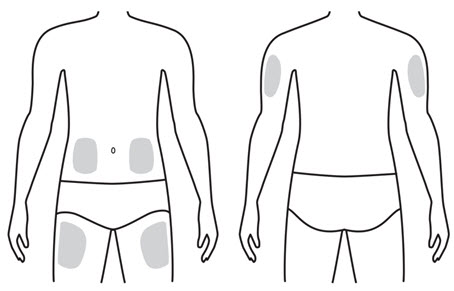
- N.
Clean the injection site
- Clean the injection site with an alcohol swab. Throw the alcohol swab away.
- Let the skin dry for 10 seconds.
- Do not touch this area again before injecting.
Step 4: Subcutaneous (under the skin) injection
- O. Withdraw the needle from the LEUKINE vial by pulling the syringe back through the rubber stopper.
- P.
Pinch your injection site to create a firm surface. Keep skin pinched while injecting.
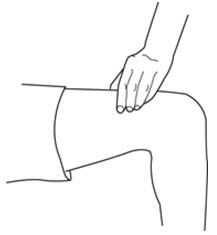
- Q.
Hold the pinch. Insert the needle into the skin at a 45 to 90-degree angle.
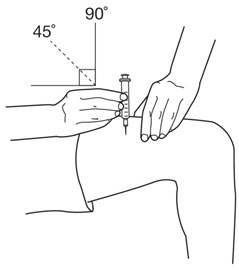
- R.
Release your grasp on the skin. Gently pull back on the plunger just a little bit (about 1/8 of an inch).
-
If you see blood in the syringe, do not inject LEUKINE. Withdraw the needle at the same angle it was inserted. Dispose of the syringe in your sharps disposal container. See Step 5.
Repeat Steps 1 through 4 to prepare a new syringe of LEUKINE. -
If you do not see blood in the syringe, slowly inject all of the LEUKINE by pushing the plunger until it reaches the bottom.
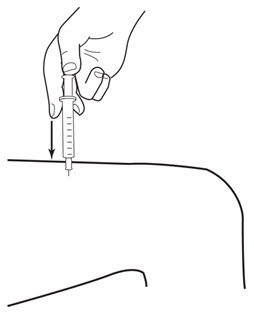
-
If you see blood in the syringe, do not inject LEUKINE. Withdraw the needle at the same angle it was inserted. Dispose of the syringe in your sharps disposal container. See Step 5.
- S. When finished injecting LEUKINE, remove the needle at the same angle as it was inserted. Do not try to re-cap the needle.
Step 5: Dispose of (throw away) needles and syringes
- Do not reuse syringes or needles. Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your sharps disposal container.
Important: Always keep the sharps disposal container out of the reach of children.
Step 6: Examine the injection site
- If there is blood at the injection site, press a cotton ball or gauze pad on the injection site. Do not rub the injection site. Apply an adhesive bandage if needed.
IMPORTANT NOTES
- Try to give yourself LEUKINE at the same time each day.
- Call your healthcare provider or pharmacist if you:
- miss a dose of LEUKINE
- notice anything unusual about your condition while you are taking LEUKINE
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by: Sanofi-aventis U.S. LLC
Bridgewater, NJ 08807
US License No. 1752
© 2018 sanofi-aventis U.S. LLC
LEUKINE® is a registered trademark licensed to Genzyme CorporationRevised: 03/2018
-
PRINCIPAL DISPLAY PANEL – 250 mcg – 5 Vial Label
NDC: 0024-5843-05
5 × 250 mcg/vial
Leukine®
sargramostim
A Recombinant GM-CSF–Yeast-Expressed1.4 × 106 IU/vial
Sterile
Lyophilized
Rx onlySANOFI
– 250 mcg
– 5 vials
– Injection

-
PRINCIPAL DISPLAY PANEL – 500 mcg – 5 Vial Label
NDC: 0024-5844-05
5 × 500 mcg/mL
Leukine®
sargramostim
A Recombinant GM-CSF–Yeast-Expressed2.8 × 106 IU/mL
Sterile
liquid injection, contains preservative
Rx onlySANOFI
– 5 multiple
use vials
-
INGREDIENTS AND APPEARANCE
LEUKINE
sargramostim liquidProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0024-5844 Route of Administration SUBCUTANEOUS, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SARGRAMOSTIM (UNII: 5TAA004E22) (sargramostim - UNII:5TAA004E22) SARGRAMOSTIM 500 ug in 1 mL Inactive Ingredients Ingredient Name Strength BENZYL ALCOHOL (UNII: LKG8494WBH) 0.011 mL in 1 mL MANNITOL (UNII: 3OWL53L36A) 40 mg in 1 mL SUCROSE (UNII: C151H8M554) 10 mg in 1 mL TROMETHAMINE (UNII: 023C2WHX2V) 1.2 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0024-5844-01 1 in 1 CARTON 12/01/1996 12/30/2021 1 1 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product 2 NDC: 0024-5844-05 5 in 1 CARTON 12/01/1996 12/30/2021 2 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103362 12/01/1996 12/30/2021 LEUKINE
sargramostim injection, powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0024-5843 Route of Administration SUBCUTANEOUS, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SARGRAMOSTIM (UNII: 5TAA004E22) (sargramostim - UNII:5TAA004E22) SARGRAMOSTIM 250 ug in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 40 mg in 1 mL SUCROSE (UNII: C151H8M554) 10 mg in 1 mL TROMETHAMINE (UNII: 023C2WHX2V) 1.2 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0024-5843-05 5 in 1 CARTON 05/01/1991 12/30/2021 1 NDC: 0024-5843-01 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103362 05/01/1991 12/30/2021 Labeler - sanofi-aventis U.S. LLC (824676584) Establishment Name Address ID/FEI Business Operations Hospira, Inc. 030606222 MANUFACTURE(0024-5843, 0024-5844) , REPACK(0024-5843, 0024-5844) , RELABEL(0024-5843, 0024-5844) , PACK(0024-5843, 0024-5844) , LABEL(0024-5843, 0024-5844) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 029487267 ANALYSIS(0024-5843, 0024-5844) , API MANUFACTURE(0024-5843, 0024-5844) Establishment Name Address ID/FEI Business Operations Bayer HealthCare LLC 127769128 ANALYSIS(0024-5843, 0024-5844)
Trademark Results [Leukine]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LEUKINE 73831918 1653426 Live/Registered |
IMMUNEX CORPORATION 1989-10-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.