VABYSMO- faricimab injection, solution
VABYSMO by
Drug Labeling and Warnings
VABYSMO by is a Prescription medication manufactured, distributed, or labeled by Genentech, Inc., Roche Diagnostics GmbH, F. Hoffmann-La Roche Ltd. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VABYSMO safely and effectively. See full prescribing information for VABYSMO.
VABYSMO® (faricimab-svoa) injection, for intravitreal use
Initial U.S. Approval: 2022RECENT MAJOR CHANGES
Dosage and Administration, Macular Edema Following Retinal Vein Occlusion (RVO) (2.4) 4/2026 INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
For intravitreal injection. (2.1)
-
Neovascular (Wet) Age-Related Macular Degeneration (nAMD)
- The recommended dose for VABYSMO is 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 ± 7 days, monthly) for the first 4 doses, followed by optical coherence tomography and visual acuity evaluations 8 and 12 weeks later to inform whether to give a 6 mg dose via intravitreal injection on one of the following three regimens: 1) Weeks 28 and 44; 2) Weeks 24, 36 and 48; or 3) Weeks 20, 28, 36 and 44. Although additional efficacy was not demonstrated in most patients when VABYSMO was dosed every 4 weeks compared to every 8 weeks, some patients may need every 4 week (monthly) dosing after the first 4 doses. Patients should be assessed regularly. (2.2)
-
Diabetic Macular Edema (DME)
- VABYSMO is recommended to be dosed by following one of these two dose regimens: 1) 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 days ± 7 days, monthly) for at least 4 doses. If after at least 4 doses, resolution of edema based on the central subfield thickness (CST) of the macula as measured by optical coherence tomography is achieved, then the interval of dosing may be modified by extensions of up to 4 week interval increments or reductions of up to 8 week interval increments based on CST and visual acuity evaluations; or 2) 6 mg dose of VABYSMO can be administered every 4 weeks for the first 6 doses, followed by 6 mg dose via intravitreal injection at intervals of every 8 weeks (2 months). Although additional efficacy was not demonstrated in most patients when VABYSMO was dosed every 4 weeks compared to every 8 weeks, some patients may need every 4 week (monthly) dosing after the first 4 doses. Patients should be assessed regularly. (2.3)
-
Macular Edema Following Retinal Vein Occlusion (RVO)
- The recommended dose for VABYSMO is 6 mg (0.05 mL of 120 mg/mL) administered by intravitreal injection every 4 weeks (approximately every 28 ± 7 days, monthly). (2.4)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Endophthalmitis and retinal detachments may occur following intravitreal injections. Patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment without delay, to permit prompt and appropriate management. (5.1)
- Increases in intraocular pressure have been seen within 60 minutes of an intravitreal injection. (5.2)
- There is a potential risk of arterial thromboembolic events (ATEs) associated with VEGF inhibition. (5.3)
ADVERSE REACTIONS
The most common adverse reactions (≥ 5%) reported in patients receiving VABYSMO were cataract (15%) and conjunctival hemorrhage (8%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2026
-
Neovascular (Wet) Age-Related Macular Degeneration (nAMD)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Neovascular (wet) Age-Related Macular Degeneration (nAMD)
1.2 Diabetic Macular Edema (DME)
1.3 Macular Edema Following Retinal Vein Occlusion (RVO)
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Neovascular (wet) Age-Related Macular Degeneration (nAMD)
2.3 Diabetic Macular Edema (DME)
2.4 Macular Edema Following Retinal Vein Occlusion (RVO)
2.5 Preparation for Administration - Prefilled Syringe
2.6 Preparation for Administration - Vial
2.7 Injection Procedure
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Active Intraocular Inflammation
4.3 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
5.2 Increase in Intraocular Pressure
5.3 Thromboembolic Events
5.4 Retinal Vasculitis and/or Retinal Vascular Occlusion
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Neovascular (wet) Age-Related Macular Degeneration (nAMD)
14.2 Diabetic Macular Edema (DME)
14.3 Macular Edema Following Retinal Vein Occlusion (RVO)
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
For intravitreal injection. VABYSMO must be administered by a qualified physician.
VABYSMO is available as:
- Prefilled syringe: A sterile injection filter needle (30-gauge × ½-inch, Extra Thin Wall) with an integrated filter in the hub is provided. Each prefilled syringe should only be used for the treatment of a single eye.
- Vial: A sterile 5-micron, blunt transfer filter needle (18-gauge × 1½-inch) is provided. Each vial should only be used for the treatment of a single eye.
2.2 Neovascular (wet) Age-Related Macular Degeneration (nAMD)
The recommended dose for VABYSMO is 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 ± 7 days, monthly) for the first 4 doses, followed by optical coherence tomography and visual acuity evaluations 8 and 12 weeks later to inform whether to give a 6 mg dose via intravitreal injection on one of the following three regimens: 1) Weeks 28 and 44; 2) Weeks 24, 36 and 48; or 3) Weeks 20, 28, 36 and 44. Although additional efficacy was not demonstrated in most patients when VABYSMO was dosed every 4 weeks compared to every 8 weeks, some patients may need every 4 week (monthly) dosing after the first 4 doses. Patients should be assessed regularly.
2.3 Diabetic Macular Edema (DME)
VABYSMO is recommended to be dosed by following one of these two dose regimens: 1) 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 days ± 7 days, monthly) for at least 4 doses. If after at least 4 doses, resolution of edema based on the central subfield thickness (CST) of the macula as measured by optical coherence tomography is achieved, then the interval of dosing may be modified by extensions of up to 4 week interval increments or reductions of up to 8 week interval increments based on CST and visual acuity evaluations; or 2) 6 mg dose of VABYSMO can be administered every 4 weeks for the first 6 doses, followed by 6 mg dose via intravitreal injection at intervals of every 8 weeks (2 months). Although additional efficacy was not demonstrated in most patients when VABYSMO was dosed every 4 weeks compared to every 8 weeks, some patients may need every 4 week (monthly) dosing after the first 4 doses. Patients should be assessed regularly.
2.4 Macular Edema Following Retinal Vein Occlusion (RVO)
The recommended dose for VABYSMO is 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 ± 7 days, monthly).
In clinical studies, patients received monthly injections of VABYSMO for 6 months [see Clinical Studies (14.3)].
2.5 Preparation for Administration - Prefilled Syringe
Before you start 
Read all the instructions carefully before using VABYSMO. The VABYSMO carton includes: 
A sterile prefilled syringe in a sealed tray. The prefilled syringe is for treatment of a single eye. 
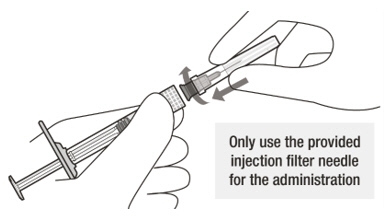
A sterile injection filter needle (30-gauge × ½ inch, Extra Thin Wall) with an integrated filter in the hub. The injection filter needle is for single use only. Only use the provided injection filter needle for the administration. 
VABYSMO should be refrigerated at temperatures between 2°C to 8°C (36°F to 46°F).
Do not freeze.
Allow VABYSMO to reach room temperature, 20°C to 25°C (68°F to 77°F) before proceeding with the administration. 
Prior to use, keep the sealed tray in the original carton to protect the prefilled syringe from light. The prefilled syringe may be kept at room temperature in the original carton for up to 24 hours. 
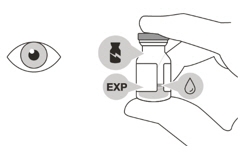
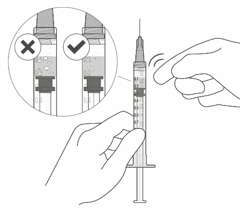
VABYSMO should be inspected visually prior to administration. Do not use if the carton seals have been tampered with. Do not use if the packaging, prefilled syringe, injection filter needle is expired, damaged, or have been tampered with. Do not use if the injection filter needle is missing. Do not remove the finger grip from the syringe. Do not use if the syringe cap is detached from the Luer lock. Do not use if particulates, cloudiness, or discoloration are visible. VABYSMO is a clear to opalescent and colorless to brownish-yellow liquid solution. Prefilled Syringe Description 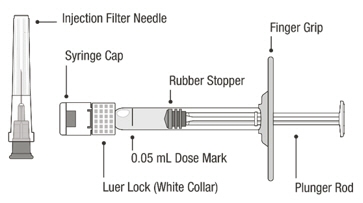
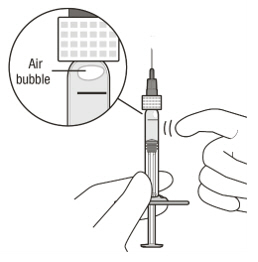
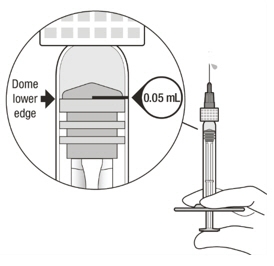
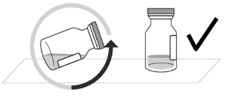
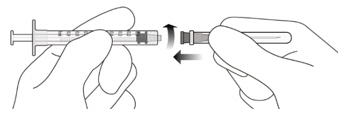
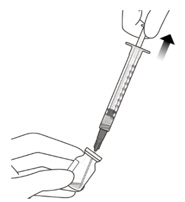
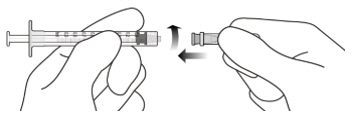
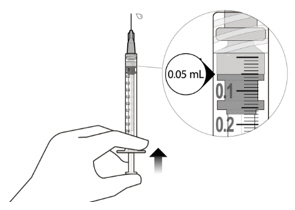
Figure A Note: the dose must be set to the 0.05 mL dose mark. Use aseptic technique to carry out the following preparation steps: Open Tray and Remove Syringe Cap 1 Peel the lid off the syringe tray and aseptically remove the prefilled syringe. 2 Hold the syringe by the white collar; snap off the syringe cap (see Figure B). Do not twist off the cap. Figure B Attach Injection Filter Needle 3 Aseptically remove the provided injection filter needle from its packaging. 4 Aseptically and firmly attach the injection filter needle onto the syringe Luer lock (see Figure C). Figure C 5 Carefully remove the needle cap by pulling it straight off. Dislodge Air Bubbles 6 Hold the syringe with the injection filter needle pointing up. Check the syringe for air bubbles. 7 If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure D). Figure D Expel Air and Adjust the Dose 8 Hold the syringe at eye level and slowly push the plunger rod until the lower edge of the rubber stopper's dome is aligned with the 0.05 mL dose mark (see Figure E). This will expel the air and the excess solution and set the dose to 0.05 mL. Ensure that the injection is given immediately after preparation of the dose. Figure E 2.6 Preparation for Administration - Vial
Before you start 
Read all the instructions carefully before using VABYSMO. 
The VABYSMO kit includes a glass vial and transfer filter needle. The glass vial is for a single dose only. The filter needle is for treatment of a single eye. 
VABYSMO should be stored refrigerated at temperatures between 2°C to 8°C (36°F to 46°F).
Do not freeze.
Do not shake.
Allow VABYSMO to reach room temperature, 20°C to 25°C (68°F to 77°F) before proceeding with the administration. Keep the vial in the original carton to protect from light. The VABYSMO vial may be kept at room temperature for up to 24 hours. 
The VABYSMO vial should be inspected visually for particulate matter and discoloration prior to administration. VABYSMO is a clear to opalescent and colorless to brownish-yellow liquid solution.
Do not use if particulates, cloudiness, or discoloration are visible.
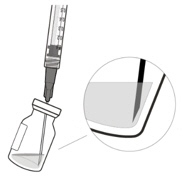
Do not use if the packaging, vial and/or transfer filter needle are expired, damaged, or have been tampered with (see Figure F).Figure F Use aseptic technique to carry out the preparation of the intravitreal injection. 1 Gather the following supplies: - One VABYSMO vial (included)
- One sterile 5-micron blunt transfer filter needle 18-gauge × 1½ inch (included)
- One sterile 1 mL Luer lock syringe with a 0.05 mL dose mark (not included)
- Note that a 30-gauge injection needle is recommended to avoid increased injection forces that could be experienced with smaller diameter needles.
- One sterile injection needle 30-gauge × ½ inch (not included)
- Alcohol swab (not included).
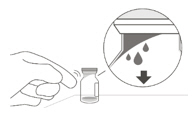
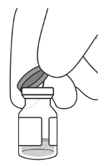
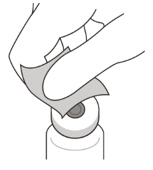
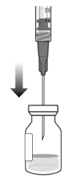
2 To ensure all liquid settles at the bottom of the vial, place the vial upright on a flat surface (for about 1 minute) after removal from packaging (see Figure G). Gently tap the vial with your finger (see Figure H), as liquid may stick to the top of the vial. Figure G Figure H 3 Remove the flip-off cap from the vial (see Figure I) and wipe the vial septum with an alcohol swab (see Figure J). Figure I Figure J 4 Aseptically and firmly attach the included 18-gauge × 1½ inch transfer filter needle onto a 1 mL Luer lock syringe (see Figure K). Figure K 5 Using aseptic technique, push the transfer filter needle into the center of the vial septum (see Figure L), push it all the way in, then tilt the vial slightly so that the needle touches the bottom edge of the vial (see Figure M). Figure L Figure M 6 Hold the vial slightly inclined and slowly withdraw all the liquid from the vial (see Figure N). Keep the bevel of the transfer filter needle submerged in the liquid, to avoid introduction of air. Figure N 7 Ensure that the plunger rod is drawn sufficiently back when emptying the vial, in order to completely empty the transfer filter needle (see Figure N). 8 Disconnect the transfer filter needle from the syringe and dispose of it in accordance with local regulations. Do not use the transfer filter needle for the intravitreal injection. 9 Aseptically and firmly attach a 30-gauge × ½ inch injection needle onto the Luer lock syringe (see Figure O). Figure O 10 Carefully remove the plastic needle shield from the needle by pulling it straight off. 11 To check for air bubbles, hold the syringe with the needle pointing up. If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure P). Figure P 12 Carefully expel the air from the syringe and needle, and slowly depress the plunger to align the rubber stopper tip to the 0.05 mL dose mark. The syringe is ready for the injection (see Figure Q). Ensure that the injection is given immediately after preparation of the dose. Figure Q 2.7 Injection Procedure
The intravitreal injection procedure must be carried out under aseptic conditions, which includes the use of surgical hand disinfection, sterile gloves, a sterile drape and a sterile eyelid speculum (or equivalent), and the availability of sterile paracentesis equipment (if required). Adequate anesthesia and a broad-spectrum microbicide should be administered prior to the injection.
Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL.
Note for the prefilled syringe: Do not recap or detach the injection filter needle from the syringe.
Any unused drug product or waste material should be disposed of in accordance with local regulations.
Immediately following the intravitreal injection, patients should be monitored for elevation in intraocular pressure. Appropriate monitoring may consist of a check for perfusion of the optic nerve head or tonometry. If required, a sterile paracentesis needle should be available. Following intravitreal injection, patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment (e.g., vision loss, eye pain, redness of the eye, photophobia, blurring of vision) without delay [see Patient Counseling Information (17)].
Each syringe should only be used for the treatment of a single eye. If the contralateral eye requires treatment, a new syringe should be used and the sterile field, syringe, gloves, drapes, eyelid speculum, filter, and injection needles should be changed before VABYSMO is administered to the other eye.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachments
Intravitreal injections, including Vabysmo, have been associated with endophthalmitis and retinal detachments [see Adverse Reactions (6.1)]. Proper aseptic injection techniques must always be used when administering VABYSMO. Patients should be instructed to report any signs or symptoms suggestive of endophthalmitis or retinal detachment without delay, to permit prompt and appropriate management [see Dosage and Administration (2.6) and Patient Counseling Information (17)].
5.2 Increase in Intraocular Pressure
Transient increases in intraocular pressure (IOP) have been seen within 60 minutes of intravitreal injection, including with VABYSMO [see Adverse Reactions (6.1)]. IOP and the perfusion of the optic nerve head should be monitored and managed appropriately [see Dosage and Administration (2.6)].
5.3 Thromboembolic Events
Although there was a low rate of arterial thromboembolic events (ATEs) observed in the VABYSMO clinical trials, there is a potential risk of ATEs following intravitreal use of VEGF inhibitors. ATEs are defined as nonfatal stroke, nonfatal myocardial infarction, or vascular death (including deaths of unknown cause).
The incidence of reported ATEs in the nAMD studies during the first year was 1% (7 out of 664) in patients treated with VABYSMO compared with 1% (6 out of 662) in patients treated with aflibercept [see Clinical Studies (14.1)].
The incidence of reported ATEs in the DME studies from baseline to week 100 was 5% (64 out of 1,262) in patients treated with VABYSMO compared with 5% (32 out of 625) in patients treated with aflibercept [see Clinical Studies (14.2)].
The incidence of reported ATEs in the RVO studies during the first 6 months was 1.1% (7 out of 641) in patients treated with VABYSMO compared with 1.4% (9 out of 635) in patients treated with aflibercept [see Clinical Studies (14.3)].
5.4 Retinal Vasculitis and/or Retinal Vascular Occlusion
Retinal vasculitis and/or retinal vascular occlusion, typically in the presence of intraocular inflammation, have been reported with the use of VABYSMO [see Adverse Reactions (6.2)]. Discontinue treatment with VABYSMO in patients who develop these events. Patients should be instructed to report any change in vision without delay.
-
6 ADVERSE REACTIONS
The following potentially serious adverse reactions are described elsewhere in the labeling:
- Hypersensitivity [see Contraindications (4)]
- Endophthalmitis and retinal detachments [see Warnings and Precautions (5.1)]
- Increase in intraocular pressure [see Warnings and Precautions (5.2)]
- Thromboembolic events [see Warnings and Precautions (5.3)]
- Retinal Vasculitis and/or Retinal Vascular Occlusion [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials of the same or another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to VABYSMO in 2,567 patients, which constituted the safety population in six Phase 3 studies [see Clinical Studies (14.1, 14.2, 14.3)].
Table 1: Common Adverse Reactions (≥ 1%) Adverse Reactions VABYSMO Active Control (aflibercept) AMD
N=664DME
N=1,262RVO
N=641AMD
N=662DME
N=625RVO
N=635- * AMD only
- † Including iridocyclitis, iritis, uveitis, vitritis
Cataract 3% 15% < 1% 2% 12% 1% Conjunctival hemorrhage 7% 8% 3% 8% 7% 4% Vitreous detachment 3% 5% 2% 3% 4% 2% Vitreous floaters 3% 4% 2% 2% 3% 2% Retinal pigment epithelial tear* 3% 1% Intraocular pressure increased 3% 4% 1% 2% 3% 3% Eye pain 3% 3% < 1% 3% 3% < 1% Intraocular inflammation† 2% 1% 1% 1% 1% < 1% Eye irritation 1% < 1% < 1% < 1% 1% < 1% Lacrimation increased 1% 1% 0 1% < 1% < 1% Ocular discomfort 1% 1% < 1% < 1% < 1% < 1% Less common adverse reactions reported in < 1% of the patients treated with VABYSMO were corneal abrasion, eye pruritus, ocular hyperemia, blurred vision, sensation of foreign body, endophthalmitis, conjunctival hyperaemia, visual acuity reduced, visual acuity reduced transiently, vitreous hemorrhage, retinal tear and rhegmatogenous retinal detachment.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of VABYSMO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of VABYSMO administration in pregnant women.
Administration of VABYSMO to pregnant monkeys throughout the period of organogenesis resulted in an increased incidence of abortions at intravenous (IV) doses 158 times the human exposure (based on Cmax) of the maximum recommended human dose [see Animal Data]. Based on the mechanism of action of VEGF and Ang-2 inhibitors, there is a potential risk to female reproductive capacity, and to embryo-fetal development. VABYSMO should not be used during pregnancy unless the potential benefit to the patient outweighs the potential risk to the fetus.
All pregnancies have a background risk of birth defect, loss, and other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies.
Data
Animal Data
An embryo fetal developmental toxicity study was performed on pregnant cynomolgus monkeys. Pregnant animals received 5 weekly IV injections of VABYSMO starting on day 20 of gestation at 1 or 3 mg/kg. A non-dose dependent increase in pregnancy loss (abortions) was observed at both doses evaluated. Serum exposure (Cmax) in pregnant monkeys at the low dose of 1 mg/kg was 158 times the human exposure at the maximum recommended intravitreal dose of 6 mg once every 4 weeks. A no observed adverse effect level (NOAEL) was not identified in this study.
8.2 Lactation
Risk Summary
There is no information regarding the presence of faricimab in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Many drugs are transferred in human milk with the potential for absorption and adverse reactions in the breastfed child.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VABYSMO and any potential adverse effects on the breastfed child from VABYSMO.
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
The safety and efficacy of VABYSMO in pediatric patients have not been established.
8.5 Geriatric Use
In the six clinical studies, approximately 58% (1,496/2,571) of patients randomized to treatment with VABYSMO were ≥ 65 years of age. No significant differences in efficacy or safety of faricimab were seen with increasing age in these studies. No dose adjustment is required in patients 65 years and above.
-
11 DESCRIPTION
Faricimab-svoa is a humanized bispecific immunoglobulin G1 (IgG1) antibody that binds both vascular endothelial growth factor A (VEGF-A) and angiopoietin-2 (Ang-2). The fragment crystallizable (Fc) region of faricimab was engineered by selected point mutations to abolish binding interactions with Fcγ and FcRn receptors. Faricimab-svoa has a total molecular weight of approximately 149 kDa and is produced by recombinant DNA technology using mammalian Chinese Hamster Ovary (CHO) cell culture.
VABYSMO (faricimab-svoa) injection is a sterile, clear to opalescent, colorless to brownish-yellow solution in a single-dose prefilled glass syringe or glass vial for intravitreal administration. Each single-dose prefilled syringe or single-dose vial is designed to deliver 0.05 mL (50 microliters) of solution containing 6 mg faricimab-svoa, L-histidine (155 mcg), L-methionine (52.2 mcg), polysorbate 20 (20 mcg), sodium chloride (73.1 mcg), D-sucrose (2.74 mg) and Water for Injection, adjusted to pH 5.5 with acetic acid. The product does not contain an anti-microbial preservative.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Faricimab is a humanized bispecific antibody that acts through inhibition of two pathways by binding to VEGF-A and Ang-2. By inhibiting VEGF-A, faricimab suppresses endothelial cell proliferation, neovascularization and vascular permeability. By inhibiting Ang-2, faricimab is thought to promote vascular stability and desensitize blood vessels to the effects of VEGF-A. Ang-2 levels are increased in some patients with nAMD, DME, and RVO. The contribution of Ang-2 inhibition to the treatment effect and clinical response for nAMD, DME, and RVO has yet to be established.
12.2 Pharmacodynamics
Increased retinal thickness, assessed by optical coherence tomography (OCT), is associated with nAMD, DME and macular edema following RVO. Leakage of blood and fluid from choroidal neovascularization, assessed by fluorescein angiography, is associated with nAMD. Reductions in CST were observed across all treatment arms throughout the six Phase 3 studies in nAMD, DME, and RVO.
12.3 Pharmacokinetics
Absorption/Distribution
Maximum faricimab plasma concentrations (Cmax) are estimated to occur approximately 2 days post-dose. Mean (±SD) free faricimab (unbound to VEGF-A and Ang-2) plasma Cmax are estimated to be 0.23 (0.07) mcg/mL and 0.22 (0.07) mcg/mL in nAMD and in DME patients, respectively. After repeated intravitreal administrations, mean plasma free faricimab trough concentrations are predicted to be 0.002-0.003 mcg/mL for every 8 weeks (Q8W) dosing and 0.021-0.029 mcg/mL for every 4 weeks (Q4W) dosing. Although not directly measured in the vitreous, no accumulation of faricimab is expected in the vitreous and no accumulation has been observed in plasma when faricimab has been administered as repeat doses in the vitreous.
Metabolism/Elimination
Metabolism and elimination of faricimab has not been fully characterized. Faricimab is expected to be catabolized in lysosomes to small peptides and amino acids, which may be excreted renally, in a similar manner to the elimination of endogenous IgG. The estimated mean apparent systemic half-life of faricimab is approximately 7.5 days.
Specific Populations
The systemic pharmacokinetics of faricimab were not influenced by gender, race, or mild to severe renal impairment (i.e., estimated normalized creatinine clearance by Cockroft-Gault equation: 15 to 89 mL/min/1.73 m2). The effect of severe renal impairment or any degree of hepatic impairment on the pharmacokinetics of VABYSMO is unknown. No special dosage modification is required for any of the populations that have been studied (e.g., elderly, gender, race).
Population pharmacokinetic analysis indicated that the pharmacokinetics of faricimab are comparable in nAMD, DME, and RVO patients.
12.6 Immunogenicity
The immunogenicity of VABYSMO was evaluated in plasma samples. The immunogenicity data reflect the percentage of patients whose test results were considered positive for antibodies to VABYSMO in immunoassays. The detection of an immune response is highly dependent on the sensitivity and specificity of the assays used, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to VABYSMO with the incidence of antibodies to other products may be misleading.
There is a potential for an immune response in patients treated with VABYSMO. In the nAMD, DME, and RVO studies, the pre-treatment incidence of anti-faricimab antibodies was approximately 0.8 to 1.8%. After initiation of dosing, the incidence of anti-faricimab antibodies was approximately 8% to 10.4% in patients treated with VABYSMO across studies. As with all therapeutic proteins, there is a potential for immunogenicity with VABYSMO.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenicity data are available for VABYSMO injection in animals or humans.
Based on the anti-VEGF and Ang-2 mechanisms of action, treatment with VABYSMO may pose a risk to reproductive capacity [see Females and Males of Reproductive Potential (8.3)].
-
14 CLINICAL STUDIES
14.1 Neovascular (wet) Age-Related Macular Degeneration (nAMD)
The safety and efficacy of VABYSMO were assessed in two randomized, multi-center, double-masked, active comparator-controlled, 2-year studies (TENAYA – NCT03823287 and LUCERNE – NCT03823300) in patients with nAMD.
A total of 1,329 newly diagnosed, treatment-naïve patients were enrolled in these studies, and 664 patients received at least one dose of VABYSMO. Patient ages ranged from 50 to 99 with a mean of 75.9 years. The studies were identically designed two year studies. Patients were randomized in a 1:1 ratio to one of two treatment arms: 1) aflibercept 2 mg administered fixed every 8 weeks (Q8W) after three initial monthly doses; and VABYSMO 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every 4 weeks (approximately every 28 ± 7 days, monthly) for the first 4 doses, followed by optical coherence tomography and visual acuity evaluations 8 and 12 weeks later to determine whether to give a 6 mg (0.05 mL of 120 mg/mL solution) dose via intravitreal injection on one of the following three regimens: 1) Weeks 28 and 44; (also referred to as Q16W dosing); 2) Weeks 24, 36 and 48 (also referred to as Q12W dosing); or 3) Weeks 20, 28, 36 and 44 (also referred to as Q8W dosing). However, the utility of these criteria to guide dosing intervals has not been established.
At week 48, after 4 initial monthly doses in the VABYSMO arm, 45% of patients received the Weeks 28 and 44 dosing, 33% of patients received the Weeks 24, 36 and 48 dosing, and the remaining 22% of patients received dosing every 8 weeks. These percentages are reflective of what happened within the conduct of these trials and indicate that some patients did well on two (2) doses spaced 16 weeks apart, or three (3) doses spaced 12 weeks apart, but the percentages may not be generalizable to a broader nAMD population for a variety of reasons. The inclusion/exclusion criteria limited enrollment to a select subset of treatment-naïve, newly diagnosed nAMD patients and there is no empirical data that a similar magnitude would be observed if eligibility criteria allowed for broader enrollment. The disease activity criteria, which was instrumental in determining dose frequency, is unvalidated. Stricter criteria would have changed how patients were treated resulting in different percentages of subjects in each dose interval cohort. There was not a similarly dosed aflibercept arm for comparison, which makes the percentages difficult to interpret.
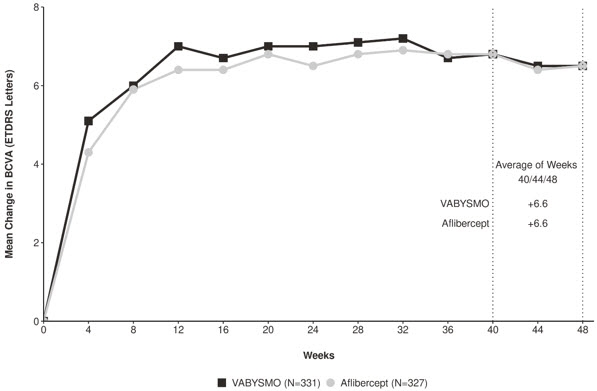
Both studies demonstrated non-inferiority to the comparator control (aflibercept) at the primary endpoint, defined as the mean change from baseline in Best Corrected Visual Acuity (BCVA) when averaged over the week 40, 44, and 48 visits and measured by the Early Treatment Diabetic Retinopathy Study (ETDRS) letter chart. The primary endpoint analysis was a non- inferiority comparison for the mean change in BCVA between the aflibercept and the VABYSMO arm. The lower bound of the 95% confidence interval for the mean change in BCVA could not be lower than minus 4 letters to declare non-inferiority. In both studies, VABYSMO treated patients had a non-inferior mean change from baseline in BCVA compared to patients treated with aflibercept. Detailed results of both studies are shown in Table 2, Figure 1, and Figure 2 below. The clinical efficacy for the second year of the study has not been reviewed.
Table 2: Primary Endpoint Results* in the TENAYA and LUCERNE Studies TENAYA LUCERNE VABYSMO
N = 334Aflibercept
N = 337VABYSMO
N = 331Aflibercept
N = 327BCVA: Best Corrected Visual Acuity ETDRS: Early Treatment Diabetic Retinopathy Study CI: Confidence Interval LS: Least Square - * Average of weeks 40, 44 and 48
Mean change in BCVA as measured by ETDRS letter score from baseline
(95% CI)5.8
(4.6, 7.1)5.1
(3.9, 6.4)6.6
(5.3, 7.8)6.6
(5.3, 7.8)Difference in LS mean
(95% CI)0.7
(-1.1, 2.5)0.0
(-1.7, 1.8)Figure 1: Mean Change in Visual Acuity from Baseline to Week 48 in TENAYA
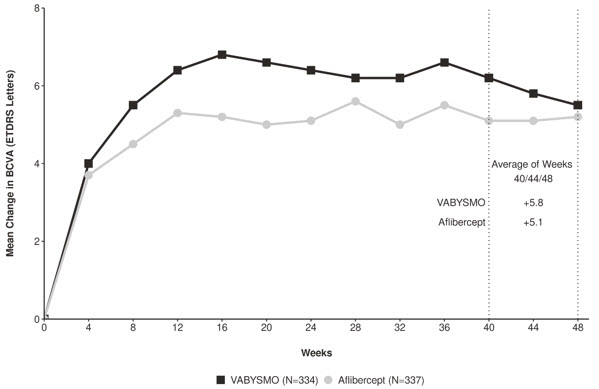
Figure 2: Mean Change in Visual Acuity from Baseline to Week 48 in LUCERNE
Treatment effects in evaluable subgroups (e.g., age, gender, race, baseline visual acuity) in each study were consistent with the results in the overall population.
14.2 Diabetic Macular Edema (DME)
The safety and efficacy of VABYSMO were assessed in two randomized, multi-center, double-masked, active comparator-controlled 2-year studies (YOSEMITE – NCT03622580 and RHINE – NCT03622593) in patients with DME.
A total of 1,891 diabetic patients were enrolled in the two studies with a total of 1,262 patients treated with at least one dose of VABYSMO. Patient ages ranged from 24 to 91 with a mean of 62.2 years. The overall population included both anti-VEGF naïve patients (78%) and patients who had been previously treated with a VEGF inhibitor prior to study participation (22%).
The studies were identically designed two year studies. Patients were randomized in a 1:1:1 ratio to one of three treatment regimens: 1) aflibercept Q8W, patients received fixed aflibercept 2 mg administered every 8 weeks (Q8W) after the first five monthly doses; 2) VABYSMO Q8W, patients received fixed VABYSMO 6 mg administered Q8W after the first six monthly doses; and 3) VABYSMO Variable, patients received VABYSMO 6 mg administered every 4 weeks for at least 4 doses and until the central subfield thickness (CST) of the macula measured by optical coherence tomography was less than approximately 325 microns, then the interval of dosing was modified by up to 4 week interval extensions or reductions of up to 8 week interval increments based on CST and visual acuity disease activity criteria at study drug dosing visits.
After 4 initial monthly doses, the patients in the VABYSMO Variable arm received between a minimum of 1 and a maximum of 21 total injections (median of 7 injections) through Week 96 inclusive. At Week 56, 32% of patients had completed at least one Q12W interval followed by one full Q16W interval. Seventeen percent (17%) of patients were treated on Q8W and/or Q4W dosing intervals through Week 56 (7% only on Q4W). These percentages are reflective of what happened within the conduct of these trials, but the percentages may not be generalizable to a broader DME population.
The inclusion/exclusion criteria limited enrollment to a select subset of DME patients and there is no empirical data that a similar magnitude would be observed if eligibility criteria allowed for broader enrollment. The disease activity criteria, which were instrumental in determining dose frequency, are unvalidated. Different criteria would have changed how patients were treated resulting in different percentages of subjects in each dose interval cohort. There was not a similarly dosed aflibercept arm for comparison which makes the percentages difficult to interpret.
Both studies demonstrated non-inferiority to the comparator control (aflibercept) at the primary endpoint, defined as the mean change from baseline in BCVA at year 1 (average of the Week 48, 52, and 56 visits), measured by the ETDRS Letter Score. The primary endpoint analysis was a non-inferiority comparison for the mean change in BCVA between the aflibercept and VABYSMO arms. The lower bound of the 97.5% confidence interval for the mean change in BCVA could not be lower than minus 4 letters to declare non-inferiority. In both studies, VABYSMO Q8W and VABYSMO Variable treated patients had a non-inferior mean change from baseline in BCVA to the patients treated with aflibercept Q8W at the year 1 primary endpoint. Detailed results of both studies are shown in Table 3, Figure 3, and Figure 4 below.
Table 3: Efficacy Results at Year 1* and at Year 2† in the YOSEMITE and RHINE Studies YOSEMITE RHINE Year 1 Year 2 Year 1 Year 2 VABYSMO
Q8W
N = 315VABYSMO
Variable
N = 313Aflibercept
Q8W
N = 312VABYSMO Q8W
N = 262VABYSMO Variable
N = 270Aflibercept Q8W
N = 259VABYSMO
Q8W
N = 317VABYSMO
Variable
N = 319Aflibercept
Q8W
N = 315VABYSMO Q8W
N = 259VABYSMO Variable
N = 282Aflibercept Q8W
N = 254BCVA: Best Corrected Visual Acuity ETDRS: Early Treatment Diabetic Retinopathy Study CI: Confidence Interval LS: Least Square - * Average of Weeks 48, 52, 56
- † Average of Weeks 92, 96, 100
- ‡ A non-inferiority margin was not available for year 2
Mean change in BCVA as measured by ETDRS letter score from baseline
(97.5% CI year 1 and 95% CI year 2)10.7
(9.4, 12.0)11.6
(10.3, 12.9)10.9
(9.6, 12.2)10.7
(9.4, 12.1)10.7
(9.4, 12.1)11.4
(10.0, 12.7)11.8
(10.6, 13.0)10.8
(9.6, 11.9)10.3
(9.1, 11.4)10.9
(9.5, 12.3)10.1
(8.7, 11.5)9.4
(7.9, 10.8)Difference in LS mean
(97.5% CI year 1 and 95% CI year 2)-0.2
(-2.0, 1.6)0.7
(-1.1, 2.5)-0.7‡ -0.7‡ 1.5
(-0.1, 3.2)0.5
(-1.1, 2.1)1.5‡ 0.7‡ Figure 3: Mean Change in Visual Acuity from Baseline to Year 2 (Week 100) in YOSEMITE
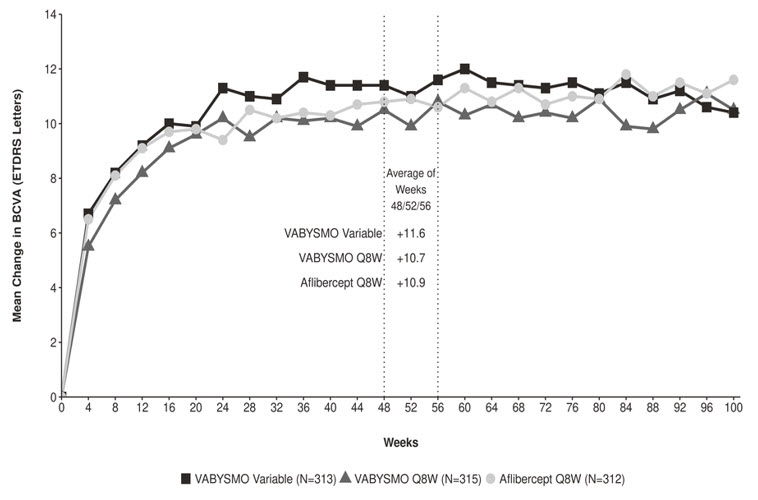
Figure 4: Mean Change in Visual Acuity from Baseline to Year 2 (Week 100) in RHINE
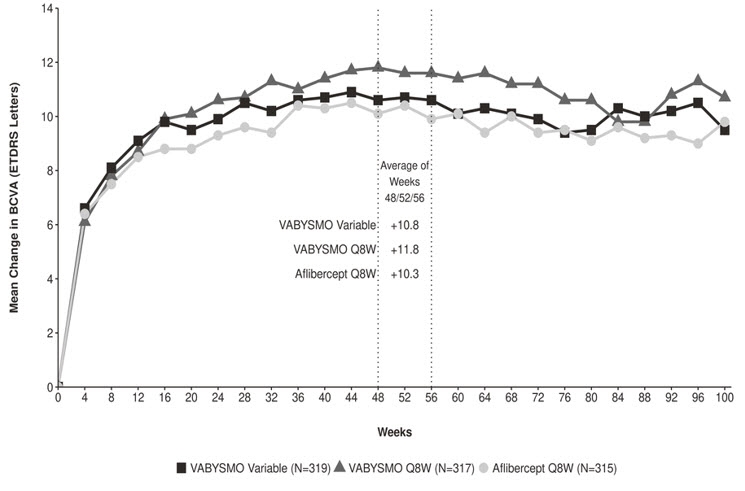
Treatment effects in the subgroup of patients who were anti-VEGF naïve prior to study participation were similar to those observed in the overall population. Treatment effects in evaluable subgroups (e.g., by age, gender, race, baseline HbA1c, baseline visual acuity) in each study were generally consistent with the results in the overall population.
14.3 Macular Edema Following Retinal Vein Occlusion (RVO)
The safety and efficacy of VABYSMO were assessed in two randomized, multicenter, double-masked, studies (BALATON – NCT04740905 in patients with macular edema following branch retinal vein occlusion, and COMINO – NCT04740931 in patients with macular edema following central retinal vein occlusion/hemiretinal vein occlusion). Active comparator-controlled data are available through month 6.
A total of 1,282 newly diagnosed, treatment-naïve patients were enrolled in these studies, of which 641 patients received at least one dose of VABYSMO through 6 months. Patient ages ranged from 28 to 93 with a mean of 64 years, and 22 to 100 with a mean of 65 years in BALATON and COMINO, respectively.
In both studies, patients were randomized in a 1:1 ratio to either 6 mg VABYSMO administered every 4 weeks, or the control arm receiving aflibercept 2 mg injections every 4 weeks for a total of 6 injections.
In both studies, the VABYSMO 6 mg Q4W arm demonstrated non-inferiority to the comparator control (aflibercept) arm for the primary endpoint, which was defined as the change from baseline in BCVA at week 24, measured by the ETDRS Letter Score. The primary endpoint analysis was a non-inferiority comparison for the mean change in BCVA between the aflibercept and VABYSMO arms, where the lower bound of the 95% confidence interval for the mean change in BCVA could not be lower than minus 4 letters to declare non-inferiority.
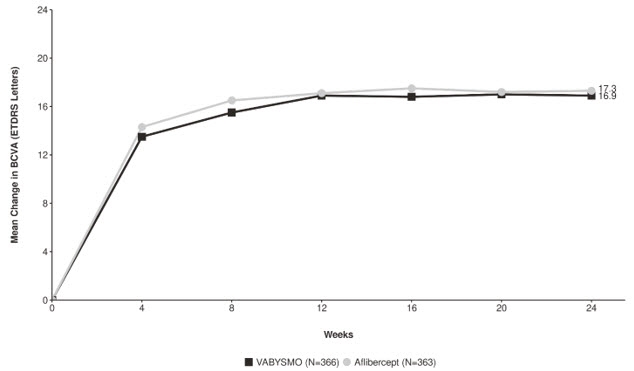
Detailed results for both BALATON and COMINO studies are shown in Table 4, Figure 5, and Figure 6 below.
Table 4: Primary Endpoint Results at Week 24 in the BALATON and COMINO Studies BALATON COMINO VABYSMO
N = 276Aflibercept
N = 277VABYSMO
N = 366Aflibercept
N = 363BCVA: Best Corrected Visual Acuity ETDRS: Early Treatment Diabetic Retinopathy Study CI: Confidence Interval LS: Least Square Mean change in BCVA as measured by ETDRS letter score from baseline
(95% CI)16.9
(15.7, 18.1)17.5
(16.3, 18.6)16.9
(15.4, 18.3)17.3
(15.9, 18.8)Difference in LS mean
(95% CI)-0.6
(-2.2, 1.1)-0.4
(-2.5, 1.6)Figure 5: Mean Change in Visual Acuity from Baseline to Week 24 in BALATON
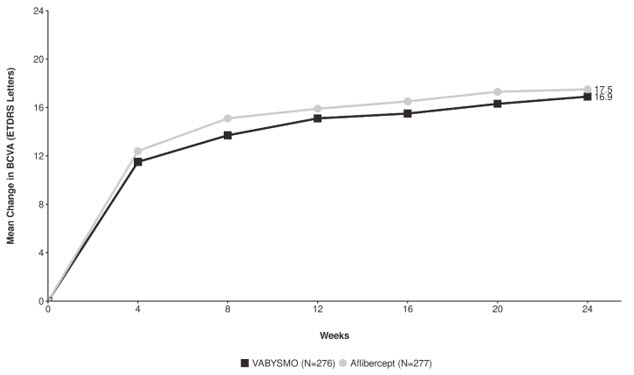
Figure 6: Mean Change in Visual Acuity from Baseline to Week 24 in COMINO
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
VABYSMO (faricimab-svoa) injection is supplied as a clear to opalescent, colorless to brownish-yellow solution as 6 mg (0.05 mL of 120 mg/mL solution) in a single-dose prefilled syringe or single-dose vial. Each prefilled syringe or vial is for treatment of a single eye.
VABYSMO is supplied in the following presentations:
NDC NUMBER CARTON TYPE CARTON CONTENTS 50242-096-06 Prefilled Syringe one 6 mg (0.05 mL of 120 mg/mL solution) single-dose prefilled glass syringe, in a sealed tray
one sterile injection filter needle (30-gauge × ½ inch, 0.30 mm × 12.7 mm, Extra Thin Wall)
one Prescribing Information50242-096-01 Vial one 6 mg (0.05 mL of 120 mg/mL solution) single-dose glass vial
one sterile 5-micron blunt transfer filter needle (18-gauge × 1½ inch, 1.2 mm × 40 mm)
one Prescribing Information16.2 Storage and Handling
Store VABYSMO in the refrigerator between 2°C to 8°C (36°F to 46°F). Do not freeze. Do not shake. Keep the sealed tray containing the prefilled syringe or the vial in the original carton to protect from light.
Prior to use, the unopened prefilled syringe or glass vial of VABYSMO may be kept at room temperature, 20°C to 25°C (68°F to 77°F), for up to 24 hours. Ensure that the injection is given immediately after preparation of the dose.
-
17 PATIENT COUNSELING INFORMATION
Advise patients that in the days following VABYSMO administration, patients are at risk of developing endophthalmitis, retinal detachment, intraocular inflammation and retinal vasculitis with or without retinal vascular occlusion. If the eye becomes red, sensitive to light, painful, or develops a change in vision, advise the patient to seek immediate care from an ophthalmologist [see Warnings and Precautions (5)].
Patients may experience temporary visual disturbances after an intravitreal injection with VABYSMO and the associated eye examinations [see Adverse Reactions (6)]. Advise patients not to drive or use machinery until visual function has recovered sufficiently.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 6 mg Vial Carton
Vabysmo®
(faricimab-svoa)
InjectionNDC: 50242-096-01
6 mg (0.05 mL of 120 mg/mL solution)
For Intravitreal Injection. Single-Dose Vial.
Discard Unused Portion.1 single-dose vial
1 transfer filter needleRx only
Genentech11028025

-
PRINCIPAL DISPLAY PANEL - 6 mg Syringe Carton
Vabysmo®
(faricimab-svoa)
InjectionNDC: 50242-096-06
6 mg (0.05 mL of 120 mg/mL solution)
For Intravitreal Injection
Single-Dose Prefilled SyringeATTENTION:
Only use the provided injection filter needle for the administration.
The dose must be set to the 0.05 mL dose mark (see enclosed prescribing information).1 Sterile Prefilled Syringe
1 Sterile Injection Filter NeedleRx only
Genentech11005157

-
INGREDIENTS AND APPEARANCE
VABYSMO
faricimab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50242-096 Route of Administration INTRAVITREAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FARICIMAB (UNII: QC4F7FKK7I) (FARICIMAB - UNII:QC4F7FKK7I) FARICIMAB 6 mg in 0.05 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 155 ug in 0.05 mL METHIONINE (UNII: AE28F7PNPL) 52.2 ug in 0.05 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 73.1 ug in 0.05 mL SUCROSE (UNII: C151H8M554) 2.74 mg in 0.05 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 20 ug in 0.05 mL WATER (UNII: 059QF0KO0R) ACETIC ACID (UNII: Q40Q9N063P) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50242-096-01 1 in 1 CARTON 01/28/2022 1 NDC: 50242-096-03 0.05 mL in 1 VIAL; Type 1: Convenience Kit of Co-Package 2 NDC: 50242-096-86 1 in 1 CARTON 01/28/2022 2 NDC: 50242-096-77 0.05 mL in 1 VIAL; Type 1: Convenience Kit of Co-Package 3 NDC: 50242-096-06 1 in 1 CARTON 07/03/2024 3 0.05 mL in 1 SYRINGE; Type 1: Convenience Kit of Co-Package 4 NDC: 50242-096-45 1 in 1 CARTON 07/03/2024 4 0.05 mL in 1 SYRINGE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761235 01/28/2022 Labeler - Genentech, Inc. (080129000) Registrant - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Roche Diagnostics 323105205 ANALYSIS(50242-096) , API MANUFACTURE(50242-096) Establishment Name Address ID/FEI Business Operations Genentech, Inc. (Oceanside) 146373191 ANALYSIS(50242-096) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd. 485244961 ANALYSIS(50242-096) , LABEL(50242-096) , PACK(50242-096) , MANUFACTURE(50242-096)
Trademark Results [VABYSMO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VABYSMO 97099363 not registered Live/Pending |
Genentech, Inc. 2021-10-29 |
 VABYSMO 97024936 not registered Live/Pending |
Genentech, Inc. 2021-09-13 |
 VABYSMO 90013584 not registered Live/Pending |
Genentech, Inc. 2020-06-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.