LAGEVRIO by A-S Medication Solutions LAGEVRIO
LAGEVRIO by
Drug Labeling and Warnings
LAGEVRIO by is a Prescription medication manufactured, distributed, or labeled by A-S Medication Solutions. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
LAGEVRIO- molnupiravir capsule
A-S Medication Solutions
----------
LAGEVRIO
| FACT SHEET FOR HEALTHCARE PROVIDERS: EMERGENCY USE AUTHORIZATION FOR LAGEVRIO™ (molnupiravir) CAPSULES | |
|---|---|
|
HIGHLIGHTS OF EMERGENCY USE AUTHORIZATION (EUA) | |
| MANDATORY REQUIREMENTS FOR ADMINISTRATION OF LAGEVRIO UNDER EMERGENCY USE AUTHORIZATION
Refer to FULL FACTSHEET for details. | |
|
--------------RECENT MAJOR CHANGES----------- Use in Specific Populations (Section 8.3), Nonclinical Toxicology (Section 13.1): Updated 06/2024 Dosage and Administration (Section 2.3): Addition of administration via gastrostomy and gastrojejunostomy feeding tubes 06/2024 Mandatory Requirements Box, Emergency Use Authorization (Section 1): Updated 10/2023 Limitations of Authorized Use (Section 1): Updated 10/2023 Adverse Reactions: Removal of Section 6.5 10/2023 Adverse Reactions (Section 6.2): update to post-authorization experience section 07/2023 ------------EUA FOR LAGEVRIO----------- The U.S. Food and Drug Administration (FDA) has issued an EUA for the emergency use of the unapproved LAGEVRIO, a nucleoside analogue that inhibits SARS-CoV-2 replication by viral mutagenesis for the treatment of adults with mild-to-moderate coronavirus disease 2019 (COVID-19):
LAGEVRIO is not FDA-approved for any use including for use for the treatment of COVID-19. Prior to initiating treatment with LAGEVRIO, carefully consider the known and potential risks and benefits. (1)
LAGEVRIO may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs in the therapeutic class to which LAGEVRIO belongs (i.e., anti-infectives).
------DOSAGE AND ADMINISTRATION------
|
-----DOSAGE FORMS AND STRENGTHS----- Capsules: 200 mg (3) ------------CONTRAINDICATIONS------------ No contraindications have been identified based on the limited available data on the emergency use of LAGEVRIO authorized under this EUA. (4) ------WARNINGS AND PRECAUTIONS------
-----------ADVERSE REACTIONS----------- Most common adverse reactions (incidence ≥ 1%) are diarrhea, nausea, and dizziness. (6.1)
-------------DRUG INTERACTIONS-------------- No drug interactions have been identified based on the limited available data on the emergency use of LAGEVRIO authorized under this EUA. (7) ----------USE IN SPECIFIC POPULATIONS---------
See FACT SHEET FOR PATIENTS AND CAREGIVERS. |
MANDATORY REQUIREMENTS FOR ADMINISTRATION OF LAGEVRIO UNDER EMERGENCY USE AUTHORIZATION
In order to mitigate the risks of using this unapproved product under the EUA and to optimize the potential benefit of LAGEVRIO, the following steps are required. Use of LAGEVRIO under this EUA is limited to the following (all requirements must be met):
- Treatment of adults with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death and for whom alternative COVID-19 treatment options approved or authorized by FDA are not accessible or clinically appropriate [see Limitations of Authorized Use (1)].
- As the prescribing healthcare provider, review the information contained within the “Fact Sheet for Patients and Caregivers” with your patient or caregiver prior to the patient receiving LAGEVRIO. Healthcare providers must provide the patient/caregiver with an electronic or hard copy of the “Fact Sheet for Patients and Caregivers” prior to the patient receiving LAGEVRIO and must document that the patient/caregiver has been given an electronic or hard copy of the “Fact Sheet for Patients and Caregivers”.
- The prescribing healthcare providers must inform the patient/caregiver that:
- LAGEVRIO is an unapproved drug that is authorized for use under this Emergency Use Authorization.
- Other therapeutics are currently approved for the same use as LAGEVRIO [see Emergency Use Authorization (1) - Information Regarding Available Alternatives for the EUA Authorized Use].
- There are benefits and risks of taking LAGEVRIO as outlined in the “Fact Sheet for Patients and Caregivers.”
- There is a pregnancy registry.
- Females of childbearing potential should use a reliable method of contraception correctly and consistently, as applicable, for the duration of treatment and for 4 days after the last dose of LAGEVRIO.
- Males of reproductive potential who are sexually active with females of childbearing potential should use a reliable method of contraception correctly and consistently during treatment and for at least 3 months after the last dose.
- The prescribing healthcare provider must assess whether a female of childbearing potential is pregnant or not, if clinically indicated [see Warnings and Precautions (5.1) and Use in Specific Populations (8.3)].
- Based on findings from animal reproduction studies, LAGEVRIO may cause fetal harm when administered to pregnant individuals. If LAGEVRIO is used during pregnancy, prescribing healthcare providers must communicate to the patient the known and potential benefits and the potential risks of LAGEVRIO use during pregnancy, as outlined in the “Fact Sheet for Patients and Caregivers” [see Warnings and Precautions (5.1, 5.3), Use in Specific Populations (8.1, 8.3) and Nonclinical Toxicology (13.1)].
- If the decision is made to use LAGEVRIO during pregnancy, the prescriber must document that the known and potential benefits and the potential risks of LAGEVRIO use during pregnancy, as outlined in the “Fact Sheet for Patients and Caregivers,” were discussed with the patient.
- The prescribing healthcare provider must document that a pregnant individual was made aware of the pregnancy registry at https://covid-pr.pregistry.com or 1-800-616-3791.
- The prescribing healthcare provider and/or the provider’s designee is/are responsible for mandatory reporting of all medication errors and serious adverse events potentially related to LAGEVRIO within 7 calendar days from the healthcare provider’s awareness of the event [see Adverse Reactions (6.4)].
For information on clinical studies of LAGEVRIO and other therapies for the treatment of COVID-19, see www.clinicaltrials.gov .
1 EMERGENCY USE AUTHORIZATION
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved product LAGEVRIO™ for treatment of adults with mild-to-moderate coronavirus disease 2019 (COVID-19):
- who are at high risk for progression to severe COVID-19, including hospitalization or death. Refer to CDC website1 for additional details, and for
- whom alternative COVID-19 treatment options approved or authorized by FDA are not accessible or clinically appropriate.
LIMITATIONS OF AUTHORIZED USE
- LAGEVRIO is not authorized for use in patients who are less than 18 years of age [see Warnings and Precautions (5.3)].
- LAGEVRIO is not authorized for initiation of treatment in patients hospitalized due to COVID-192. Benefit of treatment with LAGEVRIO has not been observed in subjects when treatment was initiated after hospitalization due to COVID-19 [see Dosing and Administration (2.1)].
- LAGEVRIO is not authorized for use for longer than 5 consecutive days.
- LAGEVRIO is not authorized for pre-exposure or post-exposure prophylaxis for prevention of COVID-19.
LAGEVRIO may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs in the therapeutic class to which LAGEVRIO belongs (i.e., anti-infectives).
LAGEVRIO is not approved for any use, including for use for the treatment of COVID-19.
Prior to initiating treatment with LAGEVRIO, carefully consider the known and potential risks and benefits [see Warnings and Precautions (5.1, 5.3), Use in Specific Populations (8.1, 8.3) and Nonclinical Toxicology (13.1)].
LAGEVRIO is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of LAGEVRIO under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Justification for Emergency Use of Drugs During the COVID-19 Pandemic
There is currently an outbreak of Coronavirus Disease 2019 (COVID-19) caused by SARS-CoV-2, a novel coronavirus. The Secretary of HHS has:
- Determined that there is a public health emergency, or significant potential for a public health emergency, related to COVID-193.
- Declared that circumstances exist justifying the authorization of emergency use of drugs and biological products for the prevention or treatment of COVID-194.
An EUA is a FDA authorization for the emergency use of an unapproved product or unapproved use of an approved product (i.e., drug, biological product, or device) in the United States under certain circumstances including, but not limited to, when the Secretary of HHS declares that there is a public health emergency that affects the national security or the health and security of United States citizens living abroad, and that involves biological agent(s) or a disease or condition that may be attributable to such agent(s). Criteria for issuing an EUA include:
- The biological agent(s) can cause a serious or life-threatening disease or condition;
- Based on the totality of the available scientific evidence (including data from adequate and well-controlled clinical trials, if available), it is reasonable to believe that
- the product may be effective in diagnosing, treating, or preventing the serious or life-threatening disease or condition; and
- the known and potential benefits of the product - when used to diagnose, prevent, or treat such disease or condition - outweigh the known and potential risks of the product, taking into consideration the material threat posed by the biological agent(s);
- There is no adequate, approved, and available alternative to the product for diagnosing, preventing, or treating the serious or life-threatening disease or condition.
APPROVED AVAILABLE ALTERNATIVES
Veklury (remdesivir) is FDA-approved for the treatment of COVID-19 in adults and pediatric patients (at least 28 days old and weighing at least 3 kg) who are not hospitalized and have mild-to-moderate COVID-19, and who are at high risk for progression to severe COVID-19, including hospitalization or death. Veklury is administered via intravenous infusion for a total treatment duration of 3 days.
Although Veklury is an approved alternative to LAGEVRIO for the treatment of mild-to-moderate COVID-19 in adults and who are at high risk for progression to severe COVID-19, including hospitalization or death, FDA does not consider Veklury to be an adequate alternative to LAGEVRIO for this authorized use because it may not be feasible or clinically appropriate for certain patients (e.g., it requires a 3-day intravenous treatment duration).
Paxlovid (nirmatrelvir tablets; ritonavir tablets co-packaged for oral use) is FDA-approved for the treatment of mild-to-moderate COVID-19 in adults who are at high risk for progression to severe COVID-19, including hospitalization or death. Paxlovid is administered orally for a total treatment duration of 5 days. Although Paxlovid is an approved alternative to LAGEVRIO for the treatment of mild-to-moderate COVID-19 in adults and who are at high risk for progression to severe COVID-19, including hospitalization or death, FDA does not consider Paxlovid to be an adequate alternative to LAGEVRIO for this authorized use because it may not be clinically appropriate for patients on medications that are primarily metabolized by CYP3A and/or that are strong CYP3A inducers.
For additional information on all products authorized for treatment or prevention of COVID-19, please see https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization .
For information on clinical studies of LAGEVRIO and other therapies for the treatment of COVID-19, see www.clinicaltrials.gov.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage for Emergency Use of LAGEVRIO in Adult Patients
The dosage in adult patients is 800 mg (four 200 mg capsules) taken orally every 12 hours for 5 days, with or without food [see Clinical Pharmacology (12.3)]. Take LAGEVRIO as soon as possible after a diagnosis of COVID-19 has been made, and within 5 days of symptom onset [see Emergency Use Authorization (1) and Clinical Studies (14)].
Completion of the full 5-day treatment course and continued isolation in accordance with public health recommendations are important to maximize viral clearance and minimize transmission of SARS-CoV-2 [see Patient Counseling Information (17)].
LAGEVRIO is not authorized for use for longer than 5 consecutive days because the safety and efficacy have not been established.
If the patient misses a dose of LAGEVRIO within 10 hours of the time it is usually taken, the patient should take it as soon as possible and resume the normal dosing schedule. If the patient misses a dose by more than 10 hours, the patient should not take the missed dose and instead take the next dose at the regularly scheduled time. The patient should not double the dose to make up for a missed dose.
Should a patient require hospitalization after starting treatment with LAGEVRIO, the patient may complete the full 5 day treatment course per the healthcare provider’s discretion.
2.2 Dosage Adjustments in Specific Populations
No dosage adjustment is recommended based on renal or hepatic impairment or in geriatric patients [see Use in Specific Populations (8.5, 8.6, 8.7)].
2.3 Administration via Nasogastric (NG), Orogastric (OG) and Gastrostomy (G) Tubes that are 12F or Larger or via Gastrojejunostomy (GJ) Tubes that are 14F or Larger
- Open four (4) capsules and transfer contents into a clean container with a lid.
- Add 40 mL of water to the container.
- Put the lid on the container and shake to mix the capsule contents and water thoroughly for 3 minutes.
- NOTE: Capsule contents may not dissolve completely.
- The prepared mixture may have visible undissolved particulates and are acceptable for administration.
- Flush the tube with 5 mL of water prior to administration.
- Using an appropriate syringe, draw up the entire contents from the container and administer immediately through the tube. If using a GJ tube, administer through the gastric port. Do not keep the mixture for future use.
- If any portion of the capsule contents are left in the container, add 10 mL of water to the container, mix, and using the same syringe draw up the entire contents of the container and administer through the tube. Repeat as needed until no capsule contents are left in the container or syringe.
- Flush the tube with 5 mL of water twice (10 mL total) after administration of the mixture.
3 DOSAGE FORMS AND STRENGTHS
Capsules: 200 mg, Swedish Orange opaque size 0 capsules. The capsules have the corporate logo and “82” printed in white ink.
4 CONTRAINDICATIONS
No contraindications have been identified based on the limited available data on the emergency use of LAGEVRIO authorized under this EUA.
5 WARNINGS AND PRECAUTIONS
There are limited clinical data available for LAGEVRIO. Serious and unexpected adverse events may occur that have not been previously reported with LAGEVRIO use.
5.1 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, LAGEVRIO may cause fetal harm when administered to pregnant individuals. There are no available human data on the use of LAGEVRIO in pregnant individuals to evaluate the risk of major birth defects, miscarriage or adverse maternal or fetal outcomes; therefore, LAGEVRIO is not recommended for use during pregnancy. When considering LAGEVRIO for a pregnant individual, the prescribing healthcare provider must communicate the known and potential benefits and the potential risks of using LAGEVRIO during pregnancy to the pregnant individual. LAGEVRIO is authorized to be prescribed to a pregnant individual only after the healthcare provider has determined that the benefits would outweigh the risks for that individual patient. If the decision is made to use LAGEVRIO during pregnancy, the prescribing healthcare provider must document that the known and potential benefits and the potential risks of using LAGEVRIO during pregnancy were communicated to the pregnant individual.
Advise individuals of childbearing potential of the potential risk to a fetus and to use an effective method of contraception correctly and consistently, as applicable, during treatment with LAGEVRIO and for 4 days after the final dose [see Use in Specific Populations (8.1, 8.3 and Nonclinical Toxicology (13.1)].
Prior to initiating treatment with LAGEVRIO, assess whether an individual of childbearing potential is pregnant or not, if clinically indicated. Pregnancy status does not need to be confirmed in patients who have undergone permanent sterilization, are currently using an intrauterine system or contraceptive implant, or in whom pregnancy is not possible. In all other patients, assess whether the patient is pregnant based on the first day of last menstrual period in individuals who have regular menstrual cycles, is using a reliable method of contraception correctly and consistently or have had a negative pregnancy test. A pregnancy test is recommended if the individual has irregular menstrual cycles, is unsure of the first day of last menstrual period or is not using effective contraception correctly and consistently [see Box].
5.2 Hypersensitivity Including Anaphylaxis
Hypersensitivity reactions, including anaphylaxis, have been reported with LAGEVRIO. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue LAGEVRIO and initiate appropriate medications and/or supportive care.
5.3 Bone and Cartilage Toxicity
LAGEVRIO is not authorized for use in patients less than 18 years of age because it may affect bone and cartilage growth. Bone and cartilage toxicity was observed in rats after repeated dosing [see Nonclinical Toxicity (13.2)]. The safety and efficacy of LAGEVRIO have not been established in pediatric patients [see Use in Specific Populations (8.4)].
6 ADVERSE REACTIONS
6.1 Adverse Reactions from Clinical Studies
The following adverse reactions have been observed in the clinical study of LAGEVRIO that supported the EUA. The adverse reaction rates observed in these clinical trials cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. Additional adverse events associated with LAGEVRIO may become apparent with more widespread use.
Overall, more than 900 subjects have been exposed to LAGEVRIO 800 mg twice daily in clinical trials. The safety assessment of LAGEVRIO is primarily based on an analysis from subjects followed through Day 29 in the Phase 3 study in non-hospitalized subjects with COVID-19 (MOVe-OUT) [see Clinical Studies (14)].
The safety of LAGEVRIO was evaluated based on an analysis of a Phase 3 double-blind trial (MOVe-OUT) in which 1,411 non-hospitalized subjects with COVID-19 were randomized and treated with LAGEVRIO (N=710) or placebo (N=701) for up to 5 days. Adverse events were those reported while subjects were on study intervention or within 14 days of study intervention completion/discontinuation.
Discontinuation of study intervention due to an adverse event occurred in 1% of subjects receiving LAGEVRIO and 3% of subjects receiving placebo. Serious adverse events occurred in 7% of subjects receiving LAGEVRIO and 10% receiving placebo; most serious adverse events were COVID-19 related. Adverse events leading to death occurred in 2 (<1%) subjects receiving LAGEVRIO and 12 (2%) of subjects receiving placebo.
The most common adverse reactions in the LAGEVRIO treatment group in MOVe-OUT are presented in Table 1, all of which were Grade 1 (mild) or Grade 2 (moderate).
| LAGEVRIO N=710 | Placebo N=701 |
|
|---|---|---|
|
|
||
| Diarrhea | 2% | 2% |
| Nausea | 1% | 1% |
| Dizziness | 1% | 1% |
Laboratory Abnormalities
Selected Grade 3 and 4 laboratory abnormalities in chemistry (alanine aminotransferase, aspartate aminotransferase, creatinine, and lipase) and hematology (hemoglobin, platelets, and leukocytes) parameters all occurred at a rate of less than or equal to 2% and occurred at a similar rate across arms in MOVe-OUT.
6.2 Post-Authorization Experience
The following adverse reactions have been identified during post-authorization use of LAGEVRIO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders
vomiting
Immune System Disorders
hypersensitivity, anaphylaxis, angioedema [see Warnings and Precautions (5.2)]
Skin and Subcutaneous Tissue Disorders
erythema, pruritus, rash, urticaria
6.4 Required Reporting for Serious Adverse Events and Medication Errors
The prescribing healthcare provider and/or the provider’s designee is/are responsible for mandatory reporting of all serious adverse events* and medication errors potentially related to LAGEVRIO within 7 calendar days from the healthcare provider’s awareness of the event, using FDA Form 3500 (for information on how to access this form, see below). The FDA requires that such reports, using FDA Form 3500, include the following:
- Patient demographics and baseline characteristics (e.g., patient identifier, age or date of birth, gender, weight, ethnicity, and race)
- A statement "LAGEVRIO use for COVID-19 under Emergency Use Authorization (EUA)” under the “Describe Event, Problem, or Product Use/Medication Error” heading
- Information about the serious adverse event or medication error (e.g., signs and symptoms, test/laboratory data, complications, timing of drug initiation in relation to the occurrence of the event, duration of the event, treatments required to mitigate the event, evidence of event improvement/disappearance after stopping or reducing the dosage, evidence of event reappearance after reintroduction, clinical outcomes).
- Patient’s preexisting medical conditions and use of concomitant products
- Information about the product (e.g., dosage, route of administration, NDC #).
Submit adverse event and medication error reports, using Form 3500, to FDA MedWatch using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax to 1-800-FDA-0178, or
- Call 1-800-FDA-1088 to request a reporting form
In addition, please provide a copy of all FDA MedWatch forms to:
Merck Sharp & Dohme LLC, Rahway, NJ USA
Fax: 215-616-5677
E-mail: dpoc.usa@msd.com
The prescribing healthcare provider and/or the provider’s designee is/are responsible for mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of LAGEVRIO.
*Serious adverse events are defined as:
- Death;
- A life-threatening adverse event;
- Inpatient hospitalization or prolongation of existing hospitalization;
- A persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- A congenital anomaly/birth defect;
- Other important medical event, which may require a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
7 DRUG INTERACTIONS
No drug interactions have been identified based on the limited available data on the emergency use of LAGEVRIO authorized under this EUA. No clinical drug-drug interaction trials of LAGEVRIO with concomitant medications, including other treatments for mild-to-moderate COVID-19, have been conducted [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Registry
There is a pregnancy registry that monitors pregnancy outcomes in individuals exposed to LAGEVRIO during pregnancy. The prescribing healthcare provider must document that a pregnant individual was made aware of the pregnancy registry at https://covid-pr.pregistry.com or 1-800-616-3791. Pregnant individuals exposed to LAGEVRIO or their healthcare providers can also report the exposure by contacting Merck Sharp & Dohme LLC, Rahway, NJ USA at 1-877-888-4231.
Risk Summary
Based on animal data, LAGEVRIO may cause fetal harm when administered to pregnant individuals. There are no available human data on the use of LAGEVRIO in pregnant individuals to evaluate the risk of major birth defects, miscarriage or adverse maternal or fetal outcomes; therefore, LAGEVRIO is not recommended during pregnancy [see Box and Warnings and Precautions (5.1)]. In an animal reproduction study, oral administration of molnupiravir to pregnant rats during the period of organogenesis resulted in embryofetal lethality and teratogenicity at 8 times the human NHC (N4-hydroxycytidine) exposures at the recommended human dose (RHD) and reduced fetal growth at ≥ 3 times the human NHC exposure at the RHD. Oral administration of molnupiravir to pregnant rabbits during the period of organogenesis resulted in reduced fetal body weights at 18 times the human NHC exposure at the RHD (see Data). When considering LAGEVRIO for a pregnant individual, the prescribing healthcare provider must communicate the known and potential benefits and the potential risks of using LAGEVRIO during pregnancy to the pregnant individual. LAGEVRIO may only be prescribed to a pregnant individual after the prescribing healthcare provider has determined that the benefits would outweigh the risks for that individual patient. If the decision is made to use LAGEVRIO during pregnancy, the prescribing healthcare provider must document that the known and potential benefits and potential risks of using LAGEVRIO during pregnancy were communicated to the pregnant individual [see Box]. There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
COVID-19 in pregnancy is associated with adverse maternal and fetal outcomes, including preeclampsia, eclampsia, preterm birth, premature rupture of membranes, venous thromboembolic disease, and fetal death.
Data
Animal Data
In an embryofetal development (EFD) study in rats, molnupiravir was administered orally to pregnant rats at 0, 100, 250, or 500 mg/kg/day from gestation days (GDs) 6 to 17. Molnupiravir was also administered orally to pregnant rats at up to 1,000 mg/kg/day from GDs 6 to 17 in a preliminary EFD study. Developmental toxicities included post-implantation losses, malformations of the eye, kidney, and axial skeleton, and rib variations at 1,000 mg/kg/day (8 times the human NHC exposure at the RHD) and decreased fetal body weights and delayed ossification at ≥500 mg/kg/day (3 times the human NHC exposure at the RHD). There were no developmental toxicities at ≤250 mg/kg/day (less than the human NHC exposure at the RHD). Maternal toxicities included decreased food consumption and body weight losses, resulting in the early sacrifice of two of sixteen animals at 1,000 mg/kg/day, and decreased body weight gain at 500 mg/kg/day.
In an EFD study in rabbits, molnupiravir was administered orally to pregnant rabbits at 0, 125, 400, or 750 mg/kg/day from GDs 7 to 19. Developmental toxicity was limited to reduced fetal body weights at 750 mg/kg/day (18 times the human NHC exposures at the RHD). There was no developmental toxicity at ≤400 mg/kg/day (7 times the human NHC exposures at the RHD). Maternal toxicities included reduced food consumption and body weight gains, and abnormal fecal output at 750 mg/kg/day.
In a pre- and post-natal developmental study, molnupiravir was administered orally to female rats at doses up to 500 mg/kg/day (similar to the human NHC exposure at the RHD) from GD6 through lactation day 20. No effects were observed in offspring.
8.2 Lactation
Risk Summary
There are no data on the presence of molnupiravir or its metabolites in human milk. NHC was detected in the plasma of nursing pups from lactating rats administered molnupiravir (see Data). It is unknown whether molnupiravir has an effect on the breastfed infant or effects on milk production.
Based on the potential for adverse reactions in the infant from LAGEVRIO, breastfeeding is not recommended during treatment with LAGEVRIO and for 4 days after the final dose. A lactating individual may consider interrupting breastfeeding and may consider pumping and discarding breast milk during treatment and for 4 days after the last dose of LAGEVRIO [see Warnings and Precautions (5.1, 5.3)].
Data
When molnupiravir was administered to lactating rats at ≥250 mg/kg/day in the pre- and post-natal development study, NHC was detected in plasma of nursing pups.
8.3 Females and Males of Reproductive Potential
Based on animal studies, LAGEVRIO may cause fetal harm when administered to a pregnant individual.
Pregnancy Testing
Prior to initiating treatment with LAGEVRIO, assess whether an individual of childbearing potential is pregnant or not, if clinically indicated [see Warnings and Precautions (5.1)].
Contraception
Females
Advise individuals of childbearing potential to use a reliable method of contraception correctly and consistently, as applicable for the duration of treatment and for 4 days after the last dose of LAGEVRIO [see Warnings and Precautions (5.1)].
Males
While the risk is regarded as low, there is a theoretical risk for LAGEVRIO to affect offspring of treated males based on its mechanism of action. Advise sexually active individuals with partners of childbearing potential to use a reliable method of contraception correctly and consistently during treatment and for at least 3 months after the last dose of LAGEVRIO.
The risk beyond three months after the last dose of LAGEVRIO is unknown.
Molnupiravir was equivocal (neither clearly positive nor negative) in one in vivo mutagenicity assay of reticulocytes and RBCs which are used to reflect prior effects on hematopoietic stem cells in bone marrow. Molnupiravir was not mutagenic when assessed in in vivo assays of liver (somatic cells), bone marrow (somatic cells and stem cells), and sperm (male germ cells) from transgenic rats administered molnupiravir for 28 days [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
LAGEVRIO is not authorized for use in patients less than 18 years of age.
Bone and cartilage toxicity were observed in a 3-month, repeat-dose toxicology study in rats. The safety and efficacy of LAGEVRIO have not been established in pediatric patients [see Warnings and Precautions (5.3) and Nonclinical Toxicology (13.2)].
8.5 Geriatric Use
In MOVe-OUT, there was no difference in safety and tolerability between patients ≥65 years of age and younger patients who were treated with LAGEVRIO. No dosage adjustment is recommended based on age. The PK of NHC was similar in geriatric patients compared to younger patients [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment in patients with any degree of renal impairment is recommended. Renal clearance is not a meaningful route of elimination for NHC. Mild or moderate renal impairment did not have a meaningful impact on the PK of NHC. While the PK of NHC has not been evaluated in patients with eGFR less than 30 mL/min/1.73m2 or on dialysis, severe renal impairment, and end-stage renal disease (ESRD) are not expected to have a significant effect on NHC exposure [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment in patients with hepatic impairment is recommended. Preclinical data indicate that hepatic elimination is not expected to be a major route of NHC elimination therefore, hepatic impairment is unlikely to affect NHC exposure [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
There is no human experience of overdosage with LAGEVRIO. Treatment of overdose with LAGEVRIO should consist of general supportive measures including the monitoring of the clinical status of the patient. Hemodialysis is not expected to result in effective elimination of NHC.
11 DESCRIPTION
LAGEVRIO capsules contain molnupiravir, a nucleoside analogue that inhibits SARS-CoV-2 replication by viral mutagenesis and is the 5´-isobutyrate ester of the ribonucleoside analog N4-hydroxycytidine (NHC).
The chemical name for molnupiravir is {(2R,3S,4R,5R)-3,4-Dihydroxy-5-[(4Z)-4-(hydroxyimino)-2-oxo-3,4-dihydropyrimidin-1(2H)-yl]oxolan-2-yl}methyl 2-methylpropanoate. It has an empirical formula of C13H19N3O7 and its molecular weight is 329.31 g/mol. Its structural formula is:
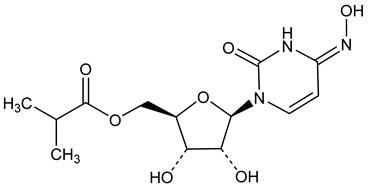
Molnupiravir is a white to off-white powder that is soluble in water.
Each LAGEVRIO capsule, for oral use, contains 200 mg of molnupiravir and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose and purified water. The capsule shell is made of hypromellose, red iron oxide and titanium dioxide. The capsule is printed with white ink made of butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, shellac, strong ammonia solution and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Molnupiravir is a prodrug with antiviral activity against SARS-CoV-2. It is metabolized to the cytidine nucleoside analogue, NHC which distributes into cells where NHC is phosphorylated to form the pharmacologically active ribonucleoside triphosphate (NHC-TP). NHC-TP incorporation (as NHC-monophosphate [NHC-MP]) into SARS-CoV-2 RNA by the viral RNA polymerase (nsp12) results in an accumulation of errors in the viral genome leading to inhibition of replication. The mechanism of action (known as viral error catastrophe or viral lethal mutagenesis) is supported by biochemical and cell culture data, studies of SARS-CoV-2 infection in animal models, and analyses of SARS-CoV-2 genome sequences in human subjects treated with LAGEVRIO.
12.2 Pharmacodynamics
The relationship between NHC and intracellular NHC-TP with antiviral efficacy has not been evaluated clinically.
12.3 Pharmacokinetics
Molnupiravir is a 5´-isobutyrate prodrug of NHC that is hydrolyzed during or after absorption. NHC, the primary circulating analyte, is taken up by cells and anabolized to NHC-TP. NHC is eliminated by metabolism to uridine and/or cytidine through the same pathways involved in endogenous pyrimidine metabolism. NHC pharmacokinetics are shown in Table 2.
Plasma NHC concentrations in patients (N=5) following administration of molnupiravir via nasogastric or orogastric tube fell within the range of NHC concentrations following oral molnupiravir capsule administration under the same dosing regimen.
| NHC Geometric Mean (%CV) | |
|---|---|
| Values were obtained from a Phase 1 study of healthy subjects, unless otherwise indicated. | |
|
|
|
| Pharmacokinetics in Patients | |
| AUC0-12hr (ng*hr/mL)* | 8260 (41.0) |
| Cmax (ng/mL)* | 2330 (36.9) |
| C12hr (ng/mL)* | 31.1 (124) |
| Pharmacokinetics in Healthy Subjects | |
| AUC0-12hr (ng*hr/mL) | 8330 (17.9) |
| Cmax (ng/mL) | 2970 (16.8) |
| C12hr (ng/mL) | 16.7 (42.8) |
| AUC Accumulation Ratio | 1.09 (11.8) |
| Absorption | |
| Tmax (hr)† | 1.50 [1.00 – 2.02] |
| Effect of Food | 35% reduction in Cmax, no effect on AUC |
| Distribution | |
| Plasma Protein Binding (in vitro) | 0% |
| Apparent Volume of Distribution (L)* | 142 |
| Elimination | |
| Effective t1/2 (hr) | 3.3 |
| Apparent Clearance (L/hr)* | 76.9 |
| Fraction of dose excreted in urine over the time interval of 0-12 hours | 3% (81.6%) |
Specific Populations
Population PK analysis results indicated that age, sex, race, ethnicity, or disease severity do not meaningfully influence the PK of NHC.
Pediatric Patients
LAGEVRIO has not been studied in pediatric patients.
Patients with Renal Impairment
Renal clearance is not a meaningful route of elimination for NHC. In a population PK analysis, mild or moderate renal impairment did not have a meaningful impact on the PK of NHC. The PK of molnupiravir and NHC has not been evaluated in patients with eGFR less than 30 mL/min/1.73m2 or on dialysis.
Patients with Hepatic Impairment
The PK of molnupiravir and NHC has not been evaluated in patients with moderate and severe hepatic impairment. Preclinical data indicate that hepatic elimination is not expected to be a major route of NHC elimination; therefore, hepatic impairment is unlikely to affect NHC exposure.
Drug Interaction Studies
In vitro study results indicated that molnupiravir and NHC are not substrates of CYP enzymes or human P-gp and BCRP transporters. In vitro study results also indicated that molnupiravir and NHC are not inhibitors of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4 or inhibitors of OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, MATE1, MATE2K, MRP2, MDR1 and BCRP or inducers of CYP1A2, 2B6, and 3A4. The interaction between molnupiravir with concomitant medications, including other treatments for mild-to-moderate COVID-19, has not been evaluated.
12.4 Microbiology
Antiviral Activity
NHC, the nucleoside analogue metabolite of molnupiravir, was active in cell culture assays against SARS-CoV-2 (USA-WA1/2020 isolate) with 50% effective concentrations (EC50 values) ranging between 0.67 to 2.7 µM in A-549 cells and 0.32 to 2.0 µM in Vero E6 cells. NHC had similar antiviral activity against SARS-CoV-2 variants Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), Lambda (C.37), Mu (B.1.621) and Omicron (B.1.1.529/BA.1, BA.1.1, BA.2, BA.4 and BA.5), with mean EC50 values of 0.55-3.0 μM. NHC had non-antagonistic antiviral activity with remdesivir against SARS-CoV-2 in cell culture.
Resistance
No amino acid substitutions in SARS-CoV-2 associated with resistance to NHC have been identified in Phase 2 clinical trials evaluating LAGEVRIO for the treatment of COVID-19. Studies to evaluate selection of resistance to NHC with SARS-CoV-2 in cell culture have not been completed. Resistance selection studies have been conducted with other coronaviruses (MHV and MERS-CoV) and showed a low likelihood of resistance development to NHC. Following 30 passages in cell culture, only a 2-fold decrease in susceptibility was observed and no NHC resistance-associated amino acid substitutions were identified.
In clinical trials, encoded amino acid changes (substitutions, deletions or insertions) were more likely to be detected in viral sequences in subjects treated with LAGEVRIO compared to placebo. In a small number of subjects amino acid changes in the spike protein occurred at positions targeted by monoclonal antibodies and vaccines. The clinical and public health significance of these changes are unknown.
Cross-Resistance
NHC retained activity in cell culture against virus with polymerase (nsp 12) substitutions (e.g., F480L, V557L and E802D) associated with decreased remdesivir susceptibility, indicating a lack of cross-resistance.
Activity against SARS-CoV-2 in animal models
The antiviral activity of molnupiravir has been demonstrated in mouse, hamster, and ferret models of SARS-CoV-2 infection when dosing was administered prior to or within 1-2 days after viral challenge. In SARS-CoV-2 infected ferrets, molnupiravir significantly reduced SARS-CoV-2 viral titers in the upper respiratory tract and completely inhibited viral spread to untreated contact animals. In SARS-CoV-2 infected Syrian hamsters, molnupiravir reduced viral RNA and infectious virus titers in the lungs of animals. Histopathological analysis of lung tissue harvested after infection showed significantly reduced SARS-CoV-2 viral antigen levels and a lower abundance of pulmonary lesions in molnupiravir-treated animals compared with controls.
In Vitro Cytotoxicity
NHC, the nucleoside analogue metabolite of molnupiravir, had variable cytotoxicity against different mammalian cell types with CC50 values ranging from 7.5 µM (human lymphoid CEM cell line) to >100 µM, in 3-day exposure assays. Molnupiravir inhibited the proliferation of human bone marrow progenitor cells with CC50 values of 24.9 µM and 7.7 µM for erythroid and myeloid progenitor proliferation, respectively, in 14-day colony formation assays.
Viral RNA Rebound
Post-treatment increases in SARS-CoV-2 RNA shedding levels (i.e., viral RNA rebound) in nasopharyngeal samples were observed on Day 10, Day 15, and/or Day 29 in a subset of LAGEVRIO and placebo recipients in the Phase 3 MOVe-OUT trial. Approximately 1% of both LAGEVRIO and placebo recipients had evidence of recurrent COVID-19 symptoms coinciding with a rebound in viral RNA levels in nasopharyngeal samples.
Post-treatment viral RNA rebound was not associated with the primary clinical outcome of hospitalization or death through Day 29 following the single 5-day course of LAGEVRIO treatment. Post-treatment viral RNA rebound also was not associated with the detection of cell culture infectious virus in nasopharyngeal swab samples.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Molnupiravir was not carcinogenic in a 6-month oral carcinogenicity study in RasH2 transgenic (Tg.RasH2) mice at any dose tested (30, 100 or 300 mg/kg/day).
Mutagenesis
Molnupiravir and NHC were positive in the in vitro bacterial reverse mutation assay (Ames assay) with and without metabolic activation. Molnupiravir was studied in two in vivo rodent mutagenicity models. The in vivo Pig-a mutagenicity assay gave equivocal results. Molnupiravir was negative in in vivo Big Blue® (cII Locus) transgenic rodent mutagenicity assays in somatic and germ cells. Molnupiravir was negative for induction of chromosomal damage in in vitro micronucleus (with and without metabolic activation) and in vivo rat micronucleus assays.
Based on the totality of the available genotoxicity data and the duration of treatment (5 days), molnupiravir is low risk for genotoxicity.
Impairment of Fertility
There were no effects on fertility, mating performance or early embryonic development when molnupiravir was administered to female or male rats at NHC exposures approximately 2 and 6 times, respectively, the human NHC exposure at the RHD.
13.2 Animal Toxicology and/or Pharmacology
Bone and cartilage toxicity changes resulting in impaired transformation of growth cartilage into new bone were observed in the femur and tibia of rats in a 3-month toxicity study at ≥ 500 mg/kg/day (5 times the human NHC exposure at the RHD). There was no bone or cartilage toxicity in a 1-month toxicity study in rats up to 500 mg/kg/day (4 and 8 times the human NHC exposure at the RHD in females and males, respectively), in dogs dosed for 14 days up to 50 mg/kg/day (similar to the human NHC exposure at the RHD), or in a 1-month toxicity study in mice up to 2,000 mg/kg/day (19 times the human NHC exposure at the RHD).
Growth cartilage is not present in mature skeletons, therefore the bone and cartilage findings are not relevant for adult humans but may be relevant for pediatric patients [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
Reversible, dose-related bone marrow toxicity affecting all hematopoietic cell lines was observed in dogs at ≥17 mg/kg/day (less than the human NHC exposure at the RHD). Mild decreases in peripheral blood cell and platelet counts were seen after 7 days of molnupiravir treatment progressing to more severe hematological changes after 14 days of treatment. Neither bone marrow nor hematological toxicity was observed in a 1-month toxicity study in mice up to 2,000 mg/kg/day (19 times the human NHC exposure at the RHD) and a 3-month toxicity study in rats up to 1,000 mg/kg/day (9 and 15 times the human NHC exposure at the RHD in females and males, respectively).
14 CLINICAL STUDIES
Clinical data supporting this EUA are based on data from 1,433 randomized subjects in the Phase 3 MOVe-OUT trial (NCT04575597). MOVe-OUT is a randomized, placebo-controlled, double-blind clinical trial studying LAGEVRIO for the treatment of non-hospitalized patients with mild-to-moderate COVID-19 who are at risk for progressing to severe COVID-19 and/or hospitalization. Eligible subjects were 18 years of age and older and had one or more pre-defined risk factors for disease progression: over 60 years of age, diabetes, obesity (BMI ≥30), chronic kidney disease, serious heart conditions, chronic obstructive pulmonary disease, or active cancer. The study included symptomatic subjects not vaccinated against SARS-CoV-2 and who had laboratory confirmed SARS-CoV-2 infection and symptom onset within 5 days of randomization. Subjects were randomized 1:1 to receive 800 mg of LAGEVRIO or placebo orally twice daily for 5 days.
At baseline, in all randomized subjects, the median age was 43 years (range:18 to 90); 17% of subjects were over 60 years of age and 3% were 75 years of age or older; 49% of subjects were male; 57% were White, 5% Black or African American, 3% Asian, 50% Hispanic or Latino. The majority of subjects were enrolled from sites in Latin America (46%) and Europe (33%); 12% were enrolled in Africa, 6% were enrolled in North America and 3% were enrolled in Asia. Forty-eight percent of subjects received LAGEVRIO or placebo within 3 days of COVID-19 symptom onset. The most common risk factors were obesity (74%), over 60 years of age (17%), and diabetes (16%). Among 792 subjects (55% of total randomized population) with available baseline SARS-CoV-2 variant/clade identification results, 58% were infected with Delta (B.1.617.2 and AY lineages), 20% were infected with Mu (B.1.621), 11% were infected with Gamma (P.1), and the remainder were infected with other variants/clades. Overall, baseline demographic and disease characteristics were well balanced between the treatment arms.
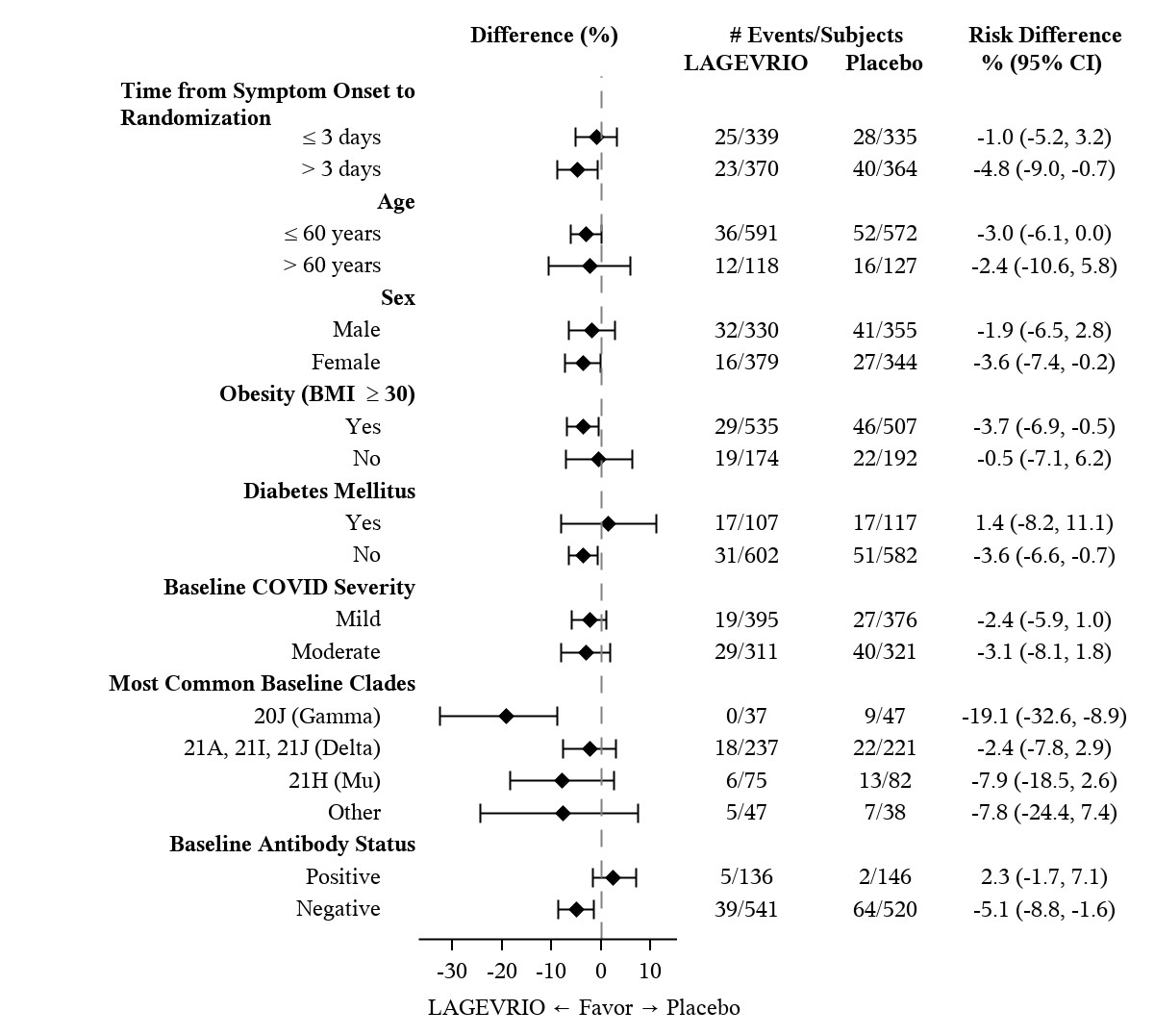
Table 3 provides the results of the primary endpoint (the percentage of subjects who were hospitalized or died through Day 29 due to any cause). The efficacy results are based on unvaccinated adults who were 18 years of age and older and had one or more pre-defined risk factors for disease progression: over 60 years of age, diabetes, obesity (BMI ≥30), chronic kidney disease, serious heart conditions, chronic obstructive pulmonary disease, or active cancer. Please refer to Figure 1 for results by certain subgroups. These subgroup analyses are considered exploratory. Data are not available in certain subgroups of subjects who are at high risk for progression to severe COVID-19 as defined by CDC.
| LAGEVRIO (N=709) n (%) | Placebo (N=699) n (%) | Adjusted Risk Difference % (95% CI) |
|---|---|---|
| Adjusted relative risk reduction of LAGEVRIO compared to placebo for all randomized subjects was 30% (95% CI: 1%, 51%). | ||
| Analyses are adjusted by the stratification factor of time of COVID-19 symptom onset (≤3 days vs. >3 [4-5] days). | ||
|
|
||
| All-cause hospitalization ≥24 hours for acute care or death through Day 29 | ||
| 48 (6.8%) | 68 (9.7%) | -3.0% (-5.9%, -0.1%) |
| All-cause mortality through Day 29 | ||
| 1 (0.1%) | 9 (1.3%) | |
| The corresponding confidence interval is based on Miettinen & Nurminen method. |
| The modified intent-to-treat population is the efficacy analysis population. |
| Baseline serum samples were evaluated with the Roche Elecsys anti-N assay to test for the presence of antibodies (IgM, IgG and IgA) against the SARS-CoV-2 nucleocapsid protein. |
| The findings of these subgroup analyses are considered exploratory. |
 |
16 HOW SUPPLIED/STORAGE AND HANDLING
Product: 50090-6841
NDC: 50090-6841-0 40 CAPSULE in a BOTTLE, PLASTIC
17 PATIENT COUNSELING INFORMATION
As a prescribing healthcare practitioner, you must communicate to the patient and/or caregiver information consistent with the “FACT SHEET FOR PATIENTS AND CAREGIVERS” and document that information was provided. A copy of this Fact Sheet should be provided to the patient and/or caregiver prior to receiving LAGEVRIO [see Box].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions have been reported, even following a single dose of LAGEVRIO, and to discontinue the drug and to inform their healthcare provider at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction [see Warnings and Precautions (5.2)].
Risk of Fetal Toxicity
Advise patients that LAGEVRIO is not recommended for use in pregnancy because it may cause fetal harm. Advise individuals of childbearing potential to inform their healthcare provider of a known or suspected pregnancy [see Box, Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
Advise individuals of childbearing potential to use effective contraception correctly and consistently while taking LAGEVRIO and for 4 days after the last dose.
While the risk is regarded as low, there is a theoretical risk for LAGEVRIO to affect offspring of treated males based on its mechanism of action. Advise sexually active individuals with partners of childbearing potential to use a reliable method of contraception correctly and consistently while taking LAGEVRIO and for at least 3 months after the last dose of LAGEVRIO. The risk beyond 3 months after the last dose of LAGEVRIO is unknown [see Use in Specific Populations (8.3)].
Risk of Bone and Cartilage Toxicity
LAGEVRIO is not authorized for use in patients less than 18 year of age as it may affect bone growth and cartilage formation [see Warnings and Precautions (5.3) and Use in Specific Populations (8.4)].
Pregnancy Registry
There is a pregnancy registry that monitors pregnancy outcomes in individuals exposed to LAGEVRIO during pregnancy. Encourage participation and advise patients about how they may enroll in the pregnancy registry at https://covid-pr.pregistry.com or 1-800-616-3791 [see Use in Specific Populations (8.1)].
Lactation
Breastfeeding is not recommended while taking LAGEVRIO and for 4 days after the last dose of LAGEVRIO. Advise lactating individuals to consider interrupting breastfeeding and to consider pumping and discarding breast milk during treatment and for 4 days after the last dose of LAGEVRIO [see Use in Specific Populations (8.2)].
Administration Instructions
Inform patients to take LAGEVRIO with or without food. Advise patients to swallow LAGEVRIO capsules whole, and to not open, break, or crush the capsules. Instruct patients that if they miss a dose of LAGEVRIO and it is within 10 hours of the time it is usually taken, the patient should take it as soon as possible and resume the normal dosing schedule. If the patient misses a dose by more than 10 hours, the patient should not take the missed dose and instead take the next dose at the regularly scheduled time. Advise the patient to not double the dose to make up for a missed dose [see Dosage and Administration (2.2)].
LAGEVRIO capsule contents can be mixed with water and given via NG/OG, G and GJ tubes. Inform patients to follow the instructions as described in the fact sheet for patients and caregivers [see Dosage and Administration (2.3)].
Alert the patient of the importance of completing the full 5-day treatment course and to continuing isolation in accordance with public health recommendations to maximize viral clearance and minimize transmission of SARS-CoV-2 [see Dosage and Administration (2.1)].
18 MANUFACTURER INFORMATION
For additional information visit: www.molnupiravir.com
If you have questions, please contact
1-800-672-6372
Manuf. for: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
For patent information: www.msd.com/research/patent
Copyright © 2021-2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates.
All rights reserved.
usfshcp-mk4482-c-2406r010
| Fact Sheet for Patients and Caregivers
Emergency Use Authorization (EUA) Of LAGEVRIO™ (molnupiravir) capsules For Coronavirus Disease 2019 (COVID-19) |
What is the most important information I should know about LAGEVRIO?
LAGEVRIO may cause serious side effects, including:
-
LAGEVRIO may cause harm to your unborn baby. It is not known if LAGEVRIO will harm your baby if you take LAGEVRIO during pregnancy.
- LAGEVRIO is not recommended for use in pregnancy.
- LAGEVRIO has not been studied in pregnancy. LAGEVRIO was studied in pregnant animals only. When LAGEVRIO was given to pregnant animals, LAGEVRIO caused harm to their unborn babies.
- You and your healthcare provider may decide that you should take LAGEVRIO during pregnancy if there are no other COVID-19 treatment options approved or authorized by the FDA that are accessible or clinically appropriate for you.
- If you and your healthcare provider decide that you should take LAGEVRIO during pregnancy, you and your healthcare provider should discuss the known and potential benefits and the potential risks of taking LAGEVRIO during pregnancy.
For individuals who are able to become pregnant:
- You should use a reliable method of birth control (contraception) correctly and consistently during treatment with LAGEVRIO and for 4 days after the last dose of LAGEVRIO. Talk to your healthcare provider about reliable birth control methods.
- Before starting treatment with LAGEVRIO your healthcare provider may do a pregnancy test to see if you are pregnant before starting treatment with LAGEVRIO.
- Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with LAGEVRIO.
Pregnancy Registry:
- There is a pregnancy registry for individuals who take LAGEVRIO during pregnancy. The purpose of this program is to collect information about the health of you and your baby.
- If you are pregnant or become pregnant during treatment with LAGEVRIO, you are encouraged to report your use of LAGEVRIO during pregnancy to this pregnancy registry at https://covid-pr.pregistry.com or 1-800-616-3791.
For individuals who are sexually active with partners who are able to become pregnant:
- It is not known if LAGEVRIO can affect sperm, but the risk is regarded as low based on animal studies.
- You should use a reliable method of birth control (contraception) correctly and consistently during treatment with LAGEVRIO and for at least 3 months after the last dose. Talk to your healthcare provider about reliable birth control methods.
- The risk to sperm beyond 3 months is not known. Talk to your healthcare provider if you have questions or concerns about how LAGEVRIO may affect sperm.
You are being given this fact sheet because your healthcare provider believes it is necessary to provide you with LAGEVRIO for the treatment of adults with mild-to-moderate coronavirus disease 2019 (COVID-19) who are at high risk for progression to severe COVID-19, including hospitalization or death, and for whom other COVID-19 treatment options approved or authorized by the FDA are not accessible or clinically appropriate.
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to make LAGEVRIO available during the COVID-19 pandemic (for more details about an EUA please see “What is an Emergency Use Authorization?” at the end of this document). LAGEVRIO is not an FDA-approved medicine in the United States. Read this Fact Sheet for information about LAGEVRIO. Talk to your healthcare provider about your options if you have any questions. It is your choice to take LAGEVRIO.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus. You can get COVID-19 through close contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild-to-severe, including illness resulting in death. While information so far suggests that most COVID-19 illness is mild, serious illness can happen and may cause some of your other medical conditions to become worse. Older people and people of all ages with severe, long lasting (chronic) medical conditions like heart disease, lung disease and diabetes, for example seem to be at higher risk of being hospitalized for COVID-19.
What is LAGEVRIO?
LAGEVRIO is an investigational medicine used to treat adults with mild to moderate COVID-19:
- who are at high risk for progression to severe COVID-19 including hospitalization or death, and for
- whom other COVID-19 treatment options approved or authorized by the FDA are not accessible or clinically appropriate.
The FDA has authorized the emergency use of LAGEVRIO for the treatment of mild-to-moderate COVID-19 in adults under an EUA. For more information on EUA, see the “What is an Emergency Use Authorization (EUA)?” section at the end of this Fact Sheet.
LAGEVRIO is not authorized:
- for use in people less than 18 years of age.
- for prevention of COVID-19.
- for people needing hospitalization for COVID-19.
- for use for longer than 5 consecutive days.
What should I tell my healthcare provider before I take LAGEVRIO?
Tell your healthcare provider if you:
- have any allergies
- are breastfeeding or plan to breastfeed
- have any serious illnesses
- take any medicines including prescription, over-the-counter medicines, vitamins, and herbal products.
How do I take LAGEVRIO?
- Take LAGEVRIO exactly as your healthcare provider tells you to take it.
- Take 4 capsules of LAGEVRIO every 12 hours (for example, at 8 am and at 8 pm)
- Take LAGEVRIO for 5 days. It is important that you complete the full 5 days of treatment with LAGEVRIO. Do not stop taking LAGEVRIO before you complete the full 5 days of treatment, even if you feel better.
- Take LAGEVRIO with or without food.
- You should stay in isolation for as long as your healthcare provider tells you to. Talk to your healthcare provider if you are not sure about how to properly isolate while you have COVID-19.
- Swallow LAGEVRIO capsules whole. Do not open, break, or crush the capsules. If you cannot swallow capsules whole, tell your healthcare provider.
- If your healthcare provider prescribes LAGEVRIO and tells you to take or give a dose through a nasogastric (NG), orogastric (OG), gastrostomy (G) or gastrojejunostomy (GJ) feeding tube, follow the instructions below: “How to take or give a dose of LAGEVRIO through a nasogastric (NG), orogastric (OG), gastrostomy (G) or gastrojejunostomy (GJ) feeding tube.” You must have an NG, OG, or G feeding tube that is size 12 French or larger, or a GJ feeding tube that is 14 French or larger.
-
If you miss a dose of LAGEVRIO:
- If it has been less than 10 hours since the missed dose, take it as soon as you remember.
- If it has been more than 10 hours since the missed dose, skip the missed dose and take your dose at the next scheduled time.
- Do not double the dose of LAGEVRIO to make up for a missed dose.
How to take or give a dose of LAGEVRIO through a nasogastric (NG), orogastric (OG), gastrostomy (G) or gastrojejunostomy (GJ) feeding tube:
- Wash your hands well with soap and water.
- Gather the supplies you will need to take or give the prescribed dose of LAGEVRIO.
- 4 LAGEVRIO capsules
- 1 liquid measuring cup with mL markings to measure 40 mL of room temperature water
- 1 clean container with a lid
- 1 syringe. Your healthcare provider should tell you what size syringe you will need to take or give a dose of LAGEVRIO.
- Place the needed supplies on a clean work surface.
- Follow your healthcare provider’s instructions on how to flush the feeding tube. Flush the feeding tube with 5 mL of water before taking or giving a dose of LAGEVRIO.
- Carefully open 4 LAGEVRIO capsules, one at a time, and empty the contents into a clean container. Discard (throw away) the capsules shells in the household trash.
- Use the liquid measuring cup to measure 40 mL of room temperature water and add to the container containing the capsule contents.
- Place the lid on the container. Shake to mix the capsule contents and water well for 3 minutes. The capsule contents may not dissolve completely.
- Remove the lid from the container and draw up all the LAGEVRIO and water mixture into a syringe.
- Give all of the mixture right away through the feeding tube. If using a GJ tube, give the mixture through the gastric port. Do not keep the mixture for future use.
-
If any capsule contents are left in the container:
- Add 10 mL of water to the container and mix to loosen any capsule contents that are left in the container.
- Use the syringe to draw up all of the mixture in the container.
- Give the mixture through the feeding tube.
- Repeat this process as needed until you no longer see any capsule contents left in the container or syringe.
- Use the same syringe to flush the feeding tube 2 times with 5 mL of water (10 mL total).
- Rinse the container, lid and syringe well with clean water after use. Place on a clean paper towel until next use.
What are the important possible side effects of LAGEVRIO?
- See, “What is the most important information I should know about LAGEVRIO?”
-
Allergic Reactions. Allergic reactions can happen in people taking LAGEVRIO, even after only 1 dose. Stop taking LAGEVRIO and call your healthcare provider right away if you get any of the following symptoms of an allergic reaction:
- hives
- rapid heartbeat
- trouble swallowing or breathing
- swelling of the mouth, lips, or face
- throat tightness
- hoarseness
- skin rash
The most common side effects of LAGEVRIO are:
- diarrhea
- nausea
- dizziness
These are not all the possible side effects of LAGEVRIO. Not many people have taken LAGEVRIO. Serious and unexpected side effects may happen. This medicine is still being studied, so it is possible that all of the risks are not known at this time.
What other treatment choices are there?
Veklury (remdesivir) is FDA-approved as an intravenous (IV) infusion for the treatment of mild-to-moderate COVID-19 in certain adults and children. Paxlovid (nirmatrelvir/ritonavir) is FDA-approved as tablets for the treatment of mild-to-moderate COVID-19 in certain adults. Talk with your doctor to see if Veklury or Paxlovid are appropriate for you.
Like LAGEVRIO, FDA may also allow for the emergency use of other medicines to treat people with COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for more information.
It is your choice to be treated or not to be treated with LAGEVRIO. Should you decide not to take it, it will not change your standard medical care.
What if I am breastfeeding?
Breastfeeding is not recommended during treatment with LAGEVRIO and for 4 days after the last dose of LAGEVRIO. If you are breastfeeding or plan to breastfeed, talk to your healthcare provider about your options and specific situation before taking LAGEVRIO.
How do I report side effects with LAGEVRIO?
Contact your healthcare provider if you have any side effects that bother you or do not go away.
Report side effects to FDA MedWatch at www.fda.gov/medwatch or call 1-800-FDA-1088 (1-800-332-1088).
How should I store LAGEVRIO?
- Store LAGEVRIO capsules at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep LAGEVRIO and all medicines out of the reach of children.
How can I learn more about COVID-19?
- Ask your healthcare provider.
- Visit www.cdc.gov/COVID19.
- Contact your local or state public health department.
- Call Merck Sharp & Dohme at 1-800-672-6372 (toll free in the U.S.)
- Visit www.molnupiravir.com.
What Is an Emergency Use Authorization (EUA)?
The United States FDA has made LAGEVRIO available under an emergency access mechanism called an Emergency Use Authorization (EUA) The EUA is supported by a Secretary of Health and Human Service (HHS) declaration that circumstances exist to justify emergency use of drugs and biological products during the COVID-19 pandemic.
LAGEVRIO is authorized for emergency use for the treatment of adults with mild-to-moderate COVID-19 who are at high risk for progression to severe COVID-19, including hospitalization or death, and for whom alternative COVID-19 treatment options approved or authorized by FDA are not accessible or clinically appropriate.
LAGEVRIO as a treatment for COVID-19, has not undergone the same type of review as an FDA-approved product. The FDA may issue an EUA when certain criteria are met, which includes that there are no adequate, approved, and available alternatives. In addition, the FDA decision is based on the totality of scientific evidence available showing that it is reasonable to believe that the product meets certain criteria for safety, performance, and labeling and may be effective in treatment of patients during the COVID-19 pandemic.
All of these criteria must be met to allow for the product to be used in the treatment of patients during the COVID-19 pandemic. The EUA for LAGEVRIO is in effect for the duration of the COVID-19 declaration justifying emergency use of LAGEVRIO, unless terminated or revoked (after which LAGEVRIO may no longer be used under the EUA).
Manuf. for: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
For patent information: www.msd.com/research/patent
Copyright © 2021-2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates.
All rights reserved.
usfsp-mk4482-c-2406r006
Revised: June 2024

| LAGEVRIO
molnupiravir capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - A-S Medication Solutions (830016429) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| A-S Medication Solutions | 830016429 | RELABEL(50090-6841) | |
Trademark Results [LAGEVRIO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LAGEVRIO 98101056 not registered Live/Pending |
Merck Sharp & Dohme LLC 2023-07-25 |
 LAGEVRIO 90230082 not registered Live/Pending |
Merck Sharp & Dohme Corp. 2020-10-01 |
 LAGEVRIO 88276559 not registered Live/Pending |
Merck Sharp & Dohme Corp. 2019-01-25 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.