LUXTURNA- voretigene neparvovec-rzyl kit
LUXTURNA by
Drug Labeling and Warnings
LUXTURNA by is a Prescription medication manufactured, distributed, or labeled by Spark Therapeutics, Inc., PPD Development, LP. (Middleton, WI Laboratory), Absorption Systems, Eurofins Lancaster Laboratories, Inc, Intertek USA Inc, Nova Laboratories Ltd, Catalent CTS (Edinburgh) Limited, Synergy Health Sterilisation UK Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LUXTURNA safely and effectively. See full prescribing information for LUXTURNA.
LUXTURNA (voretigene neparvovec-rzyl) intraocular suspension for subretinal injection
Initial U.S. Approval: 2017INDICATIONS AND USAGE
LUXTURNA is an adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Patients must have viable retinal cells as determined by the treating physician(s). (1)
DOSAGE AND ADMINISTRATION
For subretinal injection only.
- The recommended dose of LUXTURNA for each eye is 1.5 x 1011 vector genomes (vg), administered by subretinal injection in a total volume of 0.3 mL. (2.1)
- Perform subretinal administration of LUXTURNA to each eye on separate days within a close interval, but no fewer than 6 days apart. (2.1)
- Recommend systemic oral corticosteroids equivalent to prednisone at 1 mg/kg/day (maximum of 40 mg/day) for a total of 7 days (starting 3 days before administration of LUXTURNA to each eye), and followed by a tapering dose during the next 10 days. (2.1)
DOSAGE FORMS AND STRENGTHS
LUXTURNA is a suspension for subretinal injection, supplied in a 0.5 mL extractable volume in a single-dose 2 mL vial for a single administration in one eye. The supplied concentration (5x1012vg/mL) requires a 1:10 dilution prior to administration. The Diluent is supplied in two single-use 2-mL vials. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Endophthalmitis: Use proper aseptic injection technique and monitor for signs and symptoms of infection. (5.1)
- Permanent decline in visual acuity: Monitor for visual disturbances. (5.2)
- Retinal abnormalities: Monitor for macular abnormalities, retinal tears or breaks. Do not inject in the immediate vicinity of the fovea. (5.3)
- Increased intraocular pressure: Monitor and manage intraocular pressure elevations. (5.4)
- Expansion of intraocular air bubbles: Air travel and/or scuba diving is not recommended until any intraocular air bubbles have been absorbed. (5.5)
- Cataract: Subretinal injection of LUXTURNA may result in cataract formation or increase in the rate of cataract progression. (5.6)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ 5%) in the clinical trials were conjunctival hyperemia, cataract, increased intraocular pressure, retinal tear, dellen (thinning of the corneal stroma), macular hole, subretinal deposits, eye inflammation, eye irritation, eye pain, and maculopathy (wrinkling on the surface of the macula). (6)
To report SUSPECTED ADVERSE REACTIONS, contact Spark Therapeutics, Inc. at 1-855-SPARKTX, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Pediatric use: Use in infants under 12 months of age is not recommended because of potential dilution or loss of LUXTURNA after administration due to the active retinal cells proliferation occurring in this age group. (8.4)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Preparation
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis
5.2 Permanent decline in visual acuity
5.3 Retinal abnormalities
5.4 Increased intraocular pressure
5.5 Expansion of intraocular air bubbles
5.6 Cataract
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dose
- The recommended dose of LUXTURNA for each eye is 1.5 x 1011 vector genomes (vg), administered by subretinal injection in a total volume of 0.3 mL.
- Perform subretinal administration of LUXTURNA to each eye on separate days within a close interval, but no fewer than 6 days apart.
- Recommend systemic oral corticosteroids equivalent to prednisone at 1 mg/kg/day (maximum of 40 mg/day) for a total of 7 days (starting 3 days before administration of LUXTURNA to the first eye), and followed by tapering the dose during the following 10 days. The same corticosteroid dosing regimen applies for the administration of LUXTURNA to the second eye. If the corticosteroid taper following LUXTURNA administration to the first eye is not complete three days prior to the planned LUXTURNA administration to the second eye, then the corticosteroid regimen for the second eye replaces the taper for the first eye.
2.2 Preparation
Prepare LUXTURNA within 4 hours of administration using sterile technique under aseptic conditions in a Class II vertical laminar flow biological safety cabinet (BSC). Below is the list of items required for dilution and administration syringe preparation:
- One single-dose vial of Luxturna
- Two vials of Diluent
- One 3-mL sterile syringe
- One 20G 1-inch sterile needle
- Three 1-mL sterile syringes
- Three 27G ½-inch sterile needles
- Two sterile syringe caps
- One 10-mL sterile empty glass vial
- One sterile utility drape
- One sterile plastic bag
- Two sterile labels for administration syringes
- One sterile plain label
- One sterile skin marker
Dilution of LUXTURNA
- Thaw one single-dose vial of LUXTURNA and two vials of Diluent at room temperature.
- Mix the contents of the thawed Diluent vials by gently inverting them approximately 5 times.
- Inspect the Diluent vials. If particulates, cloudiness, or discoloration are visible, do not use the vial(s); new vial(s) of Diluent should be used.
- Obtain a 3-mL sterile syringe, a 20G 1-inch sterile needle, and a 10-mL sterile empty glass vial.
- Using the 3-mL syringe with 20G 1-inch needle, transfer 2.7 mL of Diluent to the 10-mL glass vial. Dispose of the needle and syringe in an appropriate container.
- Mix the contents of the thawed LUXTURNA single-dose vial by gently inverting approximately 5 times.
- Inspect the LUXTURNA single-dose vial. If particulates, cloudiness, or discoloration are visible, do not use the vial; a new single-dose vial of LUXTURNA should be used.
- Draw 0.3 mL of LUXTURNA into a 1-mL sterile syringe with a 27G ½-inch sterile needle. (Figure 1)
Figure 1. Syringe with 0.3 mL LUXTURNA
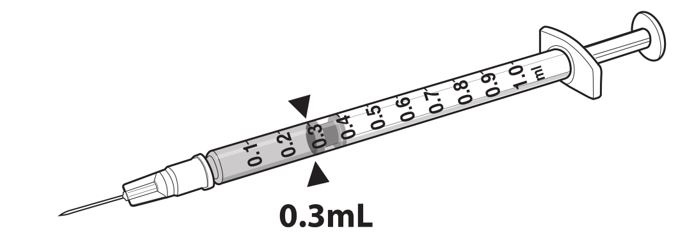
9. Transfer 0.3 mL of LUXTURNA to the glass vial containing 2.7 mL of Diluent from Step 5. Gently invert the 10-mL glass vial approximately 5 times to mix the contents.
10. Using the sterile plain label and sterile skin marker, label the 10-mL glass vial containing the diluted LUXTURNA as follows: ‘Diluted LUXTURNA’.
11. Remove all items from the BSC except the glass vial labeled ‘Diluted LUXTURNA’ and the sterile skin marker.
12. Re-sanitize the BSC prior to the next steps and place the glass vial and the sterile marker to the left side in the BSC.Preparation of LUXTURNA for Injection
To keep the syringes sterile, two operators are required for transfer of the contents of the 10-mL glass vial labeled ‘Diluted LUXTURNA’ into each of two sterile 1-mL syringes.
13. Place a sterile utility drape, a sterile plastic bag, and two sterile labels into the BSC.
14. Place the sterile drape near the Primary Operator on the right side of the sanitized BSC surface, away from the diluted LUXTURNA.
15. The Secondary Operator unwraps two 1-mL syringes, two 27G ½-inch needles, and two syringe caps in the BSC, ensuring that the Primary Operator touches only sterile surfaces while transferring the items onto the sterile drape.
16. The Secondary Operator changes to a new pair of sterile gloves and stands or sits to the left of the Primary Operator. The Secondary Operator holds the 10-mL glass vial containing the diluted LUXTURNA (Figure 2a).Figure 2a. First Position of the Operators During Preparation of LUXTURNA Syringes
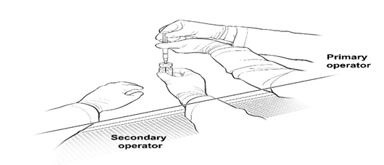
17. The Primary Operator withdraws 0.8 mL of the diluted LUXTURNA into a sterile 1-mL syringe using a 27G ½-inch sterile needle while the secondary operator holds the 10-mL glass vial. After the insertion of the needle, the Secondary Operator inverts the 10-mL glass vial enabling the Primary Operator to withdraw 0.8 mL without touching the 10-mL glass vial (Figure 2b).
Figure 2b. Second Position of the Operators During Preparation Of LUXTURNA Syringes
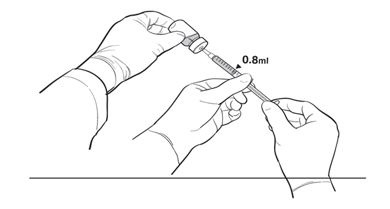
18. The Primary Operator removes the needle and affixes a sterile cap to the sterile syringe, disposes of the needle in an appropriate container, and attaches a sterile label to the administration syringe.
19. The Primary Operator repeats Steps 17 and 18 to prepare a total of two administration syringes. Label the first syringe “Diluted LUXTURNA” and label the second syringe “Back-up Diluted LUXTURNA” using the sterile skin marker. The second syringe will serve as a backup for the surgeon performing the subretinal administration procedure. Discard the back-up syringe after surgery if not used.
20. Inspect both syringes. If particulates, cloudiness, or discoloration are visible, do not use the syringe.
21. Place the syringes into the sterile plastic bag after visual inspection and seal the bag.
22. Place the sterile plastic bag with syringes containing diluted LUXTURNA into an appropriate secondary container (e.g., hard plastic cooler) for delivery to the surgical suite at room temperature.2.3 Administration
LUXTURNA should be administered in the surgical suite under controlled aseptic conditions by a surgeon experienced in performing intraocular surgery. In addition to the syringe containing the diluted LUXTURNA, the following items are required for administration:
- Subretinal injection cannula with a polyamide micro tip with an inner diameter of 41gauge.
- Extension tube made of polyvinyl chloride no longer than 6” (15.2 cm) in length and with an inner diameter no greater than 1.4mm.
Figure 3. Injection Apparatus Assembly
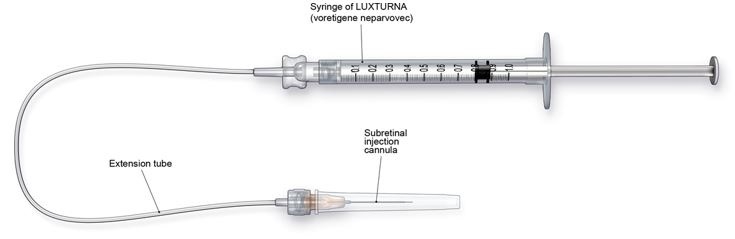
Follow the steps below for subretinal injection:
- After confirming the availability of LUXTURNA, dilate the eye and give adequate anesthesia to the patient.
- Administer a topical broad spectrum microbiocide to the conjunctiva, cornea and eyelids prior to surgery.
- Inspect LUXTURNA prior to administration. If particulates, cloudiness, or discoloration are visible, do not use the product.
- Connect the syringe containing the diluted LUXTURNA to the extension tube and subretinal injection cannula. To avoid excess priming volume, the extension tube should not exceed 15.2 cm in length and 1.4 mm in inner diameter. Inject the product slowly through the extension tube and the subretinal injection cannula to eliminate any air bubbles.
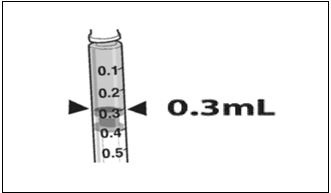
- Confirm the volume of product available in the syringe for injection, by aligning the plunger tip with the line that marks 0.3 mL. (Figure 4)
Figure 4. Volume of LUXTURNA for Injection
6. After completing a vitrectomy, identify the intended site of administration. The subretinal injection cannula can be introduced via pars plana. (Figure 5a)
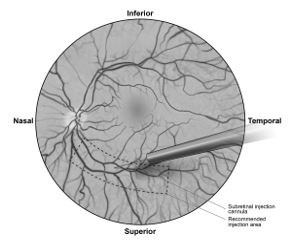
7. Under direct visualization, place the tip of the subretinal injection cannula in contact with the retinal surface. The recommended site of injection is located along the superior vascular arcade, at least 2 mm distal to the center of the fovea (Figure 5b), avoiding direct contact with the retinal vasculature or with areas of pathologic features, such as dense atrophy or intraretinal pigment migration. Inject a small amount of the product slowly until an initial subretinal bleb is observed. Then inject the remaining volume slowly until the total 0.3 mL is delivered.Figure 5a. Subretinal injection cannula introduced via pars plana
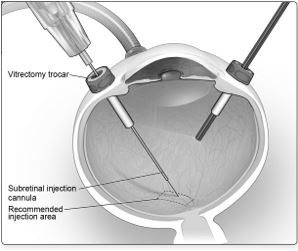
Figure 5b. Tip of the subretinal injection cannula placed within the recommended site of injection (surgeon’s point of view)
8. After completing the injection, remove the subretinal injection cannula from the eye.
9. Following injection, discard all unused product. Dispose of the back-up syringe according to local biosafety guidelines applicable for handling and disposal of the product.
10. Perform a fluid-air exchange, carefully avoiding fluid drainage near the retinotomy created for the subretinal injection.
11. Initiate supine head positioning immediately in the post-operative period.
12. Upon discharge, advise patients to rest in a supine position as much as possible for 24 hours. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis
Endophthalmitis may occur following any intraocular surgical procedure or injection. Use proper aseptic injection technique when administering LUXTURNA. Following the injection, monitor patients to permit early treatment of any infection. Advise patients to report any signs or symptoms of infection or inflammation without delay.
5.2 Permanent decline in visual acuity
Permanent decline in visual acuity may occur following subretinal injection of LUXTURNA. Monitor patients for visual disturbances.
5.3 Retinal abnormalities
Retinal abnormalities may occur during or following the subretinal injection of LUXTURNA, including macular holes, foveal thinning, loss of foveal function, foveal dehiscence, and retinal hemorrhage. Monitor and manage these retinal abnormalities appropriately. Do not administer LUXTURNA in the immediate vicinity of the fovea. [see Dosage and Administration (2.3)]
Retinal abnormalities may occur during or following vitrectomy including retinal tears, epiretinal membrane, or retinal detachment. Monitor patients during and following the injection to permit early treatment of these retinal abnormalities. Advise patients to report any signs or symptoms of retinal tears and/or detachment without delay5.4 Increased intraocular pressure
Increased intraocular pressure may occur after subretinal injection of LUXTURNA. Monitor and manage intraocular pressure appropriately.
5.5 Expansion of intraocular air bubbles
Instruct patients to avoid air travel, travel to high elevations or scuba diving until the air bubble formed following administration of LUXTURNA has completely dissipated from the eye. It may take one week or more following injection for the air bubble to dissipate. A change in altitude while the air bubble is still present can result in irreversible vision loss. Verify the dissipation of the air bubble through ophthalmic examination.
-
6 ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ 5%) were conjunctival hyperemia, cataract, increased intraocular pressure, retinal tear, dellen (thinning of the corneal stroma), macular hole, subretinal deposits, eye inflammation, eye irritation, eye pain, and maculopathy (wrinkling on the surface of the macula).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of other products and may not reflect the rates observed in practice.
The safety data described in this section reflect exposure to LUXTURNA in two clinical trials consisting of 41 subjects (81 eyes) with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Forty of the 41 subjects received sequential subretinal injections of LUXTURNA to each eye. One subject received LUXTURNA in only one eye. Seventy-two of the 81 eyes were exposed to the recommended dose of LUXTURNA at 1.5 x 1011 vg; 9 eyes were exposed to lower doses of LUXTURNA. Study 1 (n=12) was an open-label, dose-exploration safety study. Study 2 (n=29) was an open-label, randomized, controlled study for both efficacy and safety [(see Clinical Studies (14)]. The average age of the 41 subjects was 17 years ranging from 4 to 44 years. Of the 41 subjects, 25 (61%) were pediatric subjects under 18 years of age, and 23 (56%) were females.
Twenty-seven (27/41, 66%) subjects had ocular adverse reactions that involved 46 injected eyes (46/81, 57%). Adverse reactions among all subjects in Studies 1 and 2 are described in Table 1. Adverse reactions may have been related to voretigene neparvovec-rzyl, the subretinal injection procedure, the concomitant use of corticosteroids, or a combination of these procedures and products.Table 1. Ocular Adverse Reactions Following Treatment with LUXTURNA (N=41) Adverse Reactions Subjects n=41 Treated Eyes n=81 Any ocular adverse reaction 27 (66%) 46 (57%) Conjunctival hyperemia 9 (22%) 9 (11%) Cataract 8 (20%) 15 (19%) Increased intraocular pressure 6 (15%) 8 (10%) Retinal tear 4 (10%) 4 (5%) Dellen (thinning of the corneal stroma) 3 (7%) 3 (4%) Macular hole 3 (7%) 3 (4%) Subretinal deposits* 3 (7%) 3 (4%) Eye inflammation 2 (5%) 4 (5%) Eye irritation 2 (5%) 2 (2%) Eye pain 2 (5%) 2 (2%) Maculopathy (wrinkling on the surface of the macula) 2 (5%) 3 (4%) Foveal thinning and loss of foveal function 1 (2%) 2 (2%) Endophthalmitis 1 (2%) 1 (1%) Foveal dehiscence (separation of the retinal layers in the center of the macula) 1 (2%) 1 (1%) Retinal hemorrhage 1 (2%) 1 (1%) *Transient appearance of asymptomatic subretinal precipitates inferior to the retinal injection site 1-6 days after injection
Immunogenicity
At all doses of LUXTURNA evaluated in Studies 1 and 2, immune reactions and extra-ocular exposure were mild. In Study 1 (n=12), the interval between the subretinal injections into the two eyes ranged from 1.7 to 4.6 years. In Study 2, the interval between the subretinal injections into the two eyes ranged from 7 to 14 days. No subject had a clinically significant cytotoxic T-cell response to either AAV2 or RPE65.
Subjects received systemic corticosteroids before and after subretinal injection of LUXTURNA to each eye. The corticosteroids may have decreased the potential immune reaction to either vector capsid (adeno-associated virus serotype 2 [AAV2] vector) or transgene product (retinal pigment epithelial 65 kDa protein [RPE65]). -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Adequate and well-controlled studies with LUXTURNA have not been conducted in pregnant women. Animal reproductive studies have not been conducted with LUXTURNA. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of LUXTURNA in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for LUXTURNA and any potential adverse effects on the breastfed infant from LUXTURNA.8.3 Females and Males of Reproductive Potential
No nonclinical or clinical studies were performed to evaluate the effect of LUXTURNA on fertility.
8.4 Pediatric Use
Treatment with LUXTURNA is not recommended for patients younger than 12 months of age, because the retinal cells are still undergoing cell proliferation, and LUXTURNA would potentially be diluted or lost during cell proliferation.
The safety and efficacy of LUXTURNA have been established in pediatric patients. Use of LUXTURNA is supported by Study 1 and Study 2 [see Clinical Studies (14)] that included 25 pediatric patients with biallelic RPE65 mutation-associated retinal dystrophy in the following age groups: 21 children (age 4 years to less than 12 years) and 4 adolescents (age 12 years to less than 17 years). There were no significant differences in safety between the different age subgroups.
-
11 DESCRIPTION
LUXTURNA (voretigene neparvovec-rzyl) is a suspension of an adeno-associated virus vector-based gene therapy for subretinal injection. LUXTURNA is a live, non-replicating adeno-associated virus serotype 2 which has been genetically modified to express the human RPE65 gene. LUXTURNA is derived from naturally occurring adeno-associated virus using recombinant DNA techniques.
Each single-dose vial of LUXTURNA contains 5 x 1012 vector genomes (vg) per mL, and the excipients 180 mM sodium chloride, 10 mM sodium phosphate, and 0.001% Poloxamer 188 (pH 7.3), in a 0.5-mL extractable volume. LUXTURNA requires a 1:10 dilution prior to administration. After dilution, each dose of LUXTURNA consists of 1.5 x 1011 vg in a deliverable volume of 0.3 mL.
The Diluent, supplied in 1.7 mL extractable volume per vial in two 2-mL vials, is composed of sterile water containing 180 mM sodium chloride, 10 mM sodium phosphate, and 0.001% Poloxamer 188 (pH 7.3).
LUXTURNA may also contain residual components of HEK293 cells including DNA and protein and trace quantities of fetal bovine serum.
The product contains no preservative. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
LUXTURNA is designed to deliver a normal copy of the gene encoding the human retinal pigment epithelial 65 kDa protein (RPE65) to cells of the retina in persons with reduced or absent levels of biologically active RPE65. The RPE65 is produced in the retinal pigment epithelial (RPE) cells and converts all-trans-retinol to 11-cis-retinol, which subsequently forms the chomophore, 11-cis-retinal, during the visual (retinoid) cycle. The visual cycle is critical in phototransduction, which refers to the biological conversion of a photon of light into an electrical signal in the retina. Mutations in the RPE65 gene lead to reduced or absent levels of RPE65 isomerohydrolase activity, blocking the visual cycle and resulting in impairment of vision.
12.2 Pharmacodynamics
Injection of LUXTURNA into the subretinal space results in transduction of some retinal pigment epithelial cells with a cDNA encoding normal human RPE65 protein, thus providing the potential to restore the visual cycle.
12.3 Pharmacokinetics
Biodistribution (within the body) and Vector Shedding (excretion/secretion)
LUXTURNA vector DNA levels in various tissues and secretions were determined using a quantitative polymerase chain reaction (qPCR) assay.
Nonclinical data
Biodistribution of LUXTURNA was evaluated at three months following subretinal administration in non-human primates. The highest levels of vector DNA sequences were detected in intraocular fluids (anterior chamber fluid and vitreous) of vector-injected eyes. Low levels of vector DNA sequences were detected in the optic nerve of the vector-injected eye, optic chiasm, spleen and liver, and sporadically in the lymph nodes. Vector DNA sequences were not detected in the gonads.
Clinical data
LUXTURNA vector shedding and biodistribution were investigated in a study measuring LUXTURNA DNA in tears from both eyes, and from serum, and whole blood of subjects in Study 2. In summary, LUXTURNA vector was shed transiently and at low levels in tears from the injected eye in 45% of the subjects in Study 2, and occasionally (7%) from the uninjected eye until Day 3 post-injection.
In 29 subjects who received bilateral administrations, LUXTURNA vector DNA was present in tear samples of 13 subjects (45%). Peak levels of vector DNA were detected in the tear samples on Day 1 post-injection, after which no vector DNA was detected in a majority of the subjects (8 of 13). Three subjects (10%) had vector DNA in tear samples until Day 3 post-injection, and two subjects (7%) had vector DNA in tear samples for around two weeks post-injection. In another two subjects (7%), vector DNA was detected in tear samples from the uninjected (or previously injected) eye until Day 3 post-injection. Vector DNA was detected in serum in 3/29 (10%) subjects, including two with vector DNA in tear samples up to Day 3 following each injection.
Specific Populations
No pharmacokinetic studies with LUXTURNA have been conducted.
Drug Interaction Studies
No interaction studies have been performed with LUXTURNA. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been conducted to evaluate the effects of LUXTURNA on carcinogenesis, mutagenesis, and impairment of fertility.
13.2 Animal Toxicology and/or Pharmacology
Bilateral, simultaneous subretinal administration of LUXTURNA was well tolerated at dose levels up to 8.25 x 1010 vg per eye in dogs with a naturally occurring RPE-65 mutation and 7.5 x 1011 vg (5 times higher than the recommended human dose level) per eye in non-human primates (NHPs) with normal-sighted eyes. In both animal models, bilateral, sequential subretinal administrations, where the contralateral eye was injected following the first eye, were well tolerated at the recommended human dose level of 1.5 x 1011 vg per eye. In addition, dogs with with the RPE-65 mutation displayed improved visual behavior and pupillary responses. Ocular histopathology showed only mild changes, which were mostly related to healing from the surgical administration procedure. Other findings observed following subretinal injection of LUXTURNA in dogs and NHPs included occasional and isolated inflammatory cells in the retina, with no apparent retinal degeneration. Dogs not previously exposed to AAV2 vectors developed antibodies to the AAV2 capsid following a single administration of LUXTURNA, whereas NHPs did not.
-
14 CLINICAL STUDIES
The efficacy of LUXTURNA in pediatric and adult patients with biallelic RPE65 mutation-associated retinal dystrophy was evaluated in an open-label, two-center, randomized trial (Study 2). Of the 31 enrolled subjects, 21 subjects were randomized to receive subretinal injection of LUXTURNA. One subject discontinued from the study prior to treatment. Ten subjects were randomized to the control (non-intervention) group. One subject in the control group withdrew consent and was discontinued from the study. The nine subjects who were randomized to the control group were crossed over to receive subretinal injection of LUXTURNA after one year of observation. The average age of the 31 randomized subjects was 15 years (range 4 to 44 years), including 64% pediatric subjects (n=20, age from 4 to 17 years) and 36% adults (n=11). The 31 randomized subjects included 13 males and 18 females. Sixty-eight percent (68%) of the subjects were White, 16% were Asian, 10% were American Indian or Alaska Native, and 6% were Black or African-American. Bilateral subretinal injections of LUXTURNA were administered sequentially in two separate surgical procedures with an interval of 6 to 18 days.
The efficacy of LUXTURNA was established on the basis of multi-luminance mobility testing (MLMT) score change from Baseline to Year 1. The MLMT was designed to measure changes in functional vision, as assessed by the ability of a subject to navigate a course accurately and at a reasonable pace at different levels of environmental illumination. The MLMT was assessed using both eyes and each eye separately at one or more of seven levels of illumination, ranging from 400 lux (corresponding to a brightly lit office) to 1 lux (corresponding to a moonless summer night). Each light level was assigned a score code ranging from 0 to 6. A higher score indicated that a subject was able to pass the MLMT at a lower light level. A score of -1 was assigned to subjects who could not pass MLMT at a light level of 400 lux. The MLMT of each subject was videotaped and assessed by independent graders. The MLMT score was determined by the lowest light level at which the subject was able to pass the MLMT. The MLMT score change was defined as the difference between the score at Baseline and the score at Year 1. A positive MLMT score change from Baseline to Year 1 visit indicated that the subject was able to complete the MLMT at a lower light level.
Additional clinical outcomes were also evaluated, including full-field light sensitivity threshold (FST) testing, visual acuity, and visual fields.
Table 2 summarizes the median MLMT score change from Baseline to Year 1 in the LUXTURNA treatment group as compared to the control group. A median MLMT score of 2 or greater was observed in the LUXTURNA treatment group, while a median MLMT score change of 0 was observed in the control group, when using both eyes or the first-treated eye. An MLMT score change of two or greater is considered a clinically meaningful benefit in functional vision.
Table 2. Efficacy Results of Study 2 at Year 1, Compared to Baseline Efficacy Outcomes LUXTURNA
n=21Control
n=10Difference
(LUXTURNA
minus Control)p-value MLMT score change for bilateral eyes, median (min, max) 2 (0, 4) 0 (-1, 2) 2 0.001 MLMT score change for first-treated eye, median (min, max) 2 (0, 4) 0 (-1, 1) 2 0.003 Table 3 shows the number and percentage of subjects with different magnitudes of MLMT score change using both eyes at Year 1. Eleven of the 21 (52%) subjects in the LUXTURNA treatment group had an MLMT score change of two or greater, while one of the ten (10%) subjects in the control group had an MLMT score change of two.
Table 3. Magnitude of MLMT Score Change Using Both Eyes at Year 1 (Study 2) Score Change LUXTURNA
n=21Control
n=10-1 0 3 (30%) 0 2 (10%) 3 (30%) 1 8 (38%) 3 (30%) 2 5 (24%) 1 (10%) 3 5 (24%) 0 4 1 (4%) 0 Figure 6 shows MLMT performance of individual subjects using both eyes at Baseline and at Year 1.
Figure 6. MLMT Score Using Both Eyes at Baseline and One Year for Individual Subjects
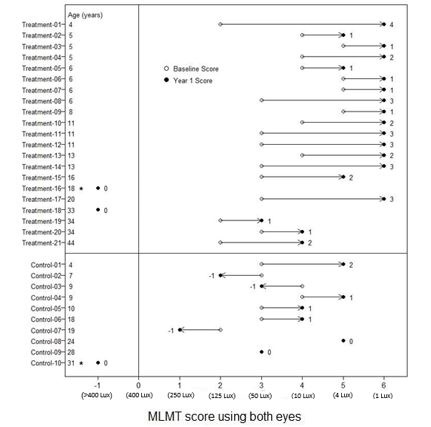
Note for Figure 6: *subjects who were withdrawn or discontinued. The open circles are the baseline scores. The closed circles are the Year 1 scores. The numbers next to the solid circle represent score change at Year 1. The horizontal lines with arrows represent the magnitude of the score change and its direction. Arrows pointing towards the right represent improvement. The top section shows the results of the 21 subjects in the treatment group. The bottom section shows the results of the 10 subjects in the control group. Subjects in each group are chronologically organized by age, with the youngest subject at the top and the oldest subject at the bottom.
Analysis of white light FST testing showed statistically significant improvement from Baseline to Year 1 in the LUXTURNA treatment group compared to the control group. The change in visual acuity from Baseline to Year 1 was not significantly different between the LUXTURNA and control groups.
Figure 7 shows the effect of LUXTURNA over the two-year period in the LUXTURNA treatment group, as well as the effect in the control group after crossing over to receive subretinal injection of LUXTURNA. A median MLMT score change of two was observed for the LUXTURNA treatment group at Day 30, and this effect was sustained over the remaining follow-up visits throughout the two-year period. For the control group, a median MLMT score change of 0 was observed at all four follow-up visits during the first year. However, after crossing-over to receive subretinal injection of LUXTURNA, the subjects in the control group showed a similar response to LUXTURNA as compared to the subjects in the LUXTURNA treatment group.
Figure 7. MLMT Time-Course over Two Years: Using Both Eyes
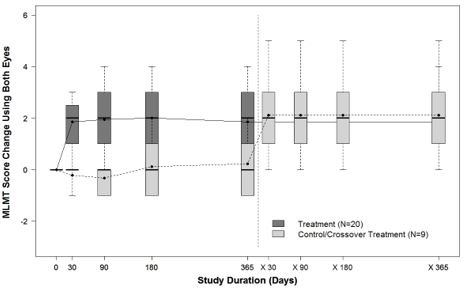
Note for Figure 7: Each box represents the middle 50% of distribution of MLMT score change. Vertical dotted lines represent additional 25% above and below the box. The horizontal bar within each box represents the median. The dot within each box represents the mean. The solid line connects the mean MLMT score changes over visits for the treatment group, including five visits during the first year and one visit at Year 2 (marked as x365). The dotted line connects the mean MLMT score change over visits for the control group, including five visits during the first year without receiving LUXTURNA, and four visits within the second year (marked as x30, x90, x180, and x365) after cross-over at Year 1 to receive LUXTURNA.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each carton of LUXTURNA (NDC 71394 – 415-01) contains one single-dose vial of the LUXTURNA (NDC 71394 – 065-01, 0.5 mL extractable volume) and two vials of Diluent (NDC 71394 – 716-01, 1.7 mL extractable volume in each vial). LUXTURNA contains 5 x 1012 vector genomes (vg) per mL, requires a 1:10 dilution prior to administration.
Store LUXTURNA and Diluent frozen at ≤ -65 °C.
Following thaw of the vials, store at room temperature. Store diluted LUXTURNA at room temperature [See Dosage and Administration 2.2].
LUXTURNA is an adeno-associated virus vector-based gene therapy. Follow universal biohazard precautions for handling. -
17 PATIENT COUNSELING INFORMATION
Advise patients and/or their caregivers of the following risks.
- Endophthalmitis and other eye infections
Serious infection can occur inside of the eye and may lead to blindness. In such cases, there is an urgent need for management without delay. Advise patients to call their healthcare provider if they experience new floaters, eye pain, or any change in vision.
- Permanent decline in visual acuity
Permanent decline in visual acuity may occur following subretinal injection of LUXTURNA. Advise patients to contact their healthcare provider if they exprience any change in vision.
- Retinal abnormalities
Treatment with LUXTURNA may cause some defects in the retina such as a small tear or a hole in the area or vicinity of the injection. Treatment may cause thinning of the central retina or bleeding in the retina. Advise patients to follow up with their healthcare provider on a regular basis and report any symptoms such as decreased vision, blurred vision, flashes of light, or floaters in their vision without delay.
- Increased intraocular pressure
Treatment with LUXTURNA may cause transient or persistent increase in intraocular pressure. If untreated, such increases in intraocular pressure may cause blindness. Advise patients to follow-up with their healthcare provider to detect and treat any increase in intraocular pressure.
- Expansion of intraocular air bubbles
Advise patients to avoid air travel, travel to high elevations or scuba diving until the air bubble formed following administration of LUXTURNA has completely dissipated from the eye. A change in altitude while the air bubble is still present may cause irreversible damage.
- Cataract
Advise patients that following treatment with LUXTURNA, they may develop a new cataract, or any existing cataract may get worse.
- Shedding of LUXTURNA
Transient and low level shedding of LUXTURNA may occur in patient tears. Advise patients and/or their caregivers on proper handling of waste material generated from dressing, tears and nasal secretion, which may include storage of waste material in sealed bags prior to disposal. These handling precautions should be followed for up to 7 days following LUXTURNA administration.
Manufactured by:
Spark Therapeutics, Inc.
3737 Market Street
Philadelphia, PA 19104US License #2056
LUXTURNA is a trademark of Spark Therapeutics, Inc.
© 2017 Spark Therapeutics, Inc. -
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Luxturna Carton Label
NDC: 71394-415-01
Rx only
voretigene neparvovec-rzyl
LUXTURNA™
5 x 1012 vector genomes/mL. No US standard of potency.
One (1) single-dose vial of voretigene neparvovec-rzyl, 0.5 mL per vial
Two (2) vials of Diluent for voretigene neparvovec-rzyl, 1.7 mL per vial
Store at ≤ -65°C. Dilute before use. Discard unused portion.
For administration by subretinal injection
See package insert for full prescribing information and instructions for
dosage and administration.Manufactured by Spark Therapeutics Inc.
3737 Market Street, Suite 1300
Philadelphia, Pennsylvania 19104
US License No. 2056
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Diluent for Voretigene Neparvovec Label
NDC: 71394-716-01
Diluent for voretigene neparvovec-rzyl
pH 7.3, containing 0.001%
poloxamer 188
For Rx only
1.7 mL per vial; Store at ≤ -65 °C
Manufactured for Spark Therapeutics
By Nova Laboratories
US License No. 2056
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Luxturna Label
NDC: 71394-065-01
voretigene neparvovec-rzyl
For subretinal injection5 x 1012 vector genomes/mL
One (1) single-dose vial, 0.5 mL per vial
For Rx only; Must dilute before use
Store at ≤ -65 °C; Discard unused portion
Manufactured by
Spark Therapeutics, Inc.
US License No. 2056
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Luxturna Pouch Label
NDC: 71394-415-01
Rx only
voretigene neparvovec-rzyl
LUXTURNA™
5 x 1012 vector genomes/mL. No US standard of potency.
One (1) single-dose vial of voretigene neparvovec-rzyl, 0.5 mL per vial
Two (2) vials of Diluent for voretigene neparvovec-rzyl, 1.7 mL per vial
Store at ≤ -65 °C. Dilute before use. Discard unused portion.
For administration by subretinal injection
See package insert for full prescribing information and instructions
for dosage and administrationNDC: 71394-415-01
Manufactured by Spark Therapeutics
3737 Market Street, Suite 1300
Philadelphia, Pennsylvania 19104
US License No. 2056
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Luxturna Pouch Label
NDC: 71394-415-01
Rx only
voretigene neparvovec-rzyl
LUXTURNA™
5 x 1012 vector genomes/mL. No US standard of potency.
One (1) single-dose vial of voretigene neparvovec-rzyl, 0.5 mL per vial
Two (2) vials of Diluent for voretigene neparvovec-rzyl, 1.7 mL per vial
Store at ≤ -65 °C. Dilute before use. Discard unused portion.
For administration by subretinal injection
See package insert for full prescribing information and instructions
for dosage and administrationNDC: 71394-415-01
Manufactured by Spark Therapeutics
3737 Market Street, Suite 1300
Philadelphia, Pennsylvania 19104
US License No. 2056
-
INGREDIENTS AND APPEARANCE
LUXTURNA
voretigene neparvovec-rzyl kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71394-415 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71394-415-01 1 in 1 CARTON 12/19/2017 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, PLASTIC 1 Part 2 2 VIAL, PLASTIC 2 Part 1 of 2 LUXTURNA
voretigene neparvovec-rzyl injection, suspensionProduct Information Item Code (Source) NDC: 71394-065 Route of Administration SUBRETINAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VORETIGENE NEPARVOVEC (UNII: 2SPI046IKD) (VORETIGENE NEPARVOVEC - UNII:2SPI046IKD) VORETIGENE NEPARVOVEC 0.05 mg Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 2.3 mmol SODIUM PHOSPHATE, DIBASIC, DIHYDRATE (UNII: 94255I6E2T) 7.7 mmol SODIUM CHLORIDE (UNII: 451W47IQ8X) 180 mmol POLOXAMER 188 (UNII: LQA7B6G8JG) 0.001 WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71394-065-01 1 in 1 VIAL, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125610 12/19/2017 Part 2 of 2 DILUENT FOR VORETIGENE NEPARVOVEC-RZYL
diluent injection, solutionProduct Information Item Code (Source) NDC: 71394-716 Route of Administration SUBRETINAL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC (UNII: 3980JIH2SW) 2.3 mmol SODIUM PHOSPHATE, DIBASIC (UNII: GR686LBA74) 7.7 mmol SODIUM CHLORIDE (UNII: 451W47IQ8X) 180 mmol POLOXAMER 188 (UNII: LQA7B6G8JG) 0.001 WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71394-716-01 1 in 1 VIAL, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125610 12/19/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125610 12/19/2017 Labeler - Spark Therapeutics, Inc. (079498241) Establishment Name Address ID/FEI Business Operations Spark Therapeutics, Inc. 079498241 API MANUFACTURE(71394-415, 71394-065, 71394-716) , ANALYSIS(71394-415, 71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations BioReliance Corporation 147227730 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Pharmaceutical Product Development, Inc. (PPD) 838082055 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Absorption Systems 007797314 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Eurofins (Lancaster Laboratories) 069777290 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Intertek Pharmaceutical Services 805606758 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Nova Laboratories Ltd 230804692 manufacture(71394-065, 71394-716) , ANALYSIS(71394-065, 71394-716) , LABEL(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Catalent Pharma Solutions 232616826 PACK(71394-415, 71394-065, 71394-716) , LABEL(71394-415, 71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Hologic Ltd Tepnel Pharma Services 218122626 ANALYSIS(71394-065, 71394-716) Establishment Name Address ID/FEI Business Operations Synergy Health (part of Steris) 226067312 STERILIZE(71394-065, 71394-716)
Trademark Results [LUXTURNA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LUXTURNA 88064462 not registered Live/Pending |
Spark Therapeutics, Inc. 2018-08-03 |
 LUXTURNA 88064454 not registered Live/Pending |
Spark Therapeutics, Inc. 2018-08-03 |
 LUXTURNA 87148092 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-23 |
 LUXTURNA 87124430 not registered Live/Pending |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 87124428 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 87124426 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 87124425 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 87124423 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 87124418 not registered Dead/Abandoned |
Spark Therapeutics, Inc. 2016-08-02 |
 LUXTURNA 86910422 5503541 Live/Registered |
Spark Therapeutics, Inc. 2016-02-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.