ALUNBRIG- brigatinib tablet, coated
Alunbrig by
Drug Labeling and Warnings
Alunbrig by is a Prescription medication manufactured, distributed, or labeled by ARIAD Pharmaceuticals Inc., PENN PHARMACEUTICAL SERVICES LTD. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ALUNBRIG safely and effectively. See full prescribing information for ALUNBRIG.
ALUNBRIG™ (brigatinib) tablets, for oral use
Initial U.S. Approval: 2017INDICATIONS AND USAGE
ALUNBRIG is a kinase inhibitor indicated for the treatment of patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib. This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. (1)
DOSAGE AND ADMINISTRATION
90 mg orally once daily for the first 7 days; if tolerated, increase to 180 mg orally once daily. May be taken with or without food. (2.1)
DOSAGE FORMS AND STRENGTHS
Tablets: 30 mg and 90 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD)/Pneumonitis: Occurred in 9.1% of patients at the recommended dose. Monitor for new or worsening respiratory symptoms, particularly during the first week of treatment. Withhold ALUNBRIG for new or worsening respiratory symptoms and promptly evaluate for ILD/pneumonitis. Upon recovery, either dose reduce or permanently discontinue ALUNBRIG. (2.2, 5.1)
- Hypertension: Monitor blood pressure after 2 weeks and then at least monthly during treatment. For severe hypertension, withhold ALUNBRIG, then dose reduce or permanently discontinue. (2.2, 5.2)
- Bradycardia: Monitor heart rate and blood pressure regularly during treatment. If symptomatic, withhold ALUNBRIG, then dose reduce or permanently discontinue. (2.2, 5.3)
- Visual Disturbance: Advise patients to report visual symptoms. Withhold ALUNBRIG and obtain ophthalmologic evaluation, then dose reduce or permanently discontinue ALUNBRIG. (2.2, 5.4)
- Creatine Phosphokinase (CPK) Elevation: Monitor CPK levels regularly during treatment. Based on the severity, withhold ALUNBRIG, then resume or reduce dose. (2.2, 5.5)
- Pancreatic Enzyme Elevation: Monitor lipase and amylase levels regularly during treatment. Based on the severity, withhold ALUNBRIG, then resume or reduce dose. (2.2, 5.6)
- Hyperglycemia: Assess fasting serum glucose prior to starting ALUNBRIG and regularly during treatment. If not adequately controlled with optimal medical management, withhold ALUNBRIG, then consider dose reduction or permanently discontinue, based on severity. (2.2, 5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use a non-hormonal method of effective contraception. (5.8, 8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (≥25%) with ALUNBRIG were nausea, diarrhea, fatigue, cough, and headache. (6)
To report SUSPECTED ADVERSE REACTIONS, contact ARIAD Pharmaceuticals, Inc. at 1-844-217-6468 or www.alunbrig.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- CYP3A Inhibitors: Avoid concomitant use of ALUNBRIG with strong CYP3A inhibitors. If concomitant use of a strong CYP3A inhibitor is unavoidable, reduce the dose of ALUNBRIG. (2.3, 7.1)
- CYP3A Inducers: Avoid concomitant use of ALUNBRIG with strong CYP3A inducers. (7.2)
- CYP3A Substrates: Hormonal contraceptives may be ineffective due to decreased exposure. (7.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Dose Modifications for Adverse Reactions
2.3 Dose Modification for Strong CYP3A Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
5.2 Hypertension
5.3 Bradycardia
5.4 Visual Disturbance
5.5 Creatine Phosphokinase (CPK) Elevation
5.6 Pancreatic Enzyme Elevation
5.7 Hyperglycemia
5.8 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
7.1 Drugs That May Increase Brigatinib Plasma Concentrations
7.2 Drugs That May Decrease Brigatinib Plasma Concentrations
7.3 Drugs That May Have Their Plasma Concentrations Altered by Brigatinib
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
ALUNBRIG is indicated for the treatment of patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
This indication is approved under accelerated approval based on tumor response rate and duration of response [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The recommended dosing regimen for ALUNBRIG is:
- 90 mg orally once daily for the first 7 days;
- if 90 mg is tolerated during the first 7 days, increase the dose to 180 mg orally once daily.
Administer ALUNBRIG until disease progression or unacceptable toxicity.
If ALUNBRIG is interrupted for 14 days or longer for reasons other than adverse reactions, resume treatment at 90 mg once daily for 7 days before increasing to the previously tolerated dose.
ALUNBRIG may be taken with or without food. Instruct patients to swallow tablets whole. Do not crush or chew tablets.
If a dose of ALUNBRIG is missed or vomiting occurs after taking a dose, do not administer an additional dose and take the next dose of ALUNBRIG at the scheduled time.
2.2 Dose Modifications for Adverse Reactions
ALUNBRIG dose modification levels are summarized in Table 1.
Table 1: Recommended ALUNBRIG Dose Reduction Levels Dose Reduction Levels Dose First Second Third - * Not applicable
90 mg once daily 60 mg once daily permanently discontinue N/A* 180 mg once daily 120 mg once daily 90 mg once daily 60 mg once daily Once reduced for adverse reactions, do not subsequently increase the dose of ALUNBRIG. Permanently discontinue ALUNBRIG if patients are unable to tolerate the 60 mg once daily dose.
Recommendations for dose modifications of ALUNBRIG for the management of adverse reactions are provided in Table 2.
Table 2: Recommended ALUNBRIG Dose Modifications for Adverse Reactions Adverse Reaction Severity* Dose Modification bpm = beats per minute; DBP = diastolic blood pressure; HR = heart rate; SBP = systolic blood pressure; ULN = upper limit of normal - * Graded per National Cancer Institute Common Terminology Criteria for Adverse Events. Version 4.0 (NCI CTCAE v4).
Interstitial Lung Disease (ILD) /Pneumonitis [see Warnings and Precautions (5.1)] Grade 1 - If new pulmonary symptoms occur during the first 7 days of treatment, withhold ALUNBRIG until recovery to baseline, then resume at same dose and do not escalate to 180 mg if ILD/pneumonitis is suspected.
- If new pulmonary symptoms occur after the first 7 days of treatment, withhold ALUNBRIG until recovery to baseline, then resume at same dose.
- If ILD/pneumonitis recurs, permanently discontinue ALUNBRIG.
Grade 2 - If new pulmonary symptoms occur during the first 7 days of treatment, withhold ALUNBRIG until recovery to baseline. Resume at next lower dose (Table 1) and do not dose escalate if ILD/pneumonitis is suspected.
- If new pulmonary symptoms occur after the first 7 days of treatment, withhold ALUNBRIG until recovery to baseline. If ILD/pneumonitis is suspected, resume at next lower dose (Table 1); otherwise, resume at same dose.
- If ILD/pneumonitis recurs, permanently discontinue ALUNBRIG.
Grade 3 or 4 Permanently discontinue ALUNBRIG for ILD/pneumonitis. Hypertension [see Warnings and Precautions (5.2)] Grade 3 hypertension (SBP greater than or equal to 160 mmHg or DBP greater than or equal to 100 mmHg, medical intervention indicated, more than one anti-hypertensive drug, or more intensive therapy than previously used indicated) - Withhold ALUNBRIG until hypertension has recovered to Grade 1 or less (SBP less than 140 mmHg and DBP less than 90 mmHg), then resume ALUNBRIG at next lower dose (Table 1).
- Recurrence: withhold ALUNBRIG until recovery to Grade 1 or less, and resume at next lower dose (Table 1) or permanently discontinue treatment.
Grade 4 hypertension (life- threatening consequences, urgent intervention indicated) - Withhold ALUNBRIG until recovery to Grade 1 or less, and resume at next lower dose (Table 1) or permanently discontinue treatment.
- Recurrence: permanently discontinue ALUNBRIG for recurrence of Grade 4 hypertension.
Bradycardia (HR less than 60 bpm) [see Warnings and Precautions (5.3)] Symptomatic bradycardia - Withhold ALUNBRIG until recovery to asymptomatic bradycardia or to a resting heart rate of 60 bpm or above.
- If a concomitant medication known to cause bradycardia is identified and discontinued or dose-adjusted, resume ALUNBRIG at same dose upon recovery to asymptomatic bradycardia or to resting heart rate of 60 bpm or above.
- If no concomitant medication known to cause bradycardia is identified, or if contributing concomitant medications are not discontinued or dose-adjusted, resume ALUNBRIG at next lower dose (Table 1) upon recovery to asymptomatic bradycardia or to resting heart rate of 60 bpm or above.
Bradycardia with life-threatening consequences, urgent intervention indicated - Permanently discontinue ALUNBRIG if no contributing concomitant medication is identified.
- If contributing concomitant medication is identified and discontinued or dose-adjusted, resume ALUNBRIG at next lower dose (Table 1) upon recovery to asymptomatic bradycardia or to a resting heart rate of 60 bpm or above, with frequent monitoring as clinically indicated.
- Recurrence: permanently discontinue ALUNBRIG.
Visual Disturbance [see Warnings and Precautions (5.4)] Grade 2 or 3 visual disturbance Withhold ALUNBRIG until recovery to Grade 1 or baseline, then resume at the next lower dose (Table 1). Grade 4 visual disturbance Permanently discontinue ALUNBRIG. Creatine Phosphokinase (CPK) Elevation [see Warnings and Precautions (5.5)] Grade 3 CPK elevation (greater than 5.0 × ULN) Withhold ALUNBRIG until recovery to Grade 1 or less (less than or equal to 2.5 × ULN) or to baseline, then resume ALUNBRIG at same dose. Grade 4 CPK elevation (greater than 10.0 × ULN) or recurrence of Grade 3 elevation Withhold ALUNBRIG until recovery to Grade 1 or less (less than or equal to 2.5 × ULN) or to baseline, then resume ALUNBRIG at next lower dose (Table 1). Lipase/Amylase Elevation [see Warnings and Precautions (5.6)] Grade 3 lipase or amylase elevation (greater than 2.0 × ULN) Withhold ALUNBRIG until recovery to Grade 1 or less (less than or equal to 1.5 × ULN) or to baseline, then resume ALUNBRIG at same dose. Grade 4 lipase or amylase elevation (greater than 5.0 × ULN) or recurrence of Grade 3 elevation Withhold ALUNBRIG until recovery to Grade 1 or less (less than or equal to 1.5 × ULN) or to baseline, then resume ALUNBRIG at next lower dose (Table 1). Hyperglycemia [see Warnings and Precautions (5.7)] Grade 3 (greater than 250 mg/dL or 13.9 mmol/L) or greater If adequate hyperglycemic control cannot be achieved with optimal medical management, withhold ALUNBRIG until adequate hyperglycemic control is achieved and consider reduction to the next dose (Table 1) or permanently discontinue ALUNBRIG. Other Grade 3 - Withhold ALUNBRIG until recovery to baseline, then resume at same dose.
- Recurrence: withhold ALUNBRIG until recovery to baseline, then resume at next lower dose or discontinue ALUNBRIG (Table 1).
Grade 4 - First occurrence: either withhold ALUNBRIG until recovery to baseline and resume at next lower dose (Table 1) or permanently discontinue.
- Permanently discontinue ALUNBRIG for recurrence.
2.3 Dose Modification for Strong CYP3A Inhibitors
Avoid concomitant use of strong CYP3A inhibitors during treatment with ALUNBRIG [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. If concomitant use of a strong CYP3A inhibitor cannot be avoided, reduce the ALUNBRIG once daily dose by approximately 50% (i.e., from 180 mg to 90 mg, or from 90 mg to 60 mg). After discontinuation of a strong CYP3A inhibitor, resume the ALUNBRIG dose that was tolerated prior to initiating the strong CYP3A inhibitor.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
Severe, life-threatening, and fatal pulmonary adverse reactions consistent with interstitial lung disease (ILD)/pneumonitis have occurred with ALUNBRIG.
In Trial ALTA (ALTA), ILD/pneumonitis occurred in 3.7% of patients in the 90 mg group (90 mg once daily) and 9.1% of patients in the 90→180 mg group (180 mg once daily with 7-day lead-in at 90 mg once daily).
Adverse reactions consistent with possible ILD/pneumonitis occurred early (within 9 days of initiation of ALUNBRIG; median onset was 2 days) in 6.4% of patients, with Grade 3 to 4 reactions occurring in 2.7%.
Monitor for new or worsening respiratory symptoms (e.g., dyspnea, cough, etc.), particularly during the first week of initiating ALUNBRIG. Withhold ALUNBRIG in any patient with new or worsening respiratory symptoms, and promptly evaluate for ILD/pneumonitis or other causes of respiratory symptoms (e.g., pulmonary embolism, tumor progression, and infectious pneumonia). For Grade 1 or 2 ILD/pneumonitis, either resume ALUNBRIG with dose reduction according to Table 1 after recovery to baseline or permanently discontinue ALUNBRIG. Permanently discontinue ALUNBRIG for Grade 3 or 4 ILD/pneumonitis or recurrence of Grade 1 or 2 ILD/pneumonitis [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.2 Hypertension
In ALTA, hypertension was reported in 11% of patients in the 90 mg group who received ALUNBRIG and 21% of patients in the 90→180 mg group. Grade 3 hypertension occurred in 5.9% of patients overall.
Control blood pressure prior to treatment with ALUNBRIG. Monitor blood pressure after 2 weeks and at least monthly thereafter during treatment with ALUNBRIG. Withhold ALUNBRIG for Grade 3 hypertension despite optimal antihypertensive therapy. Upon resolution or improvement to Grade 1 severity, resume ALUNBRIG at a reduced dose. Consider permanent discontinuation of treatment with ALUNBRIG for Grade 4 hypertension or recurrence of Grade 3 hypertension [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
Use caution when administering ALUNBRIG in combination with antihypertensive agents that cause bradycardia [see Warnings and Precautions (5.3)].
5.3 Bradycardia
Bradycardia can occur with ALUNBRIG. In ALTA, heart rates less than 50 beats per minute (bpm) occurred in 5.7% of patients in the 90 mg group and 7.6% of patients in the 90→180 mg group. Grade 2 bradycardia occurred in 1 (0.9%) patient in the 90 mg group.
Monitor heart rate and blood pressure during treatment with ALUNBRIG. Monitor patients more frequently if concomitant use of drug known to cause bradycardia cannot be avoided [see Warnings and Precautions (5.2)].
For symptomatic bradycardia, withhold ALUNBRIG and review concomitant medications for those known to cause bradycardia. If a concomitant medication known to cause bradycardia is identified and discontinued or dose adjusted, resume ALUNBRIG at the same dose following resolution of symptomatic bradycardia; otherwise, reduce the dose of ALUNBRIG following resolution of symptomatic bradycardia. Discontinue ALUNBRIG for life-threatening bradycardia if no contributing concomitant medication is identified [see Dosage and Administration (2.2)].
5.4 Visual Disturbance
In ALTA, adverse reactions leading to visual disturbance including blurred vision, diplopia, and reduced visual acuity, were reported in 7.3% of patients receiving ALUNBRIG in the 90 mg group and 10% of patients in the 90→180 mg group. Grade 3 macular edema and cataract occurred in one patient each in the 90→180 mg group.
Advise patients to report any visual symptoms. Withhold ALUNBRIG and obtain an ophthalmologic evaluation in patients with new or worsening visual symptoms of Grade 2 or greater severity. Upon recovery of Grade 2 or Grade 3 visual disturbances to Grade 1 severity or baseline, resume ALUNBRIG at a reduced dose. Permanently discontinue treatment with ALUNBRIG for Grade 4 visual disturbances [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.5 Creatine Phosphokinase (CPK) Elevation
In ALTA, creatine phosphokinase (CPK) elevation occurred in 27% of patients receiving ALUNBRIG in the 90 mg group and 48% of patients in the 90 mg→180 mg group. The incidence of Grade 3-4 CPK elevation was 2.8% in the 90 mg group and 12% in the 90→180 mg group.
Dose reduction for CPK elevation occurred in 1.8% of patients in the 90 mg group and 4.5% in the 90→180 mg group.
Advise patients to report any unexplained muscle pain, tenderness, or weakness. Monitor CPK levels during ALUNBRIG treatment. Withhold ALUNBRIG for Grade 3 or 4 CPK elevation. Upon resolution or recovery to Grade 1 or baseline, resume ALUNBRIG at the same dose or at a reduced dose as described in Table 2 [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.6 Pancreatic Enzyme Elevation
In ALTA, amylase elevation occurred in 27% of patients in the 90 mg group and 39% of patients in the 90→180 mg group. Lipase elevations occurred in 21% of patients in the 90 mg group and 45% of patients in the 90→180 mg group. Grade 3 or 4 amylase elevation occurred in 3.7% of patients in the 90 mg group and 2.7% of patients in the 90→180 mg group. Grade 3 or 4 lipase elevation occurred in 4.6% of patients in the 90 mg group and 5.5% of patients in the 90→180 mg group.
Monitor lipase and amylase during treatment with ALUNBRIG. Withhold ALUNBRIG for Grade 3 or 4 pancreatic enzyme elevation. Upon resolution or recovery to Grade 1 or baseline, resume ALUNBRIG at the same dose or at a reduced dose as described in Table 2 [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.7 Hyperglycemia
In ALTA, 43% of patients who received ALUNBRIG experienced new or worsening hyperglycemia. Grade 3 hyperglycemia, based on laboratory assessment of serum fasting glucose levels, occurred in 3.7% of patients. Two of 20 (10%) patients with diabetes or glucose intolerance at baseline required initiation of insulin while receiving ALUNBRIG.
Assess fasting serum glucose prior to initiation of ALUNBRIG and monitor periodically thereafter. Initiate or optimize anti-hyperglycemic medications as needed. If adequate hyperglycemic control cannot be achieved with optimal medical management, withhold ALUNBRIG until adequate hyperglycemic control is achieved and consider reducing the dose of ALUNBRIG as described in Table 1 or permanently discontinuing ALUNBRIG [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, ALUNBRIG can cause fetal harm when administered to pregnant women. There are no clinical data on the use of ALUNBRIG in pregnant women. Administration of brigatinib to pregnant rats during the period of organogenesis resulted in dose-related skeletal anomalies at doses as low as 12.5 mg/kg/day (approximately 0.7 times the human exposure by AUC at 180 mg once daily) as well as increased post implantation loss, malformations, and decreased fetal body weight at doses of 25 mg/kg/day (approximately 1.26 times the human exposure at 180 mg once daily) or higher.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective non-hormonal contraception during treatment with ALUNBRIG and for at least 4 months following the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment and for at least 3 months after the last dose of ALUNBRIG [see Use in Specific Populations (8.1 and 8.3) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the prescribing information:
- Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.1)]
- Hypertension [see Warnings and Precautions (5.2)]
- Bradycardia [see Warnings and Precautions (5.3)]
- Visual Disturbance [see Warnings and Precautions (5.4)]
- Creatine Phosphokinase (CPK) Elevation [see Warnings and Precautions (5.5)]
- Pancreatic Enzyme Elevation [see Warnings and Precautions (5.6)]
- Hyperglycemia[see Warnings and Precautions (5.7)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ALUNBRIG was evaluated in 219 patients with locally advanced or metastatic ALK-positive non-small cell lung cancer (NSCLC) who received at least one dose of ALUNBRIG in ALTA after experiencing disease progression on crizotinib. Patients received ALUNBRIG 90 mg once daily continuously (90 mg group) or 90 mg once daily for 7 days followed by 180 mg once daily (90→180 mg group). The median duration of treatment was 7.5 months in the 90 mg group and 7.8 months in the 90→180 mg group. A total of 150 (68%) patients were exposed to ALUNBRIG for greater than or equal to 6 months and 42 (19%) patients were exposed for greater than or equal to one year.
The study population characteristics were: median age 54 years (range: 18 to 82), age less than 65 years (77%), female (57%), White (67%), Asian (31%), Stage IV disease (98%), NSCLC adenocarcinoma histology (97%), never or former smoker (95%), ECOG Performance Status (PS) 0 or 1 (93%), and brain metastases at baseline (69%) [see Clinical Studies (14)].
Serious adverse reactions occurred in 38% of patients in the 90 mg group and 40% of patients in the 90→180 mg group. The most common serious adverse reactions were pneumonia (5.5% overall, 3.7% in the 90 mg group, and 7.3% in the 90→180 mg group) and ILD/pneumonitis (4.6% overall, 1.8% in the 90 mg group and 7.3% in the 90→180 mg group). Fatal adverse reactions occurred in 3.7% of patients and consisted of pneumonia (2 patients), sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis and urosepsis (1 patient each).
In ALTA, 2.8% of patients in the 90 mg group and 8.2% of patients in the 90→180 mg group permanently discontinued ALUNBRIG for adverse reactions. The most frequent adverse reactions that led to discontinuation were ILD/pneumonitis (0.9% in the 90 mg group and 1.8% in the 90→180 mg group) and pneumonia (1.8% in the 90→180 mg group only).
In ALTA, 14% of patients required a dose reduction due to adverse reactions (7.3% in the 90 mg group and 20% in the 90→180 mg group). The most common adverse reaction that led to dose reduction was increased creatine phosphokinase for both regimens (1.8% in the 90 mg group and 4.5% in the 90→180 mg group).
Table 3 and Table 4 summarize the common adverse reactions and laboratory abnormalities observed in ALTA.
Table 3: Adverse Reactions in ≥ 10% (All Grades*) or ≥ 2% (Grades 3-4) of Patients by Dose Group in ALTA (N=219) Adverse Reactions 90 mg once daily
N = 10990→180 mg once daily
N = 110All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)- * Per National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
- † Includes abdominal distension, abdominal pain, and epigastric discomfort
- ‡ Includes asthenia and fatigue
- § Includes dyspnea and exertional dyspnea
- ¶ Includes one Grade 5 event
- # Includes headache and sinus headache
- Þ Includes peripheral sensory neuropathy and paresthesia
- ß Includes acneiform dermatitis, exfoliative rash, rash, pruritic rash, and pustular rash
- à Includes musculoskeletal pain and myalgia
- è Includes diplopia, photophobia, blurred vision, reduced visual acuity, visual impairment, vitreous floaters, visual field defect, macular edema, and vitreous detachment
Gastrointestinal Disorders Nausea 33 0.9 40 0.9 Diarrhea 19 0 38 0 Vomiting 24 1.8 23 0 Constipation 19 0.9 15 0 Abdominal Pain† 17 0 10 0 General Disorders And Administration Site Conditions Fatigue‡ 29 1.8 36 0 Pyrexia 14 0 6.4 0.9 Respiratory, Thoracic And Mediastinal Disorders Cough 18 0 34 0 Dyspnea§ 27 2.8 21 1.8¶ ILD/Pneumonitis 3.7 1.8 9.1 2.7 Hypoxia 0.9 0 2.7 2.7 Nervous System Disorders Headache# 28 0 27 0.9 Peripheral NeuropathyÞ 13 0.9 13 1.8 Skin And Subcutaneous Tissue Disorders Rashß 15 1.8 24 3.6 Vascular Disorders Hypertension 11 5.5 21 6.4 Musculoskeletal And Connective Tissue Disorders Muscle Spasms 12 0 17 0 Back pain 10 1.8 15 1.8 Myalgiaà 9.2 0 15 0.9 Arthralgia 14 0.9 14 0 Pain in extremity 11 0 3.6 0.9 Metabolism And Nutrition Disorders Decreased Appetite 22 0.9 15 0.9 Eye Disorders Visual Disturbanceè 7.3 0 10 0.9 Infections Pneumonia 4.6 2.8¶ 10 5.5¶ Psychiatric Disorders Insomnia 11 0 7.3 0 Table 4: Laboratory Abnormalities in ≥20% (All Grades*) of Patients by Regimen in ALTA (N=219) Laboratory Abnormality 90 mg once daily
N= 10990→180 mg once daily
N=110All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)- * Per CTCAE version 4.0
- † Elevated blood insulin was also observed in both regimens
Chemistry Increased aspartate aminotransferase 38 0.9 65 0 Hyperglycemia† 38 3.7 49 3.6 Increased creatine phosphokinase 27 2.8 48 12 Increased lipase 21 4.6 45 5.5 Increased alanine aminotransferase 34 0 40 2.7 Increased amylase 27 3.7 39 2.7 Increased alkaline phosphatase 15 0.9 29 0.9 Decreased phosphorous 15 1.8 23 3.6 Prolonged activated partial thromboplastin time 22 1.8 20 0.9 Hematology Anemia 23 0.9 40 0.9 Lymphopenia 19 2.8 27 4.5 -
7 DRUG INTERACTIONS
7.1 Drugs That May Increase Brigatinib Plasma Concentrations
Strong CYP3A Inhibitors
Coadministration of itraconazole, a strong CYP3A inhibitor, increased brigatinib plasma concentrations and may result in increased adverse reactions [see Clinical Pharmacology (12.3)]. Avoid the concomitant use of strong CYP3A inhibitors with ALUNBRIG, including but not limited to certain antivirals (e.g., boceprevir, cobicistat, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir), macrolide antibiotics (e.g., clarithromycin), antifungals (e.g., itraconazole, ketoconazole, posaconazole, voriconazole), and conivaptan. Avoid grapefruit or grapefruit juice as it may also increase plasma concentrations of brigatinib [see Clinical Pharmacology (12.3)]. If concomitant use of a strong CYP3A inhibitor cannot be avoided, reduce the dose of ALUNBRIG by approximately 50% [see Dosage and Administration (2.3)].
7.2 Drugs That May Decrease Brigatinib Plasma Concentrations
Strong CYP3A Inducers
Coadministration of ALUNBRIG with rifampin, a strong CYP3A inducer, decreased brigatinib plasma concentrations and may result in decreased efficacy [see Clinical Pharmacology (12.3)]. Avoid the concomitant use of strong CYP3A inducers with ALUNBRIG, including but not limited to rifampin, carbamazepine, phenytoin, and St. John's Wort [see Clinical Pharmacology (12.3)].
7.3 Drugs That May Have Their Plasma Concentrations Altered by Brigatinib
CYP3A Substrates
Brigatinib induces CYP3A in vitro and may decrease concentrations of CYP3A substrates. Coadministration of ALUNBRIG with CYP3A substrates, including hormonal contraceptives, can result in decreased concentrations and loss of efficacy of CYP3A substrates [see Use in Specific Populations (8.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in animals, ALUNBRIG can cause fetal harm when administered to a pregnant woman [see Data and Clinical Pharmacology (12.1)]. There are no clinical data on the use of ALUNBRIG in pregnant women. Administration of brigatinib to pregnant rats during the period of organogenesis resulted in dose-related skeletal anomalies at doses as low as 12.5 mg/kg/day (approximately 0.7 times the human exposure by AUC at 180 mg once daily) as well as increased post-implantation loss, malformations, and decreased fetal body weight at doses of 25 mg/kg/day (approximately 1.26 times the human exposure at 180 mg once daily) or greater. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, advise the patient of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in which pregnant rats were administered daily doses of brigatinib during organogenesis, dose-related skeletal (incomplete ossification, small incisors) and visceral anomalies were observed at doses as low as 12.5 mg/kg/day (approximately 0.7 times the human exposure by AUC at 180 mg once daily). Malformations observed at 25 mg/kg/day (approximately 1.26 times the human AUC at 180 mg once daily) included anasarca (generalized subcutaneous edema), anophthalmia (absent eyes), forelimb hyperflexion, small, short and/or bent limbs, multiple fused ribs, bent scapulae, omphalocele (intestine protruding into umbilicus), and gastroschisis (intestines protruding through a defect in the abdominal wall) along with visceral findings of moderate bilateral dilatation of the lateral ventricles.
8.2 Lactation
Risk Summary
There are no data regarding the secretion of brigatinib in human milk or its effects on the breastfed infant or milk production. Because of the potential for adverse reactions in breastfed infants, advise lactating women not to breastfeed during treatment with ALUNBRIG and for 1 week following the final dose.
8.3 Females and Males of Reproductive Potential
Contraception
ALUNBRIG can cause fetal harm [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with ALUNBRIG and for at least 4 months after the final dose. Counsel patients to use a non-hormonal method of contraception since ALUNBRIG can render some hormonal contraceptives ineffective [see Drug Interaction (7.3]).
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment with ALUNBRIG and for at least 3 months after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in male reproductive organs in animals, ALUNBRIG may cause reduced fertility in males [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of ALUNBRIG in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of ALUNBRIG did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients. Of the 222 patients in ALTA, 19.4% were 65-74 years and 4.1% were 75 years or older. No clinically relevant differences in safety or efficacy were observed between patients ≥65 years and younger patients.
8.6 Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin within upper limit of normal [ULN] and AST greater than ULN or total bilirubin greater than 1 and up to 1.5 times ULN and any AST). The pharmacokinetics and safety of ALUNBRIG in patients with moderate or severe hepatic impairment have not been studied [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment is recommended for patients with mild and moderate renal impairment [creatinine clearance (CLcr) 30 to 89 mL/min estimated by Cockcroft-Gault)]. The pharmacokinetics and safety of ALUNBRIG in patients with severe renal impairment (CLcr 15 to 29 mL/min estimated by Cockcroft-Gault) have not been studied [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Brigatinib is a kinase inhibitor. The chemical name for brigatinib is 5-chloro-N4-[2-(dimethylphosphoryl)phenyl]-N2-{2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl}pyrimidine-2,4-diamine. The molecular formula is C29H39ClN7O2P which corresponds to a formula weight of 584.10 g/mol. Brigatinib has no chiral centers. The chemical structure is shown below:
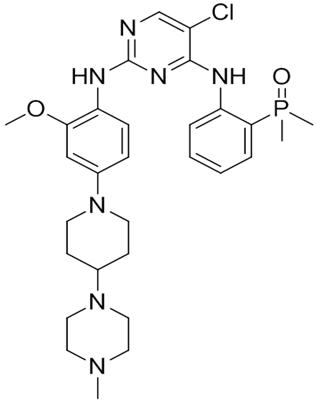
Brigatinib is an off-white to beige/tan solid. The pKas were determined to be: 1.73 ± 0.02 (base), 3.65 ± 0.01 (base), 4.72 ± 0.01 (base), and 8.04 ± 0.01 (base).
ALUNBRIG is supplied for oral use as film-coated tablets containing 30 mg or 90 mg of brigatinib and the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, sodium starch glycolate (Type A), magnesium stearate, and hydrophobic colloidal silica. The tablet coating consists of talc, polyethylene glycol, polyvinyl alcohol, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Brigatinib is a tyrosine kinase inhibitor with in vitro activity at clinically achievable concentrations against multiple kinases including ALK, ROS1, insulin-like growth factor-1 receptor (IGF-1R), and FLT-3 as well as EGFR deletion and point mutations. Brigatinib inhibited autophosphorylation of ALK and ALK-mediated phosphorylation of the downstream signaling proteins STAT3, AKT, ERK1/2, and S6 in in vitro and in vivo assays. Brigatinib also inhibited the in vitro proliferation of cell lines expressing EML4-ALK and NPM-ALK fusion proteins and demonstrated dose-dependent inhibition of EML4-ALK-positive NSCLC xenograft growth in mice.
At clinically achievable concentrations (≤ 500 nM), brigatinib inhibited the in vitro viability of cells expressing EML4-ALK and 17 mutant forms associated with resistance to ALK inhibitors including crizotinib, as well as EGFR-Del (E746-A750), ROS1-L2026M, FLT3-F691L, and FLT3-D835Y. Brigatinib exhibited in vivo anti-tumor activity against 4 mutant forms of EML4-ALK, including G1202R and L1196M mutants identified in NSCLC tumors in patients who have progressed on crizotinib. Brigatinib also reduced tumor burden and prolonged survival in mice implanted intracranially with an ALK-driven tumor cell line.
12.2 Pharmacodynamics
Brigatinib exposure-response relationships and the time course of the pharmacodynamic response are unknown.
Cardiac Electrophysiology
The QT interval prolongation potential of ALUNBRIG was assessed in 123 patients following once daily ALUNBRIG doses of 30 mg (1/6th of the approved 180 mg dose) to 240 mg (1.3 times the approved 180 mg dose). ALUNBRIG did not prolong the QT interval to a clinically relevant extent.
12.3 Pharmacokinetics
The geometric mean (CV%) steady-state maximum concentration (Cmax) of brigatinib at ALUNBRIG doses of 90 mg and 180 mg once daily was 552 (65%) ng/mL and 1452 (60%) ng/mL, respectively, and the corresponding area under the concentration-time curve (AUC0-Tau) was 8165 (57%) ng∙h/mL and 20276 (56%) ng∙h/mL. After a single dose and repeat dosing of ALUNBRIG, systemic exposure of brigatinib was dose proportional over the dose range of 60 mg (0.3 times the approved 180 mg dose) to 240 mg (1.3 times the approved 180 mg dose) once daily. The mean accumulation ratio after repeat dosing was 1.9 to 2.4.
Absorption
Following administration of single oral doses of ALUNBRIG of 30 to 240 mg, the median time to peak concentration (Tmax) ranged from 1 to 4 hours.
Distribution
Brigatinib is 66% bound to human plasma proteins and the binding is not concentration-dependent in vitro. The blood-to-plasma concentration ratio is 0.69. Following oral administration of ALUNBRIG 180 mg once daily, the mean apparent volume of distribution (Vz/F) of brigatinib at steady-state was 153 L.
Elimination
Following oral administration of ALUNBRIG 180 mg once daily, the mean apparent oral clearance (CL/F) of brigatinib at steady-state is 12.7 L/h and the mean plasma elimination half-life is 25 hours.
Metabolism
Brigatinib is primarily metabolized by CYP2C8 and CYP3A4 in vitro. Following oral administration of a single 180 mg dose of radiolabeled brigatinib to healthy subjects, N-demethylation and cysteine conjugation were the two major metabolic pathways. Unchanged brigatinib (92%) and its primary metabolite, AP26123 (3.5%), were the major circulating radioactive components. The steady-state AUC of AP26123 was less than 10% of AUC of brigatinib exposure in patients. The metabolite, AP26123, inhibited ALK with approximately 3-fold lower potency than brigatinib in vitro.
Excretion
Following oral administration of a single 180 mg dose of radiolabeled brigatinib to healthy subjects, 65% of the administered dose was recovered in feces and 25% of the administered dose was recovered in urine. Unchanged brigatinib represented 41% and 86% of the total radioactivity in feces and urine, respectively.
Specific Populations
Age, race, sex, body weight, and albumin concentration have no clinically meaningful effect on the pharmacokinetics of brigatinib.
Hepatic Impairment
As hepatic elimination is a major route of excretion for brigatinib, hepatic impairment may result in increased plasma brigatinib concentrations. Based on a population pharmacokinetic analysis, brigatinib exposures were similar between 49 subjects with mild hepatic impairment (total bilirubin within upper limit of normal [ULN] and AST greater than ULN or total bilirubin greater than 1 and up to 1.5 times ULN and any AST) and 377 subjects with normal hepatic function (total bilirubin and AST within ULN). The pharmacokinetics of brigatinib in patients with moderate (total bilirubin greater than 1.5 and up to 3.0 times ULN and any AST) to severe (total bilirubin greater than 3.0 times ULN and any AST) hepatic impairment has not been studied.
Renal Impairment
Based on a population pharmacokinetic analysis, brigatinib exposures were similar among 125 subjects with mild renal impairment (CLcr 60 to less than 90 mL/min), 34 subjects with moderate renal impairment (CLcr 30 to less than 60 mL/min) and 270 subjects with normal renal function (CLcr greater than or equal to 90 mL/min), suggesting that no dose adjustment is necessary in patients with mild to moderate renal impairment. Patients with severe renal impairment (CLcr less than 30 mL/min) were not included in clinical trials.
Drug Interactions
Effects of Other Drugs on Brigatinib
Strong CYP3A Inhibitors: Coadministration of 200 mg twice daily doses of itraconazole (a strong CYP3A inhibitor) with a single 90 mg dose of ALUNBRIG increased brigatinib Cmax by 21% and AUC0-INF by 101%, relative to a 90 mg dose of ALUNBRIG administered alone [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
Strong CYP2C8 Inhibitors: Coadministration of 600 mg twice daily doses of gemfibrozil (a strong CYP2C8 inhibitor) with a single 90 mg dose of ALUNBRIG decreased brigatinib Cmax by 41% and AUC0-INF by 12%, relative to a 90 mg dose of ALUNBRIG administered alone. The effect of gemfibrozil on the pharmacokinetics of brigatinib is not clinically meaningful and the underlying mechanism for the decreased exposure of brigatinib is unknown.
Strong CYP3A Inducers: Coadministration of 600 mg daily doses of rifampin (a strong CYP3A inducer) with a single 180 mg dose of ALUNBRIG decreased brigatinib Cmax by 60% and AUC0-INF by 80%, relative to a 180 mg dose of ALUNBRIG administered alone [see Drug Interactions (7.2)].
P-gp and BCRP Inhibitors: In vitro studies suggest that brigatinib is a substrate of the efflux transporters P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Given that brigatinib exhibits high solubility and high permeability in vitro, P-gp and BCRP inhibitors are unlikely to increase plasma concentrations of brigatinib.
Effects of Brigatinib on Other Drugs
Transporter Substrates: Brigatinib is an inhibitor of P-gp, BCRP, OCT1, MATE1, and MATE2K in vitro. Therefore, brigatinib may have the potential to increase concentrations of coadministered substrates of these transporters. Brigatinib at clinically relevant concentrations did not inhibit OATP1B1, OATP1B3, OAT1, OAT3, OCT2 or BSEP.
CYP Substrates: Brigatinib and its primary metabolite, AP26123, did not inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4/5 at clinically relevant concentrations.
Brigatinib, at clinically relevant plasma concentrations, induced CYP3A via activation of the pregnane X receptor (PXR). Brigatinib may also induce CYP2C enzymes via the same mechanism at clinically relevant concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with brigatinib.
Treatment with brigatinib resulted in chromosomal damage in an in vivo mammalian erythrocyte micronucleus in the rat, but was not mutagenic in the Ames or in vitro mammalian chromosome aberration tests.
Dedicated animal fertility studies were not conducted with brigatinib. Testicular toxicity was observed in repeat-dose animal studies at doses resulting in exposure as low as 0.2 times the exposure in patients at the 180 mg dose. In rats, findings included lower weight of testes, seminal vesicles and prostate gland, and testicular tubular degeneration; these effects were not reversible during the 2-month recovery period. In monkeys, findings included reduced size of testes along with microscopic evidence of hypospermatogenesis; these effects were reversible during the recovery period.
-
14 CLINICAL STUDIES
The efficacy of ALUNBRIG was demonstrated in a two-arm, open-label, multicenter trial (ALTA, NCT02094573) in adult patients with locally advanced or metastatic ALK-positive non-small cell lung cancer (NSCLC) who had progressed on crizotinib. The study required patients to have a documented ALK rearrangement based on an FDA-approved test or a different test with adequate archival tissue to confirm ALK arrangement by the Vysis® ALK Break-Apart fluorescence in situ hybridization (FISH) Probe Kit test. Key eligibility criteria included an ECOG Performance Status of 0-2 and progression on crizotinib. Neurologically stable patients with central nervous system (CNS) metastases were permitted to enroll. Patients with a history of interstitial lung disease or drug-related pneumonitis or who had received crizotinib within 3 days of the first dose of brigatinib were excluded. The major efficacy outcome measure was confirmed overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) as evaluated by an Independent Review Committee (IRC). Additional efficacy outcome measures included Investigator-assessed ORR, duration of response (DOR), intracranial ORR, and intracranial DOR.
A total of 222 patients were randomized to receive ALUNBRIG either 90 mg once daily (90 mg arm; n=112) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (90→180 mg arm; n=110). Randomization was stratified by brain metastases (present versus absent) and best prior response to crizotinib (complete or partial response versus any other response/unevaluable).
Baseline demographic characteristics of the overall study population were: median age 54 years (range 18 to 82, 23% 65 and over), 67% White and 31% Asian, 57% female, 36% ECOG PS 0 and 57% ECOG PS 1, and 95% never or former smokers. The disease characteristics of the overall study population were: Stage IV disease in 98%, adenocarcinoma histology in 97%, prior systemic chemotherapy in 74%, metastatic disease to the brain in 69% (61% had received prior radiation to the brain), bone metastases in 39%, and liver metastases in 26% of patients. Sixty-four percent of patients had an objective response to prior crizotinib.
The median duration of follow-up was 8 months (range: 0.1-20.2). Efficacy results from ALTA are summarized in Table 5.
Table 5: ALTA Efficacy Results Efficacy parameter IRC Assessment Investigator Assessment 90 mg once daily 90→180 mg once daily 90 mg once daily 90→180 mg once daily (N=112) (N=110) (N=112) (N=110) CI = Confidence Interval; NE = Not Estimable Overall Response Rate (95% CI) 48% (39-58) 53% (43-62) 45% (35-54) 54% (44-63) Complete Response, n (%) 4 (3.6%) 5 (4.5%) 1 (0.9%) 4 (3.6%) Partial Response, n (%) 50 (45%) 53 (48%) 49 (44%) 55 (50%) Duration of Response, median in months
(95% CI)13.8
(7.4-NE)13.8
(9.3-NE)13.8
(5.6-13.8)11.1
(9.2-13.8)IRC assessment of intracranial ORR and intracranial DOR according to RECIST v1.1 in the subgroup of 44 patients with measurable brain metastases (≥10 mm in longest diameter) at baseline are summarized in Table 6. Duration of intracranial response was measured from date of first intracranial response until intracranial disease progression (new lesions, intracranial target lesion diameter growth ≥20% from nadir, or unequivocal progression of intracranial non-target lesions) or death.
Table 6: Intracranial Objective Response in Patients with Measurable Brain Metastases in ALTA Efficacy parameter IRC Assessment 90 mg once daily
(N=26)90→180 mg once daily
(N=18)CI = Confidence Interval; NE = Not Estimable Intracranial Overall Response Rate, (95 % CI) 42% (23-63) 67% (41-87) Complete Response, n (%) 2 (7.7%) 0 Partial Response, n (%) 9 (35%) 12 (67%) Duration of Intracranial Response, median (months)
(range)NE
(1.9+ - 9.2+)5.6
(1.9+ - 9.2+)Among the 23 patients who exhibited an intracranial response, 78% of patients in the 90 mg arm and 68% of patients in the 90→180 mg arm maintained a response for at least 4 months.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
30 mg tablets: round, white to off-white film-coated tablet with "U3" debossed on one side and plain on the other side; available in:
Bottles of 21 tablets NDC: 76189-113-21 Bottles of 180 tablets NDC: 76189-113-18 90 mg tablets: oval, white to off-white film-coated tablet with "U7" debossed on one side and plain on the other side; available in:
Bottles of 7 tablets NDC: 76189-119-07 Bottles of 30 tablets NDC: 76189-119-30 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients of the following:
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the symptoms and risks of serious pulmonary adverse reactions such as ILD/pneumonitis. Advise patients to immediately report any new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Hypertension
Advise patients of risks of hypertension and to promptly report signs or symptoms of hypertension [see Warnings and Precautions (5.2)].
Bradycardia
Advise patients to report any symptoms of bradycardia and to inform their healthcare provider about the use of heart and blood pressure medications [see Warnings and Precautions (5.3)].
Visual Disturbance
Advise patients to inform their healthcare provider of any new or worsening vision symptoms [see Warnings and Precautions (5.4)].
Creatine Phosphokinase (CPK) Elevation
Inform patients of the signs and symptoms of creatinine phosphokinase (CPK) elevation and the need for monitoring during treatment. Advise patients to inform their healthcare provider of any new or worsening symptoms of unexplained muscle pain, tenderness, or weakness [see Warnings and Precautions (5.5)].
Pancreatic Enzyme Elevation
Inform patients of the signs and symptoms of pancreatitis and the need to monitor for amylase and lipase elevations during treatment [see Warnings and Precautions (5.6)].
Hyperglycemia
Inform patients of the risks of new or worsening hyperglycemia and the need to periodically monitor glucose levels. Advise patients with diabetes mellitus or glucose intolerance that anti-hyperglycemic medications may need to be adjusted during treatment with ALUNBRIG [see Warnings and Precautions (5.7)].
Females and Males of Reproductive Potential
Embryo-Fetal Toxicity
Advise females and males of reproductive potential that ALUNBRIG can cause fetal harm [see Warnings and Precautions (5.8) and Use in Specific Populations (8.1)].
- Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy and to use effective non-hormonal contraception during treatment with ALUNBRIG and for at least 4 months after the final dose [see Use in Specific Populations (8.3].
- Advise males with female partners of reproductive potential to use effective contraception during treatment with ALUNBRIG and for at least 3 months after the final dose [see Use in Specific Populations (8.3)].
Lactation
Advise females not to breastfeed during treatment with ALUNBRIG and for at least 1 week following the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential of the potential for reduced fertility from ALUNBRIG [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Drug Interactions
Advise patients to inform their health care provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Inform patients to avoid grapefruit or grapefruit juice while taking ALUNBRIG [see Drug Interactions (7)].
Dosing and Administration
Instruct patients to start with 90 mg of ALUNBRIG once daily for the first 7 days and if tolerated, increase the dose to 180 mg once daily. Advise patients to take ALUNBRIG with or without food [see Dosage and Administration (2.1)].
Missed Dose
Advise patients that if a dose of ALUNBRIG is missed or if the patient vomits after taking a dose of ALUNBRIG, not to take an extra dose, but to take the next dose at the regular time [see Dosage and Administration (2.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
ALUNBRIG (uh-lun-brig)
(brigatinib)
tabletsThis Patient Information has been approved by the U.S. Food and Drug Administration Issued: April 2017 What is the most important information I should know about ALUNBRIG?
ALUNBRIG can cause serious side effects, including:- Lung problems. ALUNBRIG may cause severe or life-threatening swelling (inflammation) of the lungs any time during treatment, and can lead to death. These lung problems happen especially within the first week of treatment with ALUNBRIG. Symptoms may be similar to those symptoms from lung cancer. Tell your healthcare provider right away if you have any new or worsening symptoms, including:
- trouble breathing or shortness of breath
- chest pain
- cough with or without mucous
- fever
- High blood pressure (hypertension). ALUNBRIG may cause high blood pressure. Your healthcare provider will check your blood pressure before starting and during treatment with ALUNBRIG. Tell your healthcare provider right away if you get headaches, dizziness, blurred vision, chest pain or shortness of breath.
- Slow heart rate (bradycardia). ALUNBRIG may cause very slow heartbeats that can be severe. Your healthcare provider will check your heart rate during treatment with ALUNBRIG. Tell your healthcare provider right away if you feel dizzy, lightheaded, or faint during treatment with ALUNBRIG. Tell your healthcare provider if you start to take or have any changes in heart or blood pressure medicines.
- Vision problems. ALUNBRIG may cause vision problems. Your healthcare provider may stop ALUNBRIG and refer you to an eye specialist if you develop severe vision problems during treatment with ALUNBRIG. Tell your healthcare provider right away if you have any loss of vision or any change in vision, including:
- double vision
- seeing flashes of light
- blurry vision
- light hurting your eyes
- new or increased floaters
- Muscle pain, tenderness, and weakness (myalgia). ALUNBRIG may increase the level of an enzyme in your blood called creatine phosphokinase (CPK), which may be a sign of muscle damage. Your healthcare provider will do blood tests to check your blood levels of CPK during treatment with ALUNBRIG. Tell your healthcare provider right away if you get new or worsening signs and symptoms of muscle problems, including unexplained muscle pain or muscle pain that does not go away, tenderness, or weakness.
- Inflammation of the pancreas (pancreatitis). ALUNBRIG may increase enzymes in your blood called amylase and lipase, which may be a sign of pancreatitis. Your healthcare provider will do blood tests to check your pancreatic enzyme blood levels during treatment with ALUNBRIG. Tell your healthcare provider right away if you get new or worsening signs and symptoms of pancreatitis, including upper abdominal pain that may spread to the back and get worse with eating, weight loss, or nausea.
- High blood sugar (hyperglycemia). ALUNBRIG may increase your blood sugar levels. Your healthcare provider will do blood tests to check your blood sugar levels before starting and during treatment with ALUNBRIG. Your healthcare provider may need to start or change your blood sugar medicine to control your blood sugar levels. Tell your healthcare provider right away if you get new or worsening signs and symptoms of hyperglycemia, including:
- feeling very thirsty
- needing to urinate more than usual
- feeling very hungry
- feeling sick to your stomach
- feeling weak or tired
- feeling confused
See "What are the possible side effects of ALUNBRIG?" for information about side effects. What is ALUNBRIG?
ALUNBRIG is a prescription medicine used to treat people with non-small cell lung cancer (NSCLC):- that has a certain type of abnormal anaplastic lymphoma kinase (ALK) gene, and
- that has spread to other parts of your body, and
- who have taken the medicine crizotinib, but their NSCLC worsened or they cannot tolerate taking crizotinib.
Before you take ALUNBRIG, tell your healthcare provider about all of your medical conditions, including if you: - have lung or breathing problems
- have high blood pressure
- have a slow heartbeat
- have any vision problems
- have or have had pancreatitis
- have diabetes mellitus or glucose intolerance
- are pregnant or plan to become pregnant. ALUNBRIG can harm your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with ALUNBRIG or think you may be pregnant.
- Females who are able to become pregnant should use effective non-hormonal birth control during treatment with ALUNBRIG and for at least 4 months after the final dose of ALUNBRIG. Birth control pills (oral contraceptives) and other hormonal forms of birth control may not be effective if used during treatment with ALUNBRIG. Talk to your healthcare provider about birth control choices that are right for you during treatment with ALUNBRIG.
- Males who have female partners that are able to become pregnant should use effective birth control during treatment with ALUNBRIG and for at least 3 months after the final dose of ALUNBRIG.
- are breastfeeding or plan to breastfeed. It is not known if ALUNBRIG passes into your breast milk. Do not breastfeed during treatment with ALUNBRIG and for 1 week after the final dose of ALUNBRIG.
How should I take ALUNBRIG? - Take ALUNBRIG exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking ALUNBRIG unless your healthcare provider tells you to.
- Your healthcare provider will start you on a low dose (90 mg) of ALUNBRIG for the first 7 days of treatment. If you tolerate this dose of ALUNBRIG well, your healthcare provider may increase your dose after the first 7 days of treatment.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with ALUNBRIG if you have side effects.
- Take ALUNBRIG 1 time each day.
- Take ALUNBRIG with or without food.
- Swallow ALUNBRIG tablets whole. Do not crush or chew tablets.
- If you miss a dose of ALUNBRIG, do not take the missed dose. Take your next dose at your regular time.
- If you vomit after taking a dose of ALUNBRIG, do not take an extra dose. Take your next dose at your regular time.
What should I avoid while taking ALUNBRIG? - Avoid eating grapefruit or drinking grapefruit juice during treatment with ALUNBRIG. Grapefruit may increase the amount of ALUNBRIG in your blood.
What are the possible side effects of ALUNBRIG?
ALUNBRIG may cause serious side effects, including: The most common side effects of ALUNBRIG include:- nausea
- diarrhea
- fatigue
- cough
- headache
ALUNBRIG may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of ALUNBRIG. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store ALUNBRIG? - Store ALUNBRIG at room temperature 20°C to 25°C (68°F to 77°F).
General information about the safe and effective use of ALUNBRIG.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information. Do not use ALUNBRIG for a condition for which it was not prescribed. Do not give ALUNBRIG to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about ALUNBRIG that is written for health professionals.What are the ingredients in ALUNBRIG?
Active ingredient: brigatinib
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, sodium starch glycolate (Type A), magnesium stearate, and hydrophobic colloidal silica. The tablet coating consists of talc, polyethylene glycol, polyvinyl alcohol, and titanium dioxide.
Manufactured for: ARIAD Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, Cambridge, MA
For more information, go to www.alunbrig.com or call 1-844-217-6468. -
PRINCIPAL DISPLAY PANEL - 30 mg Tablet Bottle Label
NDC: 76189-113-18
Alunbrig™
(brigatinib) tablets30 mg
Rx OnlyEach tablet contains 30 mg brigatinib
180 Tablets
ARIAD®

-
INGREDIENTS AND APPEARANCE
ALUNBRIG
brigatinib tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 76189-113 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Brigatinib (UNII: HYW8DB273J) (Brigatinib - UNII:HYW8DB273J) Brigatinib 30 mg Inactive Ingredients Ingredient Name Strength SILICA DIMETHYL SILYLATE (UNII: EU2PSP0G0W) Lactose Monohydrate (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE Score no score Shape OVAL Size 7mm Flavor Imprint Code U3 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 76189-113-21 21 in 1 BOTTLE; Type 0: Not a Combination Product 04/28/2017 12/12/2020 2 NDC: 76189-113-18 180 in 1 BOTTLE; Type 0: Not a Combination Product 04/28/2017 12/12/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208772 04/28/2017 12/12/2020 Labeler - ARIAD Pharmaceuticals Inc. (786227231) Registrant - PENN PHARMACEUTICAL SERVICES LTD (226277259) Establishment Name Address ID/FEI Business Operations ARIAD Pharmaceuticals Inc. 786227231 MANUFACTURE(76189-113) , ANALYSIS(76189-113) , PACK(76189-113)
Trademark Results [Alunbrig]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ALUNBRIG 86797024 5251769 Live/Registered |
ARIAD Pharmaceuticals, Inc. 2015-10-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.