BYLVAY- odevixibat capsule, coated pellets BYLVAY- odevixibat capsule
Bylvay by
Drug Labeling and Warnings
Bylvay by is a Prescription medication manufactured, distributed, or labeled by Ipsen Biopharmaceuticals, Inc., Syngene International Limited., Almac Pharma Services Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BYLVAY safely and effectively. See full prescribing information for BYLVAY.
BYLVAY (odevixibat) capsules, for oral use
BYLVAY (odevixibat) oral pellets
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BYLVAY is an ileal bile acid transporter (IBAT) inhibitor indicated for:
Progressive Familial Intrahepatic Cholestasis (PFIC)
- the treatment of pruritus in patients 3 months of age and older with progressive familial intrahepatic cholestasis (PFIC). (1.1)
Limitation of Use:
BYLVAY is not recommended in a subgroup of PFIC type 2 patients with specific ABCB11 variants resulting in non-functional or complete absence of the bile salt export pump protein. (12.5, 14.1)
Alagille Syndrome (ALGS)
- the treatment of cholestatic pruritus in patients 12 months of age and older with Alagille syndrome (ALGS). (1.2)
DOSAGE AND ADMINISTRATION
Recommended Dosage
PFIC:
- Patients 3 months and older: 40 mcg/kg taken orally once daily. (2.1)
- If there is no improvement in pruritus after 3 months, the dosage may be increased in 40 mcg/kg increments up to 120 mcg/kg once daily, not to exceed a daily dosage of 6 mg/day. (2.1)
ALGS:
- Patients 12 months and older: 120 mcg/kg taken orally once daily. (2.2)
- Preparation and Administration Instructions
CONTRAINDICATIONS
Patients with prior or active hepatic decompensation events (e.g., variceal hemorrhage, ascites, hepatic encephalopathy). (4)
WARNINGS AND PRECAUTIONS
- Hepatoxicity: Obtain baseline liver tests and monitor patients frequently for the first 6 to 8 months after starting therapy, and as clinically indicated thereafter during treatment. If liver test abnormalities or signs of clinical hepatitis occur, consider dose reduction or treatment interruption. For persistent or recurrent liver test abnormalities relative to baseline, discontinue BYLVAY. Monitor patients with compensated cirrhosis or portal hypertension more frequently. Permanently discontinue BYLVAY if hepatic decompensation occurs. (2.3, 5.1)
- Diarrhea: Treat dehydration. Treatment interruption or discontinuation may be required for persistent diarrhea. (5.2)
-
Fat-Soluble Vitamin (FSV) Deficiency: Obtain baseline levels and monitor during treatment. Supplement with FSV if deficiency is observed. If FSV deficiency persists or worsens despite FSV supplementation, consider discontinuing BYLVAY treatment.
- Fracture: Consider interrupting BYLVAY treatment. Supplement with FSV if indicated.
- Bleeding: Interrupt treatment with BYLVAY. Optimize treatment of FSV deficiency and consider restarting BYLVAY once the patient is clinically stable. (5.3)
ADVERSE REACTIONS
PFIC: Most common adverse reactions (>2%) are liver test abnormalities, diarrhea, abdominal pain, vomiting, and fat-soluble vitamin deficiency. (6.1)
ALGS: Most common adverse reactions (>5%) are diarrhea, abdominal pain, hematoma, and decreased weight. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Ipsen Biopharmaceuticals, Inc. at 1-855-463-5127, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Progressive Familial Intrahepatic Cholestasis (PFIC)
1.2 Alagille Syndrome (ALGS)
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Progressive Familial Intrahepatic Cholestasis (PFIC) in Patients Aged 3 Months and Older
2.2 Recommended Dosage for Alagille Syndrome (ALGS) in Patients Aged 12 Months and Older
2.3 Dosage Modification for Management of Adverse Reactions
2.4 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatoxicity
5.2 Diarrhea
5.3 Fat-Soluble Vitamin Deficiency
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Bile Acid Binding Resins
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 PFIC
14.2 ALGS
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Progressive Familial Intrahepatic Cholestasis (PFIC)
BYLVAY is indicated for the treatment of pruritus in patients 3 months of age and older with PFIC.
Limitations of Use
- BYLVAY is not recommended in a subgroup of PFIC type 2 patients with specific ABCB11 variants resulting in non-functional or complete absence of the bile salt export pump (BSEP) protein [see Clinical Pharmacology (12.5) and Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage for Progressive Familial Intrahepatic Cholestasis (PFIC) in Patients Aged 3 Months and Older
- The recommended dosage of BYLVAY is 40 mcg/kg taken orally once daily in the morning with a meal. Table 1 below shows the recommended once daily dosage by body weight.
- If there is no improvement in pruritus after 3 months, the dosage may be increased in 40 mcg/kg increments up to 120 mcg/kg once daily not to exceed a daily dosage of 6 mg/day.
- BYLVAY oral pellets are intended for use by patients weighing less than 19.5 kilograms.
- BYLVAY capsules are intended for use by patients weighing 19.5 kilograms or above.
Table 1. Recommended Dosage for PFIC in Patients aged 3 months and older (40 mcg/kg/day) Body Weight (kg) Once Daily Dosage (mcg) 7.4 and below 200 7.5 to 12.4 400 12.5 to 17.4 600 17.5 to 25.4 800 25.5 to 35.4 1,200 35.5 to 45.4 1,600 45.5 to 55.4 2,000 55.5 and above 2,400 2.2 Recommended Dosage for Alagille Syndrome (ALGS) in Patients Aged 12 Months and Older
- The recommended dosage of BYLVAY is 120 mcg/kg taken orally once daily in the morning with a meal. Table 2 below shows the recommended once daily dosage by body weight.
- BYLVAY oral pellets are intended for use by patients weighing less than 19.5 kilograms.
- BYLVAY capsules are intended for use by patients weighing 19.5 kilograms or above.
Table 2. Recommended Dosage for ALGS in Patients aged 12 months and older (120 mcg/kg/day) Body Weight (kg) Once Daily Dosage (mcg) 7.4 and below 600 7.5 to 12.4 1,200 12.5 to 17.4 1,800 17.5 to 25.4 2,400 25.5 to 35.4 3,600 35.5 to 45.4 4,800 45.5 to 55.4 6,000 55.5 and above 7,200 2.3 Dosage Modification for Management of Adverse Reactions
Tolerability for Alagille Syndrome (ALGS)
Dose reduction to 40 mcg/kg/day (Table 1) may be considered if tolerability issues occur in the absence of other causes. Once tolerability issues stabilize, increase to 120 mcg/kg/day.
Liver Test Abnormalities
Establish the baseline pattern of variability of liver tests prior to starting BYLVAY, so that potential signs of liver injury can be identified. Monitor liver tests (e.g., ALT [alanine aminotransferase], AST [aspartate aminotransferase], TB [total bilirubin], DB [direct bilirubin] and International Normalized Ratio [INR]) during treatment with BYLVAY. Interrupt BYLVAY if new onset liver test abnormalities occur or symptoms consistent with clinical hepatitis are observed [see Warnings and Precautions (5.1)].
Once the liver test abnormalities either return to baseline values or stabilize at a new baseline value, consider restarting BYLVAY at the recommended dosage [see Dosage and Administration (2.1, 2.2)]. Consider discontinuing BYLVAY permanently if liver test abnormalities recur.
Discontinue BYLVAY permanently if a patient experiences a hepatic decompensation event (e.g., variceal hemorrhage, ascites, hepatic encephalopathy).
2.4 Preparation and Administration Instructions
- For patients taking bile acid binding resins, take BYLVAY at least 4 hours before or 4 hours after taking a bile acid binding resin [see Drug Interactions (7.1)].
- Do not crush or chew capsules.
Oral Pellets:
- Mix the contents of the shell containing Oral Pellets into soft food or a liquid (as described below).
- Discard the emptied shells. Do not let your child swallow the unopened shells containing the Oral Pellets.
Administration Instructions for patients capable of swallowing soft food:
- Administer BYLVAY with the first morning meal.
- Place a small amount (up to 2 tablespoons [30 mL]) of soft food (apple sauce, oatmeal, banana or carrot puree, chocolate or rice pudding) in a bowl. Keep the soft food at, or cooler than, room temperature.
- Open the shell containing Oral Pellets and empty the contents into the bowl of soft food. Gently tap the Oral Pellet shell to ensure that all contents have been dispersed.
- If the dose requires more than one shell of Oral Pellets, repeat Step 3.
- Gently mix until well dispersed and administer the entire dose immediately.
- Follow the dose with breast milk, infant formula, or other age-appropriate liquid.
- Do not store the mixture for future use.
- For patients unable to swallow soft food, see the instructions below.
Administration Instructions with liquids (Using an oral dosing syringe):
- Administer BYLVAY with the first morning meal.
- Open the shell containing Oral Pellets and empty the contents into a small mixing cup. Gently tap the shell containing Oral Pellets to ensure that all contents have been emptied into the mixing cup.
- If the dose requires more than one shell of Oral Pellets, repeat Step 2.
- Add 1 teaspoon (5 mL) of an age-appropriate liquid (for example, breast milk, infant formula, or water).
- Let the pellets sit in the liquid for approximately 5 minutes to allow complete wetting REMINDER: The Oral Pellets will not dissolve in the liquid.
- After 5 minutes, place the tip of the oral syringe completely into the mixing cup. Pull the plunger of the syringe up slowly to withdraw the liquid/pellet mixture into the syringe. Gently push the plunger down again to expel the liquid/pellet mixture back into the mixing cup. Do this 2 to 3 times to ensure complete mixing of the pellets into the liquid.
- Withdraw the entire contents into the oral syringe by pulling the plunger on the end of the syringe.
- Place the tip of the syringe into the front of the patient's mouth between the tongue and the side of the mouth, and then gently push the plunger down to squirt the liquid/pellet mixture between your child's tongue and the side of the mouth. Do not squirt liquid/pellet mixture in the back of the child's throat because this could cause gagging or choking.
- Do not administer via a bottle or "sippy cup" because the Oral Pellets will not pass through the opening. The oral pellets will not dissolve in liquid.
- Follow the dose with breast milk, infant formula, or other age-appropriate liquid.
- If any pellet/liquid mixture remains in the mixing cup, repeat Step 7 and Step 8 until the entire dose has been administered.
- Check the child's mouth to make sure all of the liquid/pellet mixture has been swallowed.
Capsules:
Administration Instructions:
- Take in the morning with a meal.
- Swallow the capsule whole with a glass of water.
- Alternatively, for patients unable to swallow the capsules whole, BYLVAY capsules may be opened, and sprinkled and mixed with a small amount of soft food, or mixed with an age-appropriate liquid. Follow directions above for oral pellets to prepare and administer such a mixture.
-
3 DOSAGE FORMS AND STRENGTHS
- Oral Pellets:
- 200 mcg: capsule with ivory opaque cap and white opaque body; imprinted "A200" (black ink).
- 600 mcg: capsule with ivory opaque cap and body; imprinted "A600" (black ink).
- Capsules:
- 400 mcg: capsule with medium orange opaque cap and white opaque body; imprinted "A400" (black ink).
- 1200 mcg: capsule with medium orange opaque cap and body; imprinted "A1200" (black ink).
- Oral Pellets:
-
4 CONTRAINDICATIONS
IBAT inhibitors, including BYLVAY, are contraindicated in patients with prior or active hepatic decompensation events (e.g., variceal hemorrhage, ascites, hepatic encephalopathy) [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatoxicity
BYLVAY treatment is associated with a potential for drug-induced liver injury (DILI).
In the PFIC and ALGS trials, treatment-emergent elevations of liver tests or worsening of liver tests occurred. Of the six patients who experienced DILI, two underwent liver transplant.
Obtain baseline liver tests because some ALGS and PFIC patients have abnormal liver tests at baseline. Monitor patients frequently for the first 6 to 8 months after starting therapy and as clinically indicated thereafter during treatment with BYLVAY. Monitor for elevation in liver tests, for the development of liver-related adverse reactions, and for physical signs of hepatic decompensation. If liver test abnormalities or signs of clinical hepatitis occur in the absence of other causes, consider dose reduction or treatment interruption.
Permanently discontinue BYLVAY if a patient experiences the following:
- persistent or recurrent liver test abnormalities, or
- upon rechallenge, signs and symptoms consistent with clinical hepatitis, or
- a hepatic decompensation event.
The safety and effectiveness of BYLVAY have not been established in patients with decompensated cirrhosis. Monitor patients with compensated cirrhosis or portal hypertension more frequently and discontinue BYLVAY if hepatic decompensation occurs. IBAT inhibitors, including BYLVAY, are contraindicated in patients with prior or active hepatic decompensation events [see Contraindications (4)].
5.2 Diarrhea
In Trial 1, diarrhea in PFIC patients was reported in 2 (10%) placebo-treated patients, 9 (39%) BYLVAY-treated 40 mcg/kg/day patients and 4 (21%) BYLVAY-treated 120 mcg/kg/day patients. Treatment interruption due to diarrhea occurred in 2 patients with 3 events during treatment with BYLVAY 120 mcg/kg/day. Treatment interruption due to diarrhea ranged between 3 to 7 days [see Adverse Reactions (6.1)]. One patient treated with BYLVAY 120 mcg/kg/day withdrew from Trial 1 due to persistent diarrhea.
In Trial 3, diarrhea in ALGS patients was reported in 1 placebo-treated patient (6%) and in 10 (29%) BYLVAY-treated patients [see Adverse Reactions (6.1)]. No patients interrupted or permanently discontinued BYLVAY due to diarrhea.
If diarrhea occurs, monitor for dehydration and treat promptly. Interrupt BYLVAY dosing if a patient experiences persistent diarrhea. Restart BYLVAY at 40 mcg/kg/day when diarrhea resolves, and increase the dose as tolerated if appropriate. If diarrhea persists and no alternate etiology is identified, stop BYLVAY treatment.
5.3 Fat-Soluble Vitamin Deficiency
BYLVAY may adversely affect absorption of fat-soluble vitamins (FSV). FSV include vitamin A, D, E, and K (measured using INR levels). PFIC and ALGS patients can have FSV deficiency at baseline and are frequently supplemented with FSV.
In Trial 1 in PFIC patients, new onset or worsening of existing FSV deficiency was reported in 1 (5%) placebo-treated patient, and 3 (16%) BYLVAY-treated 120 mcg/kg/day patients; none of the patients treated with BYLVAY dosage 40 mcg/kg/day had new onset or worsening of existing FSV deficiency.
In Trial 3 in ALGS patients, new or worsening of existing FSV deficiency was reported in 3 (17.6%) placebo- treated patients and 3 (8.6%) BYLVAY-treated patients [see Adverse Reactions (6.1)].
Obtain serum FSV levels at baseline and monitor during treatment, along with any clinical manifestations of FSV deficiency. If FSV deficiency is diagnosed, supplement with FSV. If FSV deficiency persists or worsens despite adequate FSV supplementation, consider permanent discontinuation of BYLVAY depending on the benefit and risk balance.
If complications of FSV deficiency occur, consider interrupting BYLVAY treatment and reassess to ensure adequate supplementation with FSV. Consider restarting BYLVAY once the patient is clinically stable.
Bone Fracture
Fracture events have been observed with BYLVAY-treated patients in two open-label postmarketing studies (5% in PFIC patients and 4% in ALGS patients) [see Adverse Reactions (6)]. If fracture occurs, consider interrupting BYLVAY treatment and supplement with FSV if indicated.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Diarrhea [see Warnings and Precautions (5.2)]
- Fat-Soluble Vitamin Deficiency [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
PFIC Clinical Studies
Trial 1 is a randomized, double-blind, placebo-controlled, 24-week study of two dose levels of BYLVAY (40 mcg/kg and 120 mcg/kg) administered once daily [see Clinical Studies (14.1)]. Sixty-two patients were randomized (1:1:1) to receive one of the following:
- BYLVAY 40 mcg/kg/day (n=23),
- BYLVAY 120 mcg/kg/day (n=19), or
- Placebo (n=20).
Table 3 summarizes the frequency of adverse reactions reported in ≥2% and at a rate greater than placebo in patients treated with BYLVAY in Trial 1. The most common adverse reactions observed in Trial 1 included diarrhea, liver test abnormalities, vomiting, abdominal pain, and fat-soluble vitamin deficiency.
Table 3. Common Adverse Reactions* from a Clinical Study of BYLVAY in Patients with Progressive Familial Intrahepatic Cholestasis (Trial 1) Adverse Reaction Placebo
N=20
n (%)BYLVAY 40 mcg/kg/day
N=23
n (%)BYLVAY 120 mcg/kg/day
N=19
n (%)- * Adverse reactions that occurred in ≥2% of BYLVAY-treated patients
Diarrhea 2 (10%) 9 (39%) 4 (21%) Transaminases increased (ALT, AST) 1 (5%) 3 (13%) 4 (21%) Vomiting 0 4 (17%) 3 (16%) Abdominal pain 0 3 (13%) 3 (16%) Blood bilirubin increased 2 (10%) 3 (13%) 2 (11%) Fat-soluble vitamin deficiency (A, D, E) 1 (5%) 0 3 (16%) Splenomegaly 0 0 2 (11%) Cholelithiasis 0 0 1 (5%) Dehydration 0 0 1 (5%) Fracture 0 1 (4%) 0 Trial 2 is an open-label, single-arm study in 116 patients with PFIC types 1, 2, 3, 4 and 6; four patients with benign recurrent intrahepatic cholestasis (BRIC) were also enrolled. BYLVAY 40 or 120 mcg/kg/day was administered once daily for 72 weeks, with the option to continue treatment beyond 72 weeks.
Adverse reactions were similar to those observed in Trial 1. However, fractures were reported in a total of 6 patients (5%) in Trial 2. Adverse reactions observed in Trial 2 in addition to those described in Table 3 included increased INR (16%), epistaxis (9%), constipation (8%), coagulopathy (3%), headache (3%), nausea (3%), rash (3%), iron deficiency anemia (3%), gastroesophageal reflux disease (2%), prolonged prothrombin time (2%); and variceal hemorrhage, stoma hemorrhage, hematochezia, and rectal hemorrhage (<1% each). Adverse reactions leading to treatment discontinuation were increased bilirubin levels, diarrhea, progression of disease, increased INR, irritability, and decreased weight.
There was a total of 19 (16%) patients who underwent surgical intervention in Trial 2, with one patient who had surgical biliary diversion (SBD) followed by liver transplant, 15 patients who underwent liver transplant alone, and three patients who underwent SBD alone. Overall, 11 of the 19 patients had these surgical interventions prior to Week 72.
ALGS Clinical Studies
Trial 3 is a randomized, double-blind, placebo-controlled, 24-week study of a single dose level of BYLVAY (120 mcg/kg) administered once daily [see Clinical Studies (14.2)]. Fifty-two patients were randomized (2:1) to receive one of the following:
- BYLVAY 120 mcg/kg/day (n=35), or
- Placebo (n=17).
Table 4 summarizes the frequency of adverse reactions in patients with ALGS, reported in ≥5% and at a rate greater than placebo in patients treated with BYLVAY in Trial 3. No patients discontinued study treatment due to an adverse reaction. The most common adverse reactions observed in Trial 3 included diarrhea, abdominal pain, hematoma, and decreased weight.
Table 4. Common Adverse Reactions* from a Clinical Study of BYLVAY in Patients with Alagille Syndrome (Trial 3) Adverse Reaction Placebo
N=17
n (%)BYLVAY 120 mcg/kg/day
N=35
n (%)- * Adverse reactions that occurred in ≥5% of BYLVAY-treated patients
Diarrhea 1 (6%) 10 (29%) Abdominal Pain 1 (6%) 5 (14%) Hematoma 0 3 (9%) Weight decreased 0 2 (6%) Trial 4 is an open-label, single-arm study in 50 pediatric patients with ALGS. BYLVAY 120 mcg/kg/day was administered once daily for 72 weeks, with the option to continue beyond 72 weeks. Adverse reactions observed in Trial 4 in addition to those described in Table 4 included FSV deficiency (vitamin D deficiency [14%], vitamin E deficiency [10%], vitamin K deficiency [6%]), ALT increased (6%), headache (6%), increased INR (6%), increased blood bilirubin (4%), increased AST (4%), coagulopathy (4%), fracture (4%); nausea, vomiting, hematemesis, hematochezia, epistaxis, and constipation (2% each). The most common reason for BYLVAY treatment discontinuation was increased bilirubin levels. One patient underwent liver transplant in Trial 4 prior to Week 72 with no patients underwent SBD.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of BYLVAY. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal disorders: gastrointestinal hemorrhage, gingival hemorrhage, liver transplant
Investigations: gamma-glutamyltransferase increased, hemoglobin decreased
Nervous system disorders: extra-axial hemorrhage (subdural hemorrhage)
Respiratory, thoracic, and mediastinal disorders: epistaxis
-
7 DRUG INTERACTIONS
7.1 Bile Acid Binding Resins
Administer bile acid binding resins (e.g., cholestyramine, colesevelam, or colestipol) at least 4 hours before or 4 hours after administration of BYLVAY [see Dosage and Administration (2.3)]. Bile acid binding resins may bind odevixibat in the gut, which may reduce BYLVAY efficacy.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited human data on the use of BYLVAY in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage, or adverse developmental outcomes. Based on findings from animal reproduction studies, BYLVAY may cause cardiac malformations when a fetus is exposed during pregnancy. In pregnant rabbits treated orally with odevixibat during organogenesis, an increased incidence of malformations in fetal heart, great blood vessels, and other vascular sites occurred at all doses; maternal systemic exposure at the lowest dose was 2.1 times the maximum recommended dose (see Data). Odevixibat may inhibit the absorption of fat-soluble vitamins. FSV are essential for normal fetal growth and development. Monitor pregnant patients for FSV deficiency and supplement as needed. Increased supplementation of FSVs may be needed during pregnancy [see Warnings and Precautions (5.3) and Clinical Considerations]. Consider the woman's need for BYLVAY, the potential drug-related risks to the fetus, and the potential adverse outcomes from untreated maternal PFIC and ALGS.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
There is a pregnancy safety study that monitors pregnancy outcomes in women exposed to BYLVAY during pregnancy. Pregnant women exposed to BYLVAY, or their healthcare providers, should report BYLVAY exposure by calling 1-855-463-5127.
Fetal/Neonatal Adverse Reactions
Odevixibat may inhibit the absorption of fat-soluble vitamins (FSV). Monitor pregnant patients for FSV deficiency and supplement as needed. Increased supplementation of FSVs may be needed during pregnancy [see Warnings and Precautions (5.3)].
Data
Animal Data
In an embryo-fetal development study, pregnant rabbits received oral doses of 10, 30, or 100 mg/kg/day during the period of organogenesis. Fetuses from all maternal groups treated with odevixibat showed an increase in cardiovascular malformations, which included 5-chambered heart, small ventricle, large atrium, ventricular septum defect, misshapen aortic valve, dilated aortic arch, right sided and retroesophageal aortic arch, fusion of aortic arch and pulmonary trunk, ductus arteriosus atresia, and absence of subclavian artery. These malformations occurred at 2.1 times the maximum recommended dose and higher, based on AUC (area under the plasma concentration-time curve). Odevixibat was shown to cross the placenta in pregnant rats.
No adverse effects on embryo-fetal development were observed following oral administration of 100, 300, or 1,000 mg/kg/day in pregnant rats during organogenesis. An increase in skeletal variations (delayed/incomplete ossification and thick ribs) was observed at 1,000 mg/kg/day. Maternal systemic exposure to odevixibat at the maximum dose tested was 272 times the maximum recommended dose, based on AUC.
No adverse effects on postnatal development were observed in a pre- and postnatal development study, in which female rats were treated orally with up to 1,000 mg/kg/day during organogenesis through lactation. The maternal AUC for odevixibat at 1,000 mg/kg/day was 434 times the maximum recommended dose, based on AUC.
8.2 Lactation
Risk Summary
Odevixibat has low absorption following oral administration, and exposure of the infant to BYLVAY through breast milk is not expected at the recommended doses [see Clinical Pharmacology (12.3)]. There are no data on the presence of odevixibat in human milk, the effects on the breastfed infant, or the effects on milk production. BYLVAY may reduce absorption of fat-soluble vitamins [see Warning and Precautions (5.3)]. Monitor FSV levels and increase FSV intake, if FSV deficiency is observed during lactation. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BYLVAY and any potential adverse effects on the breastfed child from BYLVAY or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of BYLVAY have been established in pediatric patients 3 months to 17 years of age for the treatment of pruritus in PFIC. Use of BYLVAY in this age group is supported by evidence from one randomized, double-blind, placebo-controlled trial conducted in 62 patients with a confirmed diagnosis of PFIC type 1 or type 2 (Trial 1), and an open-label extension trial in PFIC patients (Trial 2) [see Adverse Reactions (6.1) and Clinical Studies (14.1)].
The safety and effectiveness of BYLVAY for the treatment of pruritus in PFIC in pediatric patients less than 3 months of age have not been established.
The safety and effectiveness of BYLVAY have been established in pediatric patients 12 months to 17 years of age for the treatment of pruritus in ALGS. Use of BYLVAY in this age group is supported by evidence from one randomized, double-blind, placebo-controlled trial conducted in 52 patients with a confirmed diagnosis of ALGS (Trial 3) and one open-label extension trial in ALGS patients (Trial 4) [see Adverse Reactions (6.1) and Clinical Studies (14.2)].
The safety and effectiveness of BYLVAY for the treatment of pruritus in ALGS in pediatric patients less than 12 months of age have not been established.
8.5 Geriatric Use
PFIC and ALGS are largely diseases of pediatric and young adult patients. Clinical studies in BYLVAY did not include patients 65 years of age and older.
8.6 Hepatic Impairment
Patients with PFIC and ALGS may have impaired hepatic function. The efficacy and safety of BYLVAY in PFIC and ALGS patients with clinically significant portal hypertension, and in patients with decompensated cirrhosis have not been established [see Dosage and Administration (2.3), Contraindications (4), Warning and Precautions (5.1), and Clinical Studies (14)].
-
11 DESCRIPTION
The active ingredient in BYLVAY (odevixibat) capsules and BYLVAY (odevixibat) oral pellets, an ileal bile acid transporter (IBAT) inhibitor, is (2S)-2-{[(2R)-2-(2-{[3,3-dibutyl-7-(methylsulfanyl)-1,1-dioxo-5-phenyl-2,3,4,5-tetrahydro-1H-1λ6,2,5-benzothiadiazepin-8yl]oxy}acetamido)-2-(4-hydroxyphenyl)acetly]amino}butanoic acid, which is formulated as the sesquihydrate having the following chemical structure:
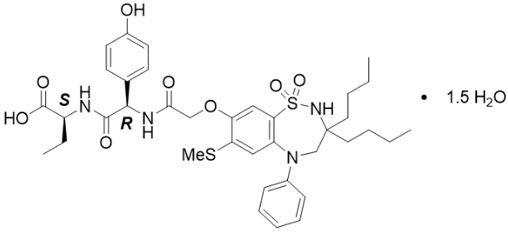
The molecular formula is C37H48N4O8S2 × 1.5 H2O, with a molecular weight of 768.0 g/mol (anhydrous 740.9 g/mol). Odevixibat sesquihydrate is a white to off-white solid. Its solubility in aqueous solutions is pH-dependent and increases with increased pH.
BYLVAY is available for oral administration as oral pellets containing odevixibat sesquihydrate equivalent to 200 mcg or 600 mcg of odevixibat, and as capsules containing odevixibat sesquihydrate equivalent to 400 mcg or 1200 mcg of odevixibat, and the following excipients: hypromellose and microcrystalline cellulose.
The capsule shells for the oral pellets contain hypromellose, titanium dioxide and yellow iron oxide.
The capsule shells for the capsules contain hypromellose, red iron oxide, titanium dioxide and yellow iron oxide.
The imprinting ink contains ferrosoferric oxide/black iron oxide and shellac glaze.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Odevixibat is a reversible inhibitor of the ileal bile acid transporter (IBAT). It decreases the reabsorption of bile acids (primarily the salt forms) from the terminal ileum.
Pruritus is a common symptom in patients with PFIC and ALGS; the pathophysiology of pruritus in patients with PFIC is not completely understood. Although the complete mechanism by which odevixibat improves pruritus in both PFIC and ALGS patients is unknown, it may involve inhibition of the IBAT, which results in decreased reuptake of bile salts, as observed by a decrease in serum bile acids [see Clinical Pharmacology (12.2)].
12.2 Pharmacodynamics
Odevixibat reduces serum bile acids in patients with PFIC and ALGS.
In Trial 1, a 24-week, randomized, double-blind, placebo-controlled trial conducted in 62 patients with a confirmed diagnosis of PFIC type 1 or type 2, the majority of patients (88.7%) had elevated serum bile acids above 100 mcmol/L at baseline [see Clinical Studies (14.1)]. Serum bile acids concentrations were reduced from baseline within 4-8 weeks of odevixibat treatment compared to placebo treatment. The decreased concentrations of serum bile acids fluctuated over time but generally were maintained during the treatment over 24 weeks. The extent of decrease in serum bile acids was similar between 40 and 120 mcg/kg.
Trial 3 is a 24-week, randomized, double-blind, placebo-controlled trial conducted in 52 patients with a confirmed diagnosis of ALGS who were administered treatment with BYLVAY 120 mcg/kg once daily [see Clinical Studies (14.2)]. At baseline, serum bile acids were variable ranging from 93 to 510 mcmol/L. Serum bile acid concentrations were reduced from baseline as early as Week 4 of odevixibat treatment and the reduction was generally maintained during treatment over 24 weeks.
12.3 Pharmacokinetics
In pediatric patients with PFIC, 6 months to 17 years of age who received BYLVAY 40 mcg/kg or 120 mcg/kg once daily with food in the morning, the measurable odevixibat concentrations ranged from 0.06 to 0.72 ng/mL, and odevixibat concentrations were below the limit of quantification (0.05 ng/mL) in the majority of plasma samples.
In pediatric patients with ALGS who received BYLVAY 120 mcg/kg once daily with food in the morning, the measurable odevixibat concentrations ranged from 0.05 to 3.4 ng/mL.
Following single and repeated oral administration of odevixibat from 0.1 to 3 mg in healthy adults, plasma concentrations of odevixibat were mostly below the limit of quantification (0.05 ng/mL); therefore, AUC and peak plasma concentration (Cmax) could not be calculated.
Following a single administration of odevixibat 7.2 mg in healthy adults, the mean (%CV) Cmax and AUC0-24h were 0.47 ng/mL (34.8) and 2.19 ng∙h/mL (36.2), respectively. No accumulation of odevixibat was observed following once-daily dosing.
Absorption
Odevixibat is minimally absorbed following oral administration. Following a single administration of odevixibat 7.2 mg in healthy adults, odevixibat Cmax is reached between 1 to 5 hours.
Sprinkle on Applesauce
When odevixibat 9.6 mg was administered after sprinkling the pellets on applesauce, decreases of 39% and 35% in Cmax and AUC0-24h, respectively, and delayed median Tmax from 3 hours to 4.5 hours were observed compared to administration of whole capsules (eight 1200 mcg capsules) under fasted conditions. The effect of sprinkling on soft food on systemic exposure is not clinically significant [see Dosage and Administration (2.3)].
Effect of Food
Concomitant administration of a high-fat meal (800-1000 calories with approximately 50% of total caloric content of the meal from fat) with a single dose of odevixibat 9.6 mg delayed median Tmax from 3 hours to 4.5 hours and resulted in decreases of 72% and 62% in Cmax and AUC0-24h, respectively, compared to administration under fasted conditions in healthy adults. The effect of food on the changes of systemic exposures to odevixibat is not clinically significant [see Dosage and Administration (2.3)].
Elimination
Following a single oral dose of 7.2 mg odevixibat in healthy adults, the mean half-life (t1/2) was 2.36 hours.
Drug Interaction Studies
Effect of Other Drugs on Odevixibat
Odevixibat is a substrate of P-glycoprotein (P-gp) but not a substrate of breast cancer resistance protein (BCRP).
Coadministration of itraconazole (a strong P-gp inhibitor) with a single dose of BYLVAY 7.2 mg increased odevixibat AUC0-24h by 66% and Cmax by 52%, which is not expected to have a clinically significant effect.
Effect of Odevixibat on Other Drugs
In in vitro studies, odevixibat was not an inhibitor of CYP isoforms 1A2, 2B6, 2C8, 2C9, 2C19, or 2D6 nor an inducer of CYP isoforms 1A2, 2B6, or 3A4.
Concomitant use of BYLVAY 7.2 mg once daily for 4 days with oral midazolam (a CYP3A4 substrate) in healthy adults decreased the AUC0-24h of midazolam and 1-OH midazolam by 29% and 13%, respectively, which is not expected to have a clinically relevant effect.
In in vitro studies, odevixibat did not inhibit the transporters P-gp; BCRP; organic anion transporter polypeptide 1B1 and 1B3(OATP1B1 and OATP1B3); organic anion transporter (OAT)1, OAT3; organic cation transporter 2 (OCT2), multidrug and toxin extrusion transporter 1 and 2K (MATE1 and MATE2K).
In a clinical drug interaction study with a lipophilic combination oral contraceptive containing ethinyl estradiol (EE) (0.03 mg) and levonorgestrel (LVN) (0.15 mg) conducted in healthy adult females, concomitant use of odevixibat had no impact on the AUC of LVN and decreased the AUC of EE by 17%, which is not expected to have a clinically relevant effect.
12.5 Pharmacogenomics
PFIC is a heterogenous disease caused by homozygous or compound heterozygous variants, with different PFIC subtypes occurring in the general population. PFIC1 is caused by variants in the ATP8B1 gene, which encodes FIC1. PFIC2 is caused by variants in the ABCB11 gene, which encodes BSEP. PFIC2 is further categorized into BSEP subgroups based on specific variants. The BSEP-1 subgroup includes patients with at least one p.D482G (c.1445A>G) or p.E297G (c.890A>G) variant, BSEP-2 includes patients with at least one missense variant other than p.D482G or p.E297G (non BSEP-1), and BSEP-3 includes patients with variants that are predicted to encode a non-functional protein. PFIC3 is caused by variants in the ABCB4 gene, which encodes MDR3. PFIC4 is caused by variants in the TJP2 gene, which encodes TJP2. PFIC6 is caused by variants in the MYO5B gene, which encodes MYO5B. Patients can be clinically diagnosed with PFIC without a known pathogenic variant.
PFIC2 is the most common subtype accounting for 37-90% of PFIC patients. The prevalence of BSEP-1, BSEP-2, and BSEP-3 subgroups are approximately 27%, 52%, and 21%, respectively, based on data from a global consortium characterizing the natural history of severe BSEP deficiency.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In 2-year carcinogenicity studies, odevixibat was not tumorigenic in rats or mice at oral doses up to 100 mg/kg/day. Systemic exposure to odevixibat (AUC) at the maximum dose studied in rats and mice was approximately 231 and 459 times the maximum recommended dose, respectively.
-
14 CLINICAL STUDIES
14.1 PFIC
The efficacy of BYLVAY was evaluated in Trial 1 (NCT03566238), a 24-week, randomized, double-blind, placebo-controlled trial. Trial 1 was conducted in 62 pediatric patients, aged 6 months to 17 years, with a confirmed molecular diagnosis of PFIC type 1 or type 2, and presence of pruritus at baseline. Patients with variants in the ABCB11 gene that predict non-function or complete absence of the bile salt export pump (BSEP) protein, who had experienced prior hepatic decompensation events, who had other concomitant liver disease, whose INR was greater than 1.4, whose ALT or total bilirubin was greater than 10-times the upper limit of normal (ULN), or who had received a liver transplant were excluded in Trial 1.
Patients were randomized to placebo (n=20), 40 mcg/kg (n=23), or 120 mcg/kg (n=19). Study drug was administered once daily with a meal in the morning. In patients weighing less than 19.5 kg or patients who could not swallow the whole capsule, study drug was sprinkled on soft food and then administered orally.
Median age (range) of the patients in Trial 1 was 3.2 (0.5 to 15.9) years; 3 patients were older than 12 years of age. Of the 62 patients, 50% were male and 84% were white; 27% had PFIC type 1, and 73% had PFIC type 2. The mean (standard error [SE]) scratching score in the 2 weeks prior to baseline was 2.9 (0.08). Baseline mean (SE) eGFR was 164 (30.6) mL/min/1.73 m2. Baseline median (range) ALT, AST, and total bilirubin were 65 (16-798) U/L, 83.5 (32-405) U/L, and 2.2 (0.2-18.6) mg/dL, respectively.
In Trial 1, a total of 13 patients discontinued from trial prematurely either due to no improvement in pruritus (n=11) or due to adverse reactions (n=2); 5/20 (25%) patients discontinued from the placebo arm and 8/42 (19%) patients discontinued from the BYLVAY arms. A total of 11 of the 13 patients rolled over to Trial 2 to receive BYLVAY 120 mcg/kg/day. One patient treated with BYLVAY 120 mcg/kg/day withdrew from the trial due to an adverse reaction of diarrhea [see Adverse Reactions (6)].
Given the patients' young age, a single-item observer-reported outcome (ObsRO) was used to measure patients' scratching severity as observed by their caregiver twice daily (once in the morning and once in the evening). Scratching severity was assessed on a 5-point ordinal response scale, with scores ranging from 0 (no scratching) to 4 (worst possible scratching). Patients were included in Trial 1 if their average scratching score was greater than or equal to 2 (medium scratching) in the 2 weeks prior to baseline.
Table 5 presents the results of the comparison between BYLVAY and placebo on the mean of patients' percentage of ObsRO assessments over the 24-week treatment period that were scored as 0 (no scratching) or 1 (a little scratching). Patients treated with BYLVAY demonstrated greater improvement in pruritus compared with placebo. Figure 1 displays the mean of patients' worst weekly average scratching scores in each treatment group for each month, where the weekly average utilized the worst score from each day (morning or evening score).
Table 5: Efficacy Results Over the 24-Week Treatment Period in Patients with PFIC Type 1 or 2 in Trial 1 Placebo
(n=20)BYLVAY 40 mcg/kg/day
(n=23)120 mcg/kg/day
(n=19)- * Based on least squares means from analysis of covariance model with daytime and nighttime baseline pruritus scores as covariates and treatment group and stratification factors (i.e., PFIC type and age category) as fixed effects.
Mean* Percentage of Assessments Over the Treatment Period Scored as 0 (No Scratching) or 1 (A Little Scratching) (%) Mean (SE) 13.2 (8.7) 35.4 (8.1) 30.1 (9.0) Mean Difference vs Placebo (95% CI) 22.2 (4.7, 39.6) 16.9 (-2.0, 35.7) - * Figure 1 presents least squares means
Based on a mixed model repeated measure (MMRM) analysis accounting for baseline score, treatment group, time (in months), treatment-by-baseline interaction, treatment-by-time interaction, and stratification factors (i.e., PFIC type and age category). Missing data were accounted for using placebo-reference multiple imputation.Figure 1: Mean* of the Worst Weekly Average Scratching Scores for Each Month in Trial 1 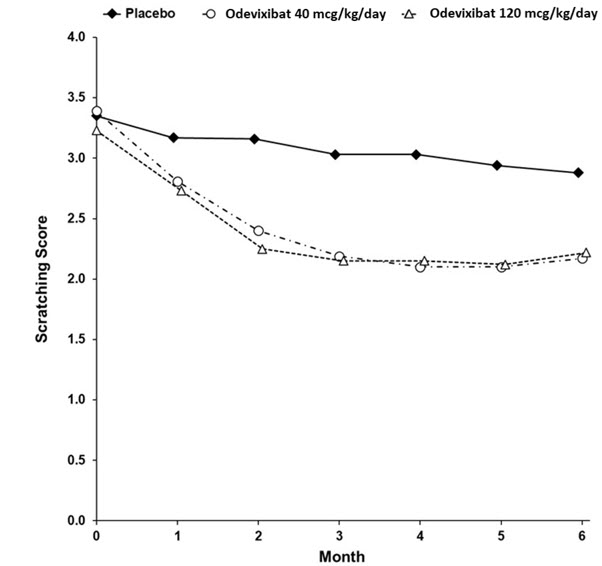
14.2 ALGS
The efficacy of BYLVAY was evaluated in Trial 3 (NCT04674761), a 24-week, randomized, double-blind, placebo-controlled trial. Trial 3 was conducted in 52 pediatric patients, aged 6 months to 15 years, with a confirmed diagnosis of ALGS and presence of pruritus at baseline. Patients who had decompensated liver disease, who had other concomitant liver disease, whose INR was greater than 1.4, whose ALT was greater than 10-times the upper limit of normal (ULN) at screening, whose total bilirubin was greater than 15-times the ULN at screening, or who had received a liver transplant were excluded from Trial 3.
Patients were randomized to placebo (n=17) or 120 mcg/kg (n=35). Study drug was administered once daily with a meal in the morning. In patients weighing less than 19.5 kg or patients who could not swallow the whole capsule, study drug was sprinkled on soft food and then administered orally.
Median age (range) of the patients in Trial 3 was 6.1 (1.7 to 15.5) years in the BYLVAY group and 4.2 (0.5 to 14.3) years in the placebo group; 5 patients were older than 12 years of age. Of the 52 patients, 52% were male and 83% were white; 92% of patients had the JAG1 mutation and 8% had the NOTCH2 mutation. The mean (standard deviation [SD]) scratching score in the 2 weeks prior to baseline was 2.9 (0.6). Baseline mean (SD) eGFR was 159 (51.4) mL/min/1.73 m2. Baseline median (range) ALT, AST, and total bilirubin were 152 (39-403) U/L, 135 (57-427) U/L, and 2.0 (0.4-11.4) mg/dL, respectively.
Given the patients' young ages, a single-item observer-reported outcome (ObsRO) was used to measure patients' scratching severity as observed by their caregiver twice daily (once in the morning and once in the evening). Scratching severity was assessed on a 5-point ordinal response scale, with scores ranging from 0 (no scratching) to 4 (worst possible scratching). Patients were included in Trial 3 if the average scratching score was greater than or equal to 2 (medium scratching) in the 14 days prior to baseline.
Table 6 presents the results of the comparison between BYLVAY and placebo on the change from baseline in average scratching score based on ObsRO assessments to Month 6 (Weeks 21 to 24). The average scratching score for each patient for each month post-baseline was calculated by: (Step 1) averaging the morning scores and averaging the evening scores within a week; (Step 2) averaging the morning and evening weekly scores to yield a single weekly score; and finally (Step 3) averaging the 4 weekly scores within the month. The baseline average scratching score for each patient was calculated by averaging the weekly scores obtained in Step 2 across the 2 weeks prior to randomization and initiation of blinded treatment. Patients treated with BYLVAY demonstrated greater improvement in pruritus compared with placebo. Figure 2 displays the means (95% confidence interval) of patients' average scratching scores in each treatment group for each month.
Table 6: Efficacy Results in Patients with ALGS in Trial 3 Placebo
(n=17)BYLVAY
120 mcg/kg/day
(n=35)- * Based on least square means from a mixed-effect model for repeated measures (MMRM) for change from baseline to each month accounting for baseline average scratching score, baseline age stratification (<10, ≥10 years), baseline direct bilirubin, treatment group, time (in months), and treatment-by-time interaction.
Baseline Average Scratching Score Mean (SD) 3.0 (0.6) 2.8 (0.5) Change from Baseline in Average Scratching Score to Month 6 (Weeks 21 to 24)* Mean (SE) -0.8 (0.2) -1.7 (0.2) Mean Difference vs Placebo (95% CI)
p-value-0.9 (-1.4, -0.3)
0.002- * Figure 2 presents means for baseline and least squares means for Month 1 to 6
Least squares means are based on a mixed model repeated measure (MMRM) analysis accounting for baseline average scratching score, baseline age stratification (<10, ≥10 years), baseline direct bilirubin, treatment group, time (in months), and treatment-by-time interaction.Figure 2: Mean* of the Average Scratching Scores for Each Month in Trial 3 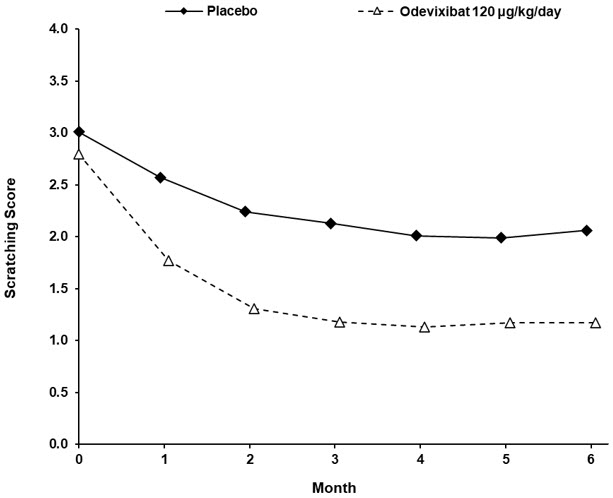
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Oral Pellets
- 200 mcg Oral Pellets: supplied as Size 0 capsule with ivory opaque cap and white opaque body; imprinted "A200" (black ink). Supplied in bottles of 30 with child-resistant closure (NDC: 15054-3301-1).
- 600 mcg Oral Pellets: supplied as Size 0 capsule with ivory opaque cap and body; imprinted "A600" (black ink). Supplied in bottles of 30 with child-resistant closure (NDC: 15054-3303-1).
Capsules
- 400 mcg Capsule: supplied as Size 3 capsule with medium orange opaque cap and white opaque body; imprinted "A400" (black ink). Supplied in bottles of 30 with child-resistant closure (NDC: 15054-3302-1).
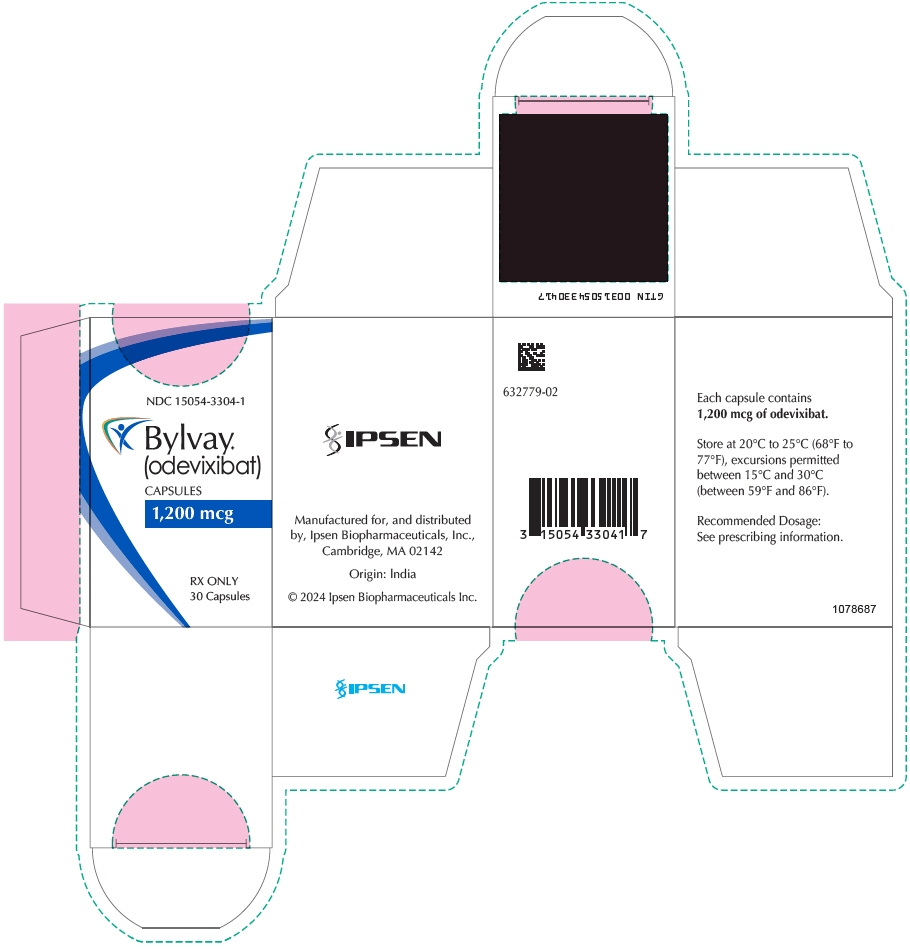
- 1,200 mcg Capsule: supplied as Size 3 capsule with medium orange opaque cap and body; imprinted "A1200" (black ink). Supplied in bottles of 30 with child-resistant closure (NDC: 15054-3304-1).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Instructions for Use).
Administration Instructions
Advise patients or their caregivers(s) to:
- Take BYLVAY in the morning with a meal. Do not swallow the 200 mcg or 600 mcg capsule(s) containing Oral Pellets whole. These are intended to be opened and the contents mixed into soft food or liquids [see Dosage and Administration (2.1, 2.2, 2.4)].
- Follow stepwise administration Instructions [see Dosage and Administration (2.4)] for Oral Pellets and Capsules for patients unable to swallow the capsules whole.
- Take BYLVAY at least 4 hours before or 4 hours after taking a bile acid binding resin (e.g., cholestyramine, colesevelam, or colestipol) [see Drug Interactions (7.1)].
Hepatotoxicity
Advise patients or their caregiver(s) that liver tests should be obtained before starting BYLVAY and periodically during BYLVAY therapy. Inform patients or their caregiver(s) of the potential risk of hepatotoxicity, and that they will need to undergo monitoring for liver injury. Instruct patients or their caregiver(s) to immediately report any signs or symptoms of severe liver injury to their healthcare provider [see Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
Diarrhea
Advise patients or their caregiver(s) to notify their healthcare provider if they experience new onset or worsening of diarrhea [see Warnings and Precautions (5.2)].
Fat Soluble Vitamin (FSV) Deficiency
Advise patients or their caregiver(s) that INR (for vitamin K) and serum levels of vitamins A, D, E will be obtained before starting and periodically during treatment to assess for FSV deficiency [see Warnings and Precautions (5.3)]. Inform patients or their caregiver(s) that they may bleed more easily, may bleed longer, or have a bone fracture. Advise patients or their caregiver(s) to call their healthcare provider for any signs or symptoms of bleeding or report any fractures.
Pregnancy
Advise patients or their caregiver(s) that there is a pregnancy safety study that collects pregnancy outcome data in women taking BYLVAY during pregnancy. Pregnant women exposed to BYLVAY, or their healthcare providers, should report BYLVAY exposure by calling 1-855-463-5127 [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
Instructions For UseBYLVAY [bil-vay] (odevixibat)Capsules, for oral useOral Pellets
This Instructions for Use contains information on how to give BYLVAY Capsules and Oral Pellets. This information does not take the place of talking to your healthcare provider about your child's medical condition or their treatment.
Important information you need to know before giving or taking BYLVAY
- Give BYLVAY along with the morning meal.
- Mix BYLVAY in a small amount of soft food (up to 2 tablespoons [30 mL]), such as apple sauce, oatmeal, banana or carrot puree, chocolate or rice pudding, in a bowl. You may also mix BYLVAY with an age-appropriate liquid and give through an oral syringe.
- If your child is taking bile acid binding resins (for example, cholestyramine, colestipol), give them BYLVAY at least 4 hours before or 4 hours after they take the bile acid binding resin.
Preparing to Give BYLVAY
You will be provided with the number of BYLVAY Capsules or Oral Pellets prescribed by your child's healthcare provider in a child-resistant closure.
Giving BYLVAY Oral Pellets with soft food:
- The shell containing Oral Pellets are to be opened and sprinkled. Do not let your child swallow the shell containing the Oral Pellets. Dispose of (throw away) the emptied shell.
- Mix the contents of the Oral Pellets with soft food as shown in Steps 1 through 9 below.
Step 1. Give BYLVAY with the first morning meal. Place a small amount of soft food (up to 2 tablespoons [30 mL], such as apple sauce, oatmeal, banana or carrot puree, chocolate or rice pudding) in a bowl. Keep the soft food at, or cooler than, room temperature.
Note: This small amount of soft food should be less than what your child would normally eat.Step 2. Hold the shell containing Oral Pellets horizontally on both ends, twist in opposite directions and pull apart (see Figure A). 
Figure AStep 3. Empty the Oral Pellets into the bowl of soft food (see Figure B). 
Figure BStep 4. Gently tap the shell containing Oral Pellets to make sure that all pellets come out (see Figure C). 
Figure CStep 5. If the dose requires more than 1 capsule shell, repeat Step 2 and Step 3. Step 6. Gently mix the Oral Pellets with a spoon into the soft food. Note that the Oral Pellets will not dissolve (see Figure D). 
Figure DStep 7. Give the entire dose right away after mixing. Do not store the BYLVAY mixture for later use. Step 8. Give water or an age-appropriate liquid, such as breast milk or infant formula, after the dose is taken. Step 9. Dispose of (throw away) the empty Oral Pellet shells in the trash. Giving BYLVAY Oral Pellets with liquids (Using an oral dosing syringe):
- The shell containing the Oral Pellets are to be opened.
- Mix the Oral Pellets with liquid as shown in Steps 1 through 14 below.
- Dispose of (throw away) the emptied shells. Do not let your child swallow the unopened shells containing the Oral Pellets.
Step 1. Give BYLVAY with the first morning meal. Step 2. Hold the shell containing the Oral Pellets horizontally on both ends, twist in opposite directions and pull apart (see Figure A). Step 3. Empty the Oral Pellets into a small mixing cup. Gently tap the Oral Pellet shell to ensure that all contents have been emptied into the mixing cup (see Figure E). 
Figure EStep 4. If the dose requires more than 1 capsule shell, repeat Step 2 and Step 3. Step 5. Add 1 teaspoon (5 mL) of an age-appropriate liquid (for example, breast milk, infant formula, or water). Step 6. Let the pellets sit in the liquid for about 5 minutes to allow complete wetting. REMINDER: The pellets will not dissolve in the liquid. Step 7. After 5 minutes, place the tip of the oral syringe completely into the mixing cup. Pull the plunger of the syringe up slowly to withdraw the liquid and pellet mixture into the syringe. Gently push the plunger down again to expel the liquid and pellet mixture back into the mixing cup. Do this 2 to 3 times to ensure complete mixing of the pellets into the liquid. Step 8. Withdraw the entire contents into the syringe by pulling the plunger on the end of the syringe (see Figure F). 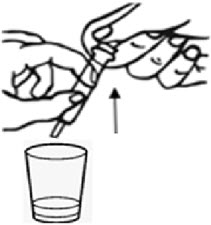
Figure FStep 9. Place the tip of the syringe into the front of the child's mouth between the tongue and the side of the mouth, and then gently push the plunger down to squirt the liquid and pellet mixture between the tongue and the side of the mouth. Do not squirt the liquid and pellet mixture in the back of the throat because this could cause gagging or choking (see Figure G). 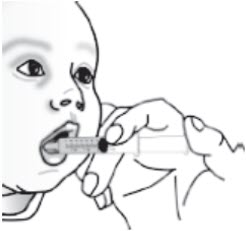
Figure GStep 10. Do not give using a bottle or "sippy cup" because the Oral Pellets will not pass through the opening. The oral pellets will not dissolve in liquid. Step 11. Give water or an age-appropriate liquid such as breast milk or infant formula after the dose is taken. Step 12. Repeat Steps 8 and 9 until the entire dose has been given. Step 13. Check to make sure all of the liquid and pellet mixture has been swallowed. Step 14. Dispose of (throw away) the empty Oral Pellet shells in the trash. Taking BYLVAY Capsules
- Take BYLVAY Capsules along with your morning meal. Swallow BYLVAY Capsules whole with a glass of water. Do not chew or crush the Capsules.
- For children unable to swallow BYLVAY Capsules whole, follow instructions under Preparing to Give BYLVAY above.
How should I store BYLVAY Capsules or Oral Pellets?
Store BYLVAY at room temperature between 68°F to 77°F (20°C to 25°C).
Disposing (throwing away) of BYLVAY Capsules or Oral Pellets shells.
Dispose of (throw away) the empty BYLVAY Capsule or Oral Pellets shells in the household trash.
What are the ingredients in BYLVAY?
Active ingredient: odevixibat.
Inactive ingredients: hypromellose and microcrystalline cellulose.
Manufactured for:
Ipsen Biopharmaceuticals, Inc.
One Main Street,
Cambridge, MA 02142© 2024 Ipsen Biopharmaceuticals, Inc. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 01/2024 - PRINCIPAL DISPLAY PANEL - 200 mcg Capsule Bottle Carton
- PRINCIPAL DISPLAY PANEL - 400 mcg Capsule Bottle Carton
- PRINCIPAL DISPLAY PANEL - 600 mcg Capsule Bottle Carton
- PRINCIPAL DISPLAY PANEL - 1,200 mcg Capsule Bottle Carton
-
INGREDIENTS AND APPEARANCE
BYLVAY
odevixibat capsule, coated pelletsProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 15054-3301 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength odevixibat (UNII: 2W150K0UUC) (odevixibat - UNII:2W150K0UUC) odevixibat 200 ug Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) Product Characteristics Color WHITE (Ivory/White Opaque) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code A200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 15054-3301-1 1 in 1 CARTON 01/10/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215498 01/10/2024 BYLVAY
odevixibat capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 15054-3302 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength odevixibat (UNII: 2W150K0UUC) (odevixibat - UNII:2W150K0UUC) odevixibat 400 ug Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) Product Characteristics Color ORANGE (Medium Orange Opaque/White Opaque) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code A400 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 15054-3302-1 1 in 1 CARTON 01/10/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215498 01/10/2024 BYLVAY
odevixibat capsule, coated pelletsProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 15054-3303 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength odevixibat (UNII: 2W150K0UUC) (odevixibat - UNII:2W150K0UUC) odevixibat 600 ug Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) Product Characteristics Color WHITE (Ivory/White Opaque) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code A600 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 15054-3303-1 1 in 1 CARTON 01/10/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215498 01/10/2024 BYLVAY
odevixibat capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 15054-3304 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength odevixibat (UNII: 2W150K0UUC) (odevixibat - UNII:2W150K0UUC) odevixibat 1200 ug Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) Product Characteristics Color ORANGE (Medium Orange Opaque) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code A1200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 15054-3304-1 1 in 1 CARTON 01/10/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215498 01/10/2024 Labeler - Ipsen Biopharmaceuticals, Inc. (118461578) Establishment Name Address ID/FEI Business Operations Syngene International Limited. 915833842 API MANUFACTURE(15054-3301, 15054-3302, 15054-3303, 15054-3304) , ANALYSIS(15054-3301, 15054-3302, 15054-3303, 15054-3304) Establishment Name Address ID/FEI Business Operations Almac Pharma Services Limited 233170864 PACK(15054-3301, 15054-3302, 15054-3303, 15054-3304) , LABEL(15054-3301, 15054-3302, 15054-3303, 15054-3304)

Trademark Results [Bylvay]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BYLVAY 88932949 not registered Live/Pending |
Albireo AB 2020-05-26 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.