MVASI- bevacizumab-awwb injection, solution
MVASI by
Drug Labeling and Warnings
MVASI by is a Prescription medication manufactured, distributed, or labeled by Amgen Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MVASI safely and effectively. See full prescribing information for MVASI.
MVASI™ (bevacizumab-awwb) injection, for intravenous use
Initial U.S. Approval: 2017
MVASI (bevacizumab-awwb) is biosimilar* to AVASTIN® (bevacizumab)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
MVASI is a vascular endothelial growth factor inhibitor indicated for the treatment of:
- Metastatic colorectal cancer, in combination with intravenous fluorouracil-based chemotherapy for first- or second-line treatment. (1.1)
- Metastatic colorectal cancer, in combination with fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy for second-line treatment in patients who have progressed on a first-line bevacizumab product-containing regimen. (1.1)
Limitations of Use: MVASI is not indicated for adjuvant treatment of colon cancer. (1.1)
- Unresectable, locally advanced, recurrent or metastatic non-squamous non-small cell lung cancer, in combination with carboplatin and paclitaxel for first-line treatment. (1.2)
- Recurrent glioblastoma in adults. (1.3)
- Metastatic renal cell carcinoma in combination with interferon-alfa. (1.4)
- Persistent, recurrent, or metastatic cervical cancer, in combination with paclitaxel and cisplatin, or paclitaxel and topotecan. (1.5)
DOSAGE AND ADMINISTRATION
Do not administer MVASI for 28 days following major surgery and until surgical wound is fully healed. (2.1)
Metastatic colorectal cancer (2.2)
- 5 mg/kg every 2 weeks with bolus-IFL
- 10 mg/kg every 2 weeks with FOLFOX4
- 5 mg/kg every 2 weeks or 7.5 mg/kg every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin based chemotherapy after progression on a first-line bevacizumab product-containing regimen
First-line non-squamous non-small cell lung cancer (2.3)
- 15 mg/kg every 3 weeks with carboplatin and paclitaxel
Recurrent glioblastoma (2.4)
- 10 mg/kg every 2 weeks
Metastatic renal cell carcinoma (2.5)
- 10 mg/kg every 2 weeks with interferon-alfa
Persistent, recurrent, or metastatic cervical cancer (2.6)
- 15 mg/kg every 3 weeks with paclitaxel and cisplatin or paclitaxel and topotecan
Administer as an intravenous infusion (2.8)
DOSAGE FORMS AND STRENGTHS
- Injection: 100 mg/4 mL (25 mg/mL) or 400 mg/16 mL (25 mg/mL) in a single-dose vial (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
-
Gastrointestinal Perforations and Fistula: Discontinue for gastrointestinal perforations, tracheoesophageal fistula, Grade 4 fistula, or fistula formation involving any organ. (5.1)
-
Surgery and Wound Healing Complications: Discontinue in patients who develop wound healing complications that require medical intervention or necrotizing fasciitis. Withhold for at least 28 days prior to elective surgery. Do not administer MVASI for at least 28 days after surgery and until the wound is fully healed. (5.2)
-
Hemorrhage: Severe or fatal hemorrhages have occurred. Do not administer for recent hemoptysis. Discontinue for Grade 3-4 hemorrhage. (5.3)
-
Arterial Thromboembolic Events (ATE): Discontinue for severe ATE. (5.4)
-
Venous Thromboembolic Events (VTE): Discontinue for Grade 4 VTE. (5.5)
-
Hypertension: Monitor blood pressure and treat hypertension. Withhold until medically controlled; resume once controlled. Discontinue for hypertensive crisis or hypertensive encephalopathy. (5.6)
-
Posterior Reversible Encephalopathy Syndrome (PRES): Discontinue. (5.7)
-
Renal Injury and Proteinuria: Monitor urine protein. Discontinue for nephrotic syndrome. Withhold until less than 2 grams of protein in urine. (5.8)
-
Infusion-Related Reactions: Decrease rate for infusion-related reactions. Discontinue for severe infusion-related reactions and administer medical therapy. (5.9)
-
Embryo-Fetal Toxicity: May cause fetal harm. Advise females of potential risk to fetus and need for use of effective contraception. (5.10, 8.1, 8.3)
-
Ovarian Failure: Advise females of the potential risk. (5.11, 8.3)
- Congestive Heart Failure (CHF): Discontinue MVASI in patients who develop CHF. (5.12)
ADVERSE REACTIONS
Most common adverse reactions incidence (incidence > 10%) are epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, rectal hemorrhage, lacrimation disorder, back pain and exfoliative dermatitis. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Medical Information at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
*Biosimilar means that the biological product is approved based on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product. Biosimilarity of MVASI has been demonstrated for the condition(s) of use (e.g., indication(s), dosing regimen(s)), strength(s), dosage form(s), and route(s) of administration described in its Full Prescribing Information.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2019
- Metastatic colorectal cancer, in combination with intravenous fluorouracil-based chemotherapy for first- or second-line treatment. (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Metastatic Colorectal Cancer
1.2 First-Line Non-Squamous Non-Small Cell Lung Cancer
1.3 Recurrent Glioblastoma
1.4 Metastatic Renal Cell Carcinoma
1.5 Persistent, Recurrent, or Metastatic Cervical Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
2.2 Metastatic Colorectal Cancer
2.3 First-Line Non-Squamous Non-Small Cell Lung Cancer
2.4 Recurrent Glioblastoma
2.5 Metastatic Renal Cell Carcinoma
2.6 Persistent, Recurrent, or Metastatic Cervical Cancer
2.7 Dosage Modifications for Adverse Reactions
2.8 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Perforations and Fistulae
5.2 Surgery and Wound Healing Complications
5.3 Hemorrhage
5.4 Arterial Thromboembolic Events
5.5 Venous Thromboembolic Events
5.6 Hypertension
5.7 Posterior Reversible Encephalopathy Syndrome
5.8 Renal Injury and Proteinuria
5.9 Infusion-Related Reactions
5.10 Embryo-Fetal Toxicity
5.11 Ovarian Failure
5.12 Congestive Heart Failure (CHF)
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Metastatic Colorectal Cancer
14.2 Lack of Efficacy in Adjuvant Treatment of Colon Cancer
14.3 First-Line Non-Squamous Non-Small Cell Lung Cancer
14.4 Recurrent Glioblastoma
14.5 Metastatic Renal Cell Carcinoma
14.6 Persistent, Recurrent, or Metastatic Cervical Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
1.1 Metastatic Colorectal Cancer
MVASI, in combination with intravenous fluorouracil-based chemotherapy, is indicated for the first- or second-line treatment of patients with metastatic colorectal cancer (mCRC).
MVASI, in combination with fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy, is indicated for the second-line treatment of patients with mCRC who have progressed on a first-line bevacizumab product-containing regimen.
Limitations of Use: MVASI is not indicated for adjuvant treatment of colon cancer [see Clinical Studies (14.2)].
1.2 First-Line Non-Squamous Non-Small Cell Lung Cancer
MVASI, in combination with carboplatin and paclitaxel, is indicated for the first-line treatment of patients with unresectable, locally advanced, recurrent or metastatic non-squamous non-small cell lung cancer (NSCLC).
1.3 Recurrent Glioblastoma
MVASI is indicated for the treatment of recurrent glioblastoma (GBM) in adults.
-
2
DOSAGE AND ADMINISTRATION
2.1 Important Administration Information
Do not administer MVASI until at least 28 days following surgery and the wound is fully healed.
2.2 Metastatic Colorectal Cancer
The recommended dosage when MVASI is administered in combination with intravenous fluorouracil-based chemotherapy is:
- 5 mg/kg intravenously every 2 weeks in combination with bolus-IFL.
- 10 mg/kg intravenously every 2 weeks in combination with FOLFOX4.
- 5 mg/kg intravenously every 2 weeks or 7.5 mg/kg intravenously every 3 weeks in combination with fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy in patients who have progressed on a first-line bevacizumab product-containing regimen.
2.3 First-Line Non-Squamous Non-Small Cell Lung Cancer
The recommended dosage is 15 mg/kg intravenously every 3 weeks in combination with carboplatin and paclitaxel.
2.5 Metastatic Renal Cell Carcinoma
The recommended dosage is 10 mg/kg intravenously every 2 weeks in combination with interferon-alfa.
2.6 Persistent, Recurrent, or Metastatic Cervical Cancer
The recommended dosage is 15 mg/kg intravenously every 3 weeks in combination with paclitaxel and cisplatin, or in combination with paclitaxel and topotecan.
2.7 Dosage Modifications for Adverse Reactions
Table 1 describes dosage modifications for specific adverse reactions [see Warnings and Precautions (5)]. No dose reductions for MVASI are recommended.
Table 1: Dosage Modifications for Adverse Reactions Adverse Reaction Severity Dosage Modification Gastrointestinal Perforations and Fistulae [see Warnings and Precautions (5.1)] - Gastrointestinal perforation, any grade
- Tracheoesophageal fistula, any grade
- Fistula, Grade 4
- Fistula formation involving any internal organ
Discontinue MVASI Wound Healing Complications [see Warnings and Precautions (5.2)] - Wound healing complications requiring medical intervention
- Necrotizing fasciitis
Discontinue MVASI Hemorrhage [see Warnings and Precautions (5.3)] - Grade 3 or 4
Discontinue MVASI - Recent history of hemoptysis of 1/2 teaspoon (2.5 mL) or more
Withhold MVASI Thromboembolic Events [see Warnings and Precautions (5.4, 5.5)] - Arterial thromboembolism, severe
Discontinue MVASI - Venous thromboembolism, Grade 4
Discontinue MVASI Hypertension [see Warnings and Precautions (5.6)] - Hypertensive crisis
- Hypertensive encephalopathy
Discontinue MVASI - Hypertension, severe (such as Grade 3 and above)
Withhold MVASI until controlled with medical management; resume once controlled Posterior Reversible Encephalopathy Syndrome (PRES) [see Warnings and Precautions (5.7)] - Any
Discontinue MVASI Renal Injury and Proteinuria [see Warnings and Precautions (5.8)] - Nephrotic syndrome
Discontinue MVASI - Proteinuria greater than or equal to 2 grams per 24 hours in absence of nephrotic syndrome
Withhold MVASI until proteinuria less than 2 grams per 24 hours Infusion-Related Reactions [see Warnings and Precautions (5.9)] - Severe
Discontinue MVASI - Clinically significant
Interrupt infusion; resume at a decreased rate of infusion after symptoms resolve - Mild, clinically insignificant
Decrease infusion rate Congestive Heart Failure [see Warnings and Precautions (5.12)] Any Discontinue MVASI 2.8 Preparation and Administration
Preparation
- Use appropriate aseptic technique.
- Visually inspect vial for particulate matter and discoloration prior to preparation and administration. Discard vial if solution is cloudy, discolored or contains particulate matter.
- Withdraw necessary amount of MVASI and dilute in a total volume of 100 mL of 0.9% Sodium Chloride Injection, USP. DO NOT ADMINISTER OR MIX WITH DEXTROSE SOLUTION.
- Discard any unused portion left in a vial, as the product contains no preservatives.
- Store diluted MVASI solution at 2°C to 8°C (36°F to 46°F) for up to 8 hours.
- No incompatibilities between MVASI and polyvinylchloride or polyolefin bags have been observed.
Administration
- Administer as an intravenous infusion.
- First infusion: Administer infusion over 90 minutes.
- Subsequent infusions: Administer second infusion over 60 minutes if first infusion is tolerated. Administer all subsequent infusions over 30 minutes if second infusion over 60 minutes is tolerated.
- 5 mg/kg intravenously every 2 weeks in combination with bolus-IFL.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Perforations and Fistulae
Serious, and sometimes fatal, gastrointestinal perforation occurred at a higher incidence in patients receiving bevacizumab products compared to patients receiving chemotherapy. The incidence ranged from 0.3% to 3% across clinical studies, with the highest incidence in patients with a history of prior pelvic radiation. Perforation can be complicated by intra-abdominal abscess, fistula formation, and the need for diverting ostomies. The majority of perforations occurred within 50 days of the first dose [see Adverse Reactions 6.1)].
Serious fistulae (including, tracheoesophageal, bronchopleural, biliary, vaginal, renal and bladder sites) occurred at a higher incidence in patients receiving bevacizumab products compared to patients receiving chemotherapy. The incidence ranged from < 1% to 1.8% across clinical studies, with the highest incidence in patients with cervical cancer. The majority of fistulae occurred within 6 months of the first dose. Patients who develop a gastrointestinal vaginal fistula may also have a bowel obstruction and require surgical intervention, as well as a diverting ostomy.
Discontinue in patients who develop gastrointestinal perforation, tracheoesophageal fistula or any Grade 4 fistula. Discontinue in patients with fistula formation involving any internal organ.
5.2 Surgery and Wound Healing Complications
In a controlled clinical study in which bevacizumab was not administered within 28 days of major surgical procedures, the incidence of wound healing complications, including serious and fatal complications, was 15% in patients with mCRC who underwent surgery while receiving bevacizumab and 4% in patients who did not receive bevacizumab. In a controlled clinical study in patients with relapsed or recurrent GBM, the incidence of wound healing events was 5% in patients who received bevacizumab and 0.7% in patients who did not receive bevacizumab [see Adverse Reactions (6.1)].
Discontinue MVASI in patients with wound healing complications requiring medical intervention. Withhold for at least 28 days prior to elective surgery. Do not administer for at least 28 days following surgery and until the wound is fully healed.
Necrotizing fasciitis including fatal cases, has been reported in patients receiving bevacizumab, usually secondary to wound healing complications, gastrointestinal perforation or fistula formation. Discontinue MVASI in patients who develop necrotizing fasciitis.
5.3 Hemorrhage
Bevacizumab products can result in two distinct patterns of bleeding: minor hemorrhage, which is most commonly Grade 1 epistaxis, and serious hemorrhage, which in some cases has been fatal. Severe or fatal hemorrhage, including hemoptysis, gastrointestinal bleeding, hematemesis, CNS hemorrhage, epistaxis, and vaginal bleeding occurred up to 5-fold more frequently in patients receiving bevacizumab compared to patients receiving chemotherapy alone. Across clinical studies, the incidence of Grades 3-5 hemorrhagic events ranged from 0.4% to 7% in patients receiving bevacizumab [see Adverse Reactions (6.1)].
Serious or fatal pulmonary hemorrhage occurred in 31% of patients with squamous NSCLC and 4% of patients with non-squamous NSCLC receiving bevacizumab with chemotherapy compared to none of the patients receiving chemotherapy alone.
Do not administer MVASI to patients with recent history of hemoptysis of 1/2 teaspoon or more of red blood. Discontinue in patients who develop a Grade 3-4 hemorrhage.
5.4 Arterial Thromboembolic Events
Serious, sometimes fatal, arterial thromboembolic events (ATE) including cerebral infarction, transient ischemic attacks, myocardial infarction, and angina occurred at a higher incidence in patients receiving bevacizumab compared to patients receiving chemotherapy. Across clinical studies, the incidence of Grades 3-5 ATE was 5% in patients receiving bevacizumab with chemotherapy compared to ≤ 2% in patients receiving chemotherapy alone; the highest incidence occurred in patients with GBM. The risk of developing ATE was increased in patients with a history of arterial thromboembolism, diabetes, or >65 years [see Use in Specific Populations (8.5)].
Discontinue in patients who develop a severe ATE. The safety of reinitiating bevacizumab products after an ATE is resolved is not known.
5.5 Venous Thromboembolic Events
An increased risk of venous thromboembolic events (VTE) was observed across clinical studies [see Adverse Reactions (6.1)]. In Study GOG-0240, Grades 3-4 VTE occurred in 11% of patients receiving bevacizumab with chemotherapy compared with 5% of patients receiving chemotherapy alone. In EORTC 26101, the incidence of Grades 3-4 VTE was 5% in patients receiving bevacizumab with chemotherapy compared to 2% in patients receiving chemotherapy alone.
Discontinue MVASI in patients with a Grade 4 VTE, including pulmonary embolism.
5.6 Hypertension
Severe hypertension occurred at a higher incidence in patients receiving bevacizumab products as compared to patients receiving chemotherapy alone. Across clinical studies the incidence of Grades 3-4 hypertension ranged from 5% to 18%.
Monitor blood pressure every two to three weeks during treatment with MVASI. Treat with appropriate anti-hypertensive therapy and monitor blood pressure regularly. Continue to monitor blood pressure at regular intervals in patients with MVASI-induced or -exacerbated hypertension after discontinuing MVASI. Withhold MVASI in patients with severe hypertension (such as Grade 3 and above) until controlled with medical management; resume once controlled with medical management. Discontinue in patients who develop hypertensive crisis or hypertensive encephalopathy.
5.7 Posterior Reversible Encephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES) was reported in < 0.5% of patients across clinical studies. The onset of symptoms occurred from 16 hours to 1 year after the first dose. PRES is a neurological disorder which can present with headache, seizure, lethargy, confusion, blindness and other visual and neurologic disturbances. Mild to severe hypertension may be present. Magnetic resonance imaging is necessary to confirm the diagnosis of PRES.
Discontinue MVASI in patients who develop PRES. Symptoms usually resolve or improve within days after discontinuing bevacizumab products, although some patients have experienced ongoing neurologic sequelae. The safety of reinitiating bevacizumab products in patients who developed PRES is not known.
5.8 Renal Injury and Proteinuria
The incidence and severity of proteinuria was higher in patients receiving bevacizumab products as compared to patients receiving chemotherapy. Grade 3 (defined as urine dipstick 4+ or > 3.5 grams of protein per 24 hours) to Grade 4 (defined as nephrotic syndrome) ranged from 0.7% to 7% in clinical studies. The overall incidence of proteinuria (all grades) was only adequately assessed in Study BO17705, in which the incidence was 20%. Median onset of proteinuria was 5.6 months (15 days to 37 months) after initiating bevacizumab. Median time to resolution was 6.1 months (95% CI: 2.8, 11.3). Proteinuria did not resolve in 40% of patients after median follow-up of 11.2 months and required discontinuation of bevacizumab in 30% of the patients who developed proteinuria [see Adverse Reactions (6.1)].
In an exploratory, pooled analysis of patients from seven randomized clinical studies, 5% of patients receiving bevacizumab with chemotherapy experienced Grades 2-4 (defined as urine dipstick 2+ or greater or > 1 gram of protein per 24 hours or nephrotic syndrome) proteinuria. Grades 2-4 proteinuria resolved in 74% of patients. Bevacizumab was reinitiated in 42% of patients. Of the 113 patients who reinitiated bevacizumab, 48% experienced a second episode of Grades 2-4 proteinuria.
Nephrotic syndrome occurred in < 1% of patients receiving bevacizumab across clinical studies, in some instances with fatal outcome. In a published case series, kidney biopsy of 6 patients with proteinuria showed findings consistent with thrombotic microangiopathy. Results of a retrospective analysis of 5805 patients who received bevacizumab with chemotherapy and 3713 patients who received chemotherapy alone, showed higher rates of elevated serum creatinine levels (between 1.5 to 1.9 times baseline levels) in patients who received bevacizumab. Serum creatinine levels did not return to baseline in approximately one-third of patients who received bevacizumab.
Monitor proteinuria by dipstick urine analysis for the development or worsening of proteinuria with serial urinalyses during MVASI therapy. Patients with a 2+ or greater urine dipstick reading should undergo further assessment with a 24-hour urine collection. Withhold for proteinuria greater than or equal to 2 grams per 24 hours and resume when less than 2 grams per 24 hours. Discontinue in patients who develop nephrotic syndrome.
Data from a postmarketing safety study showed poor correlation between UPCR (Urine Protein/Creatinine Ratio) and 24-hour urine protein [Pearson Correlation 0.39 (95% CI: 0.17, 0.57)].
5.9 Infusion-Related Reactions
Infusion-related reactions reported across clinical studies and postmarketing experience include hypertension, hypertensive crises associated with neurologic signs and symptoms, wheezing, oxygen desaturation, Grade 3 hypersensitivity, chest pain, headaches, rigors, and diaphoresis. In clinical studies, infusion-related reactions with the first dose occurred in < 3% of patients and severe reactions occurred in 0.2% of patients.
Decrease the rate of infusion for mild, clinically insignificant infusion-related reactions. Interrupt the infusion in patients with clinically significant infusion-related reactions and consider resuming at a slower rate following resolution. Discontinue in patients who develop a severe infusion-related reaction and administer appropriate medical therapy (e.g., epinephrine, corticosteroids, intravenous antihistamines, bronchodilators and/or oxygen).
5.10 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, bevacizumab products may cause fetal harm when administered to pregnant women. Congenital malformations were observed with the administration of bevacizumab to pregnant rabbits during organogenesis every 3 days at a dose as low as a clinical dose of 10 mg/kg. Furthermore, animal models link angiogenesis and VEGF and VEGFR2 to critical aspects of female reproduction, embryo-fetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with MVASI and for 6 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.11 Ovarian Failure
The incidence of ovarian failure was 34% vs. 2% in premenopausal women receiving bevacizumab with chemotherapy as compared to those receiving chemotherapy alone for adjuvant treatment of a solid tumor. After discontinuing bevacizumab, recovery of ovarian function at all time points during the post-treatment period was demonstrated in 22% of women receiving bevacizumab. Recovery of ovarian function is defined as resumption of menses, a positive serum β-HCG pregnancy test, or an FSH level < 30 mIU/mL during the post-treatment period. Long-term effects of bevacizumab products on fertility are unknown. Inform females of reproductive potential of the risk of ovarian failure prior to initiating MVASI [see Adverse Reactions (6.1), Use in Specific Populations (8.3)].
5.12 Congestive Heart Failure (CHF)
MVASI is not indicated for use with anthracycline-based chemotherapy. The incidence of Grade ≥ 3 left ventricular dysfunction was 1% in patients receiving bevacizumab compared to 0.6% of patients receiving chemotherapy alone. Among patients who received prior anthracycline treatment, the rate of CHF was 4% for patients receiving bevacizumab with chemotherapy as compared to 0.6% for patients receiving chemotherapy alone.
In previously untreated patients with a hematological malignancy, the incidence of CHF and decline in left-ventricular ejection fraction (LVEF) were increased in patients receiving bevacizumab with anthracycline-based chemotherapy compared to patients receiving placebo with the same chemotherapy regimen. The proportion of patients with a decline in LVEF from baseline of ≥ 20% or a decline from baseline of 10% to < 50%, was 10% in patients receiving bevacizumab with chemotherapy compared to 5% in patients receiving chemotherapy alone. Time to onset of left-ventricular dysfunction or CHF was 1 to 6 months after the first dose in at least 85% of the patients and was resolved in 62% of the patients who developed CHF in the bevacizumab arm compared to 82% in the placebo arm. Discontinue MVASI in patients who develop CHF.
-
6
ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Gastrointestinal Perforations and Fistulae [see Warnings and Precautions (5.1)].
- Surgery and Wound Healing Complications [see Warnings and Precautions (5.2)].
- Hemorrhage [see Warnings and Precautions (5.3)].
- Arterial Thromboembolic Events [see Warnings and Precautions (5.4)].
- Venous Thromboembolic Events [see Warnings and Precautions (5.5)].
- Hypertension [see Warnings and Precautions (5.6)].
- Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.7)].
- Renal Injury and Proteinuria [see Warnings and Precautions (5.8)].
- Infusion-Related Reactions [see Warnings and Precautions (5.9)].
- Ovarian Failure [see Warnings and Precautions (5.11)].
- Congestive Heart Failure [see Warnings and Precautions (5.12)].
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The safety data in Warnings and Precautions and described below reflect exposure to bevacizumab in 4134 patients including those with mCRC (AVF2107g, E3200), non-squamous NSCLC (E4599), GBM (EORTC 26101), mRCC (BO17705), and cervical cancer (GOG-0240), including controlled studies or other cancers, at the recommended dose and schedule for a median of 6 to 23 doses. The most common adverse reactions observed in patients receiving bevacizumab as a single agent or in combination with chemotherapy at a rate > 10% were epistaxis, headache, hypertension, rhinitis, proteinuria, taste alteration, dry skin, rectal hemorrhage, lacrimation disorder, back pain and exfoliative dermatitis.
Across clinical studies, bevacizumab was discontinued in 8% to 22% of patients because of adverse reactions [see Clinical Studies (14)].
Metastatic Colorectal Cancer
In Combination with bolus-IFL
The safety of bevacizumab was evaluated in 392 patients who received at least one dose of bevacizumab in a double-blind, active-controlled study (AVF2107g), which compared bevacizumab (5 mg/kg every 2 weeks) with bolus-IFL to placebo with bolus-IFL in patients with mCRC [see Clinical Studies (14.1)]. Patients were randomized (1:1:1) to placebo with bolus-IFL, bevacizumab with bolus-IFL, or bevacizumab with fluorouracil and leucovorin. The demographics of the safety population were similar to the demographics of the efficacy population. All Grades 3-4 adverse reactions and selected Grades 1-2 adverse reactions (i.e., hypertension, proteinuria, thromboembolic events) were collected in the entire study population. Adverse reactions are presented in Table 2.
Table 2: Grades 3-4 Adverse Reactions Occurring at Higher Incidence (≥ 2%) in Patients Receiving Bevacizumab vs. Placebo in Study AVF2107g Adverse Reactiona Bevacizumab with IFL
(N = 392)Placebo with IFL
(N = 396)Hematology Leukopenia 37% 31% Neutropenia 21% 14% Gastrointestinal Diarrhea 34% 25% Abdominal pain 8% 5% Constipation 4% 2% Vascular Hypertension 12% 2% Deep vein thrombosis 9% 5% Intra-abdominal Thrombosis 3% 1% Syncope 3% 1% General Asthenia 10% 7% Pain 8% 5% a NCI-CTC version 3.
In Combination with FOLFOX4
The safety of bevacizumab was evaluated in 521 patients in an open-label, active-controlled study (E3200) in patients who were previously treated with irinotecan and fluorouracil for initial therapy for mCRC. Patients were randomized (1:1:1) to FOLFOX4, bevacizumab (10 mg/kg every 2 weeks prior to FOLFOX4 on Day 1) with FOLFOX4, or bevacizumab alone (10 mg/kg every 2 weeks). Bevacizumab was continued until disease progression or unacceptable toxicity.
The demographics of the safety population were similar to the demographics of the efficacy population.
Selected Grades 3-5 non-hematologic and Grades 4-5 hematologic occurring at a higher incidence (≥ 2%) in patients receiving bevacizumab with FOLFOX4 compared to FOLFOX4 alone were fatigue (19% vs. 13%), diarrhea (18% vs. 13%), sensory neuropathy (17% vs. 9%), nausea (12% vs. 5%), vomiting (11% vs. 4%), dehydration (10% vs. 5%), hypertension (9% vs. 2%), abdominal pain (8% vs. 5%), hemorrhage (5% vs. 1%), other neurological (5% vs. 3%), ileus (4% vs. 1%) and headache (3% vs. 0%). These data are likely to under-estimate the true adverse reaction rates due to the reporting mechanisms.
First-Line Non-Squamous Non-Small Cell Lung Cancer
The safety of bevacizumab was evaluated as first-line treatment in 422 patients with unresectable NSCLC who received at least one dose of bevacizumab in an active-controlled, open-label, multicenter trial (E4599) [see Clinical Studies (14.3)]. Chemotherapy-naïve patients with locally advanced, metastatic or recurrent non–squamous NSCLC were randomized (1:1) to receive six 21-day cycles of paclitaxel and carboplatin with or without bevacizumab (15 mg/kg every 3 weeks). After completion or upon discontinuation of chemotherapy, patients randomized to receive bevacizumab continued to receive bevacizumab alone until disease progression or until unacceptable toxicity. The trial excluded patients with predominant squamous histology (mixed cell type tumors only), CNS metastasis, gross hemoptysis (1/2 teaspoon or more of red blood), unstable angina, or receiving therapeutic anticoagulation. The demographics of the safety population were similar to the demographics of the efficacy population.
Only Grades 3-5 non-hematologic and Grades 4-5 hematologic adverse reactions were collected. Grades 3-5 non-hematologic and Grades 4-5 hematologic adverse reactions occurring at a higher incidence (≥ 2%) in patients receiving bevacizumab with paclitaxel and carboplatin compared with patients receiving chemotherapy alone were neutropenia (27% vs. 17%), fatigue (16% vs. 13%), hypertension (8% vs. 0.7%), infection without neutropenia (7% vs. 3%), venous thromboembolism (5% vs. 3%), febrile neutropenia (5% vs. 2%), pneumonitis/pulmonary infiltrates (5% vs. 3%), infection with Grade 3 or 4 neutropenia (4% vs. 2%), hyponatremia (4% vs. 1%), headache (3% vs. 1%) and proteinuria (3% vs. 0%).
Recurrent Glioblastoma
The safety of bevacizumab was evaluated in a multicenter, randomized, open-label study (EORTC 26101) in patients with recurrent GBM following radiotherapy and temozolomide of whom 278 patients received at least one dose of bevacizumab and are considered safety evaluable [see Clinical Studies (14.4)]. Patients were randomized (2:1) to receive bevacizumab (10 mg/kg every 2 weeks) with lomustine or lomustine alone until disease progression or unacceptable toxicity. The demographics of the safety population were similar to the demographics of the efficacy population. In the bevacizumab with lomustine arm, 22% of patients discontinued treatment due to adverse reactions compared with 10% of patients in the lomustine arm. In patients receiving bevacizumab with lomustine, the adverse reaction profile was similar to that observed in other approved indications.
Metastatic Renal Cell Carcinoma
The safety of bevacizumab was evaluated in 337 patients who received at least one dose of bevacizumab in a multicenter, double-blind study (BO17705) in patients with mRCC. Patients who had undergone a nephrectomy were randomized (1:1) to receive either bevacizumab (10 mg/kg every 2 weeks) or placebo with interferon-alfa [see Clinical Studies (14.5)]. Patients were treated until disease progression or unacceptable toxicity. The demographics of the safety population were similar to the demographics of the efficacy population.
Grades 3-5 adverse reactions occurring at a higher incidence (> 2%) were fatigue (13% vs. 8%), asthenia (10% vs. 7%), proteinuria (7% vs. 0%), hypertension (6% vs. 1%; including hypertension and hypertensive crisis), and hemorrhage (3% vs. 0.3%; including epistaxis, small intestinal hemorrhage, aneurysm ruptured, gastric ulcer hemorrhage, gingival bleeding, hemoptysis, hemorrhage intracranial, large intestinal hemorrhage, respiratory tract hemorrhage, and traumatic hematoma). Adverse reactions are presented in Table 3.
Table 3: Grades 1-5 Adverse Reactions Occurring at Higher Incidence (≥ 5%) of Patients Receiving Bevacizumab vs. Placebo with Interferon-Alfa in Study BO17705 Adverse Reactiona Bevacizumab with Interferon-Alfa
(N = 337)Placebo with
Interferon-Alfa
(N = 304)Metabolism and nutrition Decreased appetite 36% 31% Weight loss 20% 15% General Fatigue 33% 27% Vascular Hypertension 28% 9% Respiratory, thoracic and mediastinal Epistaxis 27% 4% Dysphonia 5% 0% Nervous system Headache 24% 16% Gastrointestinal Diarrhea 21% 16% Renal and urinary Proteinuria 20% 3% Musculoskeletal and connective tissue Myalgia 19% 14% Back pain 12% 6% a NCI-CTC version 3.
The following adverse reactions were reported at a 5-fold greater incidence in patients receiving bevacizumab with interferon-alfa compared to patients receiving placebo with interferon-alfa and not represented in Table 2: gingival bleeding (13 patients vs. 1 patient); rhinitis (9 vs. 0); blurred vision (8 vs. 0); gingivitis (8 vs. 1); gastroesophageal reflux disease (8 vs. 1); tinnitus (7 vs. 1); tooth abscess (7 vs. 0); mouth ulceration (6 vs. 0); acne (5 vs. 0); deafness (5 vs. 0); gastritis (5 vs. 0); gingival pain (5 vs. 0) and pulmonary embolism (5 vs. 1).
Persistent, Recurrent, or Metastatic Cervical Cancer
The safety of bevacizumab was evaluated in 218 patients who received at least one dose of bevacizumab in a multicenter study (GOG-0240) in patients with persistent, recurrent, or metastatic cervical cancer [see Clinical Studies (14.6)]. Patients were randomized (1:1:1:1) to receive paclitaxel and cisplatin with or without bevacizumab (15 mg/kg every 3 weeks), or paclitaxel and topotecan with or without bevacizumab (15 mg/kg every 3 weeks). The demographics of the safety population were similar to the demographics of the efficacy population.
Grades 3-4 adverse reactions occurring at a higher incidence (≥ 2%) in 218 patients receiving bevacizumab with chemotherapy compared to 222 patients receiving chemotherapy alone were abdominal pain (12% vs. 10%), hypertension (11% vs. 0.5%), thrombosis (8% vs. 3%), diarrhea (6% vs. 3%), anal fistula (4% vs. 0%), proctalgia (3% vs. 0%), urinary tract infection (8% vs. 6%), cellulitis (3% vs. 0.5%), fatigue (14% vs. 10%), hypokalemia (7% vs. 4%), hyponatremia (4% vs. 1%), dehydration (4% vs. 0.5%), neutropenia (8% vs. 4%), lymphopenia (6% vs. 3%), back pain (6% vs. 3%), and pelvic pain (6% vs. 1%). Adverse reactions are presented in Table 4.
Table 4: Grades 1-4 Adverse Reactions Occurring at Higher Incidence (≥ 5%) in Patients Receiving Bevacizumab with Chemotherapy vs. Chemotherapy Alone in Study GOG-0240 Adverse Reactiona Bevacizumab with
Chemotherapy
(N = 218)Chemotherapy
(N = 222)General Fatigue 80% 75% Peripheral edema 15% 22% Metabolism and nutrition Decreased appetite 34% 26% Hyperglycemia 26% 19% Hypomagnesemia 24% 15% Weight loss 21% 7% Hyponatremia 19% 10% Hypoalbuminemia 16% 11% Vascular Hypertension 29% 6% Thrombosis 10% 3% Infections Urinary tract infection 22% 14% Infection 10% 5% Nervous system Headache 22% 13% Dysarthria 8% 1% Psychiatric Anxiety 17% 10% Respiratory, thoracic and mediastinal Epistaxis 17% 1% Renal and urinary Increased blood creatinine 16% 10% Proteinuria 10% 3% Gastrointestinal Stomatitis 15% 10% Proctalgia 6% 1% Anal fistula 6% 0.0% Reproductive system and breast Pelvic pain 14% 8% Hematology Neutropenia 12% 6% Lymphopenia 12% 5% a NCI-CTC version 3.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and the specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to bevacizumab in the studies described below with the incidence of antibodies in other studies or to other bevacizumab products may be misleading.
In clinical studies for adjuvant treatment of a solid tumor, 0.6% (14/2233) of patients tested positive for treatment-emergent anti-bevacizumab antibodies as detected by an electrochemiluminescent (ECL) based assay. Among these 14 patients, three tested positive for neutralizing antibodies against bevacizumab using an enzyme-linked immunosorbent assay (ELISA). The clinical significance of these anti-bevacizumab antibodies is not known.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of bevacizumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
General: Polyserositis
Cardiovascular: Pulmonary hypertension, Mesenteric venous occlusion
Gastrointestinal: Gastrointestinal ulcer, Intestinal necrosis, Anastomotic ulceration
Hemic and lymphatic: Pancytopenia
Hepatobiliary disorders: Gallbladder perforation
Musculoskeletal and Connective Tissue Disorders: Osteonecrosis of the jaw
Renal: Renal thrombotic microangiopathy (manifested as severe proteinuria)
Respiratory: Nasal septum perforation
- Gastrointestinal Perforations and Fistulae [see Warnings and Precautions (5.1)].
-
7
DRUG INTERACTIONS
Effects of MVASI on Other Drugs
No clinically meaningful effect on the pharmacokinetics of irinotecan or its active metabolite SN38, interferon-alfa, carboplatin or paclitaxel was observed when bevacizumab was administered in combination with these drugs; however, 3 of the 8 patients receiving bevacizumab with paclitaxel and carboplatin had lower paclitaxel exposure after four cycles of treatment (at Day 63) than those at Day 0, while patients receiving paclitaxel and carboplatin alone had a greater paclitaxel exposure at Day 63 than at Day 0.
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and their mechanism of action [see Clinical Pharmacology (12.1)], bevacizumab products may cause fetal harm in pregnant women. Limited postmarketing reports describe cases of fetal malformations with use of bevacizumab products in pregnancy; however, these reports are insufficient to determine drug associated risks. In animal reproduction studies, intravenous administration of bevacizumab to pregnant rabbits every 3 days during organogenesis at doses approximately 1 to 10 times the clinical dose of 10 mg/kg produced fetal resorptions, decreased maternal and fetal weight gain and multiple congenital malformations including corneal opacities and abnormal ossification of the skull and skeleton including limb and phalangeal defects (see Data). Furthermore, animal models link angiogenesis and VEGF and VEGFR2 to critical aspects of female reproduction, embryofetal development, and postnatal development. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Pregnant rabbits dosed with 10 mg/kg to 100 mg/kg bevacizumab (approximately 1 to 10 times the clinical dose of 10 mg/kg) every three days during the period of organogenesis (gestation day 6-18) exhibited decreases in maternal and fetal body weights and increased number of fetal resorptions.
There were dose-related increases in the number of litters containing fetuses with any type of malformation (42% for the 0 mg/kg dose, 76% for the 30 mg/kg dose, and 95% for the 100 mg/kg dose) or fetal alterations (9% for the 0 mg/kg dose, 15% for the 30 mg/kg dose, and 61% for the 100 mg/kg dose). Skeletal deformities were observed at all dose levels, with some abnormalities including meningocele observed only at the 100 mg/kg dose level. Teratogenic effects included: reduced or irregular ossification in the skull, jaw, spine, ribs, tibia and bones of the paws; fontanel, rib and hindlimb deformities; corneal opacity; and absent hindlimb phalanges.
8.2 Lactation
Risk Summary
No data are available regarding the presence of bevacizumab products in human milk, the effects on the breast fed infant, or the effects on milk production. Human IgG is present in human milk, but published data suggest that breast milk antibodies do not enter the neonatal and infant circulation in substantial amounts. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with MVASI and for 6 months after the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Bevacizumab products may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with MVASI and for 6 months after the last dose.
Infertility
Females
Bevacizumab products increase the risk of ovarian failure and may impair fertility. Inform females of reproductive potential of the risk of ovarian failure prior to the first dose of MVASI. Long-term effects of bevacizumab products on fertility are not known.
In a clinical study of 179 premenopausal women randomized to receive chemotherapy with or without bevacizumab, the incidence of ovarian failure was higher in patients who received bevacizumab with chemotherapy (34%) compared to patients who received chemotherapy alone (2%). After discontinuing bevacizumab with chemotherapy, recovery of ovarian function occurred in 22% of these patients [see Warnings and Precautions (5.11), Adverse Reactions (6.1)].
8.4 Pediatric Use
The safety and effectiveness of bevacizumab products in pediatric patients have not been established. In published literature reports, cases of non-mandibular osteonecrosis have been observed in patients under the age of 18 years who have received bevacizumab. Bevacizumab products are not approved for use in patients under the age of 18 years.
Antitumor activity was not observed among eight pediatric patients with relapsed GBM who received bevacizumab and irinotecan. Addition of bevacizumab to standard of care did not result in improved event-free survival in pediatric patients enrolled in two randomized clinical studies, one in high grade glioma (n = 121) and one in metastatic rhabdomyosarcoma or non-rhabdomyosarcoma soft tissue sarcoma (n = 154).
Based on the population pharmacokinetics analysis of data from 152 pediatric and young adult patients with cancer (7 months to 21 years of age), bevacizumab clearance normalized by body weight in pediatrics was comparable to that in adults.
Juvenile Animal Toxicity Data
Juvenile cynomolgus monkeys with open growth plates exhibited physeal dysplasia following 4 to 26 weeks exposure at 0.4 to 20 times the recommended human dose (based on mg/kg and exposure). The incidence and severity of physeal dysplasia were dose-related and were partially reversible upon cessation of treatment.
8.5 Geriatric Use
In an exploratory, pooled analysis of 1745 patients from five randomized, controlled studies, 35% of patients were ≥ 65 years old. The overall incidence of ATE was increased in all patients receiving bevacizumab with chemotherapy as compared to those receiving chemotherapy alone, regardless of age; however, the increase in the incidence of ATE was greater in patients ≥ 65 years (8% vs. 3%) as compared to patients < 65 years (2% vs. 1%) [see Warnings and Precautions (5.4)].
-
11
DESCRIPTION
Bevacizumab-awwb is a vascular endothelial growth factor inhibitor. Bevacizumab-awwb is a recombinant humanized monoclonal IgG1 antibody that contains human framework regions and murine complementarity-determining regions. Bevacizumab-awwb has an approximate molecular weight of 149 kDa. Bevacizumab-awwb is produced in a mammalian cell (Chinese Hamster Ovary) expression system.
MVASI (bevacizumab-awwb) injection is a sterile, preservative-free, clear to slightly opalescent, colorless to pale yellow solution in a single-dose vial for intravenous use. MVASI contains bevacizumab-awwb at a concentration of 25 mg/mL in either 100 mg/4 mL or 400 mg/16 mL, single-dose vials.
Each mL of solution contains 25 mg bevacizumab-awwb, α,α-trehalose dihydrate (60 mg), polysorbate 20 (0.4 mg), sodium phosphate dibasic, anhydrous (1.2 mg), sodium phosphate monobasic, monohydrate (5.8 mg), and Water for Injection, USP. The pH is 6.2.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bevacizumab products bind VEGF and prevent the interaction of VEGF to its receptors (Flt-1 and KDR) on the surface of endothelial cells. The interaction of VEGF with its receptors leads to endothelial cell proliferation and new blood vessel formation in in vitro models of angiogenesis. Administration of bevacizumab to xenotransplant models of colon cancer in nude (athymic) mice caused reduction of microvascular growth and inhibition of metastatic disease progression.
12.3 Pharmacokinetics
The pharmacokinetic profile of bevacizumab was assessed using an assay that measures total serum bevacizumab concentrations (i.e., the assay did not distinguish between free bevacizumab and bevacizumab bound to VEGF ligand). Based on a population pharmacokinetic analysis of 491 patients who received 1 to 20 mg/kg of bevacizumab every week, every 2 weeks, or every 3 weeks, bevacizumab pharmacokinetics are linear and the predicted time to reach more than 90% of steady state concentration is 84 days. The accumulation ratio following a dose of 10 mg/kg of bevacizumab once every 2 weeks is 2.8.
Population simulations of bevacizumab exposures provide a median trough concentration of 80.3 mcg/mL on Day 84 (10th, 90th percentile: 45, 128) following a dose of 5 mg/kg once every two weeks.
Distribution
The mean (% coefficient of variation [CV%]) central volume of distribution is 2.9 (22%) L.
Elimination
The mean (CV%) clearance is 0.23 (33) L/day. The estimated half-life is 20 days (11 to 50 days).
Specific Populations
The clearance of bevacizumab varied by body weight, sex, and tumor burden. After correcting for body weight, males had a higher bevacizumab clearance (0.26 L/day vs. 0.21 L/day) and a larger central volume of distribution (3.2 L vs. 2.7 L) than females. Patients with higher tumor burden (at or above median value of tumor surface area) had a higher bevacizumab clearance (0.25 L/day vs. 0.20 L/day) than patients with tumor burdens below the median. In Study AVF2107g, there was no evidence of lesser efficacy (hazard ratio for overall survival) in males or patients with higher tumor burden treated with bevacizumab as compared to females and patients with low tumor burden.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted to assess potential of bevacizumab products for carcinogenicity or mutagenicity.
Bevacizumab products may impair fertility. Female cynomolgus monkeys treated with 0.4 to 20 times the recommended human dose of bevacizumab exhibited arrested follicular development or absent corpora lutea as well as dose-related decreases in ovarian and uterine weights, endometrial proliferation, and the number of menstrual cycles. Following a 4- or 12-week recovery period, there was a trend suggestive of reversibility. After the 12-week recovery period, follicular maturation arrest was no longer observed, but ovarian weights were still moderately decreased. Reduced endometrial proliferation was no longer observed at the 12-week recovery time point; however, decreased uterine weight, absent corpora lutea, and reduced number of menstrual cycles remained evident.
13.2 Animal Toxicology and/or Pharmacology
Rabbits dosed with bevacizumab exhibited reduced wound healing capacity. Using full-thickness skin incision and partial thickness circular dermal wound models, bevacizumab dosing resulted in reductions in wound tensile strength, decreased granulation and re-epithelialization, and delayed time to wound closure.
-
14
CLINICAL STUDIES
14.1 Metastatic Colorectal Cancer
Study AVF2107g
The safety and efficacy of Avastin was evaluated in a double-blind, active-controlled study [AVF2107g (NCT00109070)] in 923 patients with previously untreated mCRC who were randomized (1:1:1) to placebo with bolus-IFL (irinotecan 125 mg/m2, fluorouracil 500 mg/m2, and leucovorin 20 mg/m2 given once weekly for 4 weeks every 6 weeks), bevacizumab (5 mg/kg every 2 weeks) with bolus-IFL, or bevacizumab (5 mg/kg every 2 weeks) with fluorouracil and leucovorin. Enrollment to the bevacizumab with fluorouracil and leucovorin arm was discontinued, after enrollment of 110 patients in accordance with the protocol-specified adaptive design. Bevacizumab was continued until disease progression or unacceptable toxicity or for a maximum of 96 weeks. The main outcome measure was overall survival (OS).
The median age was 60 years; 60% were male, 79% were White, 57% had an ECOG performance status of 0, 21% had a rectal primary and 28% received prior adjuvant chemotherapy. The dominant site of disease was extra-abdominal in 56% of patients and was the liver in 38% of patients.
The addition of bevacizumab improved survival across subgroups defined by age (< 65 years, ≥ 65 years) and sex. Results are presented in Table 5 and Figure 1.
Table 5: Efficacy Results in Study AVF2107g Efficacy Parameter Bevacizumab with bolus-IFL
(N = 402)Placebo with bolus-IFL
(N = 411)Overall Survival Median, in months 20.3 15.6 Hazard ratio (95% CI) 0.66 (0.54, 0.81) p-valuea < 0.001 Progression-Free Survival Median, in months 10.6 6.2 Hazard ratio (95% CI) 0.54 (0.45, 0.66) p-valuea < 0.001 Overall Response Rate Rate (%) 45% 35% p-valueb < 0.01 Duration of Response Median, in months 10.4 7.1 a by stratified log-rank test.
b by χ2 test.
Figure 1: Kaplan-Meier Curves for Duration of Survival in Metastatic Colorectal Cancer in Study AVF2107g 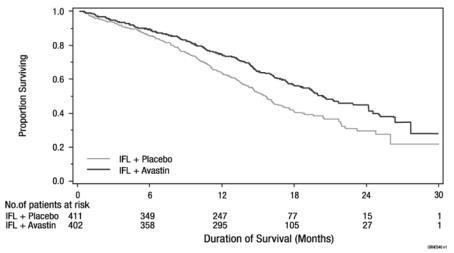
Among the 110 patients randomized to bevacizumab with fluorouracil and leucovorin, median OS was 18.3 months, median progression-free survival (PFS) was 8.8 months, overall response rate (ORR) was 39%, and median duration of response was 8.5 months.
Study E3200
The safety and efficacy of Avastin were evaluated in a randomized, open-label, active-controlled study [E3200 (NCT00025337)] in 829 patients who were previously treated with irinotecan and fluorouracil for initial therapy for metastatic disease or as adjuvant therapy. Patients were randomized (1:1:1) to FOLFOX4 (Day 1: oxaliplatin 85 mg/m2 and leucovorin 200 mg/m2 concurrently, then fluorouracil 400 mg/m2 bolus followed by 600 mg/m2 continuously; Day 2: leucovorin 200 mg/m2, then fluorouracil 400 mg/m2 bolus followed by 600 mg/m2 continuously; every 2 weeks), bevacizumab (10 mg/kg every 2 weeks prior to FOLFOX4 on Day 1) with FOLFOX4, or bevacizumab alone (10 mg/kg every 2 weeks). Bevacizumab was continued until disease progression or unacceptable toxicity. The main outcome measure was OS.
The bevacizumab alone arm was closed to accrual after enrollment of 244 of the planned 290 patients following a planned interim analysis by the data monitoring committee based on evidence of decreased survival compared to FOLFOX4 alone.
The median age was 61 years; 60% were male, 87% were White, 49% had an ECOG performance status of 0, 26% received prior radiation therapy, and 80% received prior adjuvant chemotherapy, 99% received prior irinotecan, with or without fluorouracil for metastatic disease, and 1% received prior irinotecan and fluorouracil as adjuvant therapy.
The addition of bevacizumab to FOLFOX4 resulted in significantly longer survival as compared to FOLFOX4 alone; median OS was 13.0 months vs. 10.8 months [hazard ratio (HR) 0.75 (95% CI: 0.63, 0.89), p-value of 0.001 stratified log-rank test] with clinical benefit seen in subgroups defined by age (< 65 years, ≥ 65 years) and sex. PFS and ORR based on investigator assessment were higher in patients receiving bevacizumab with FOLFOX4.
Study TRC-0301
The activity of bevacizumab with fluorouracil (as bolus or infusion) and leucovorin was evaluated in a single arm study [TRC-0301 (NCT00066846)] enrolling 339 patients with mCRC with disease progression following both irinotecan- and oxaliplatin-based chemotherapy. Seventy-three percent of patients received concurrent bolus fluorouracil and leucovorin. One objective partial response was verified in the first 100 evaluable patients for an ORR of 1% (95 % CI: 0%, 5.5%).
Study ML18147
The safety and efficacy of Avastin were evaluated in a prospective, randomized, open-label, multinational, controlled study [ML18147 (NCT00700102)] in 820 patients with histologically confirmed mCRC who had progressed on a first-line bevacizumab-containing regimen. Patients were excluded if they progressed within 3 months of initiating first-line chemotherapy and if they received bevacizumab for less than 3 consecutive months in the first-line setting. Patients were randomized (1:1) within 3 months after discontinuing bevacizumab as first-line treatment to receive fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy with or without bevacizumab (5 mg/kg every 2 weeks or 7.5 mg/kg every 3 weeks). The choice of second-line treatment was contingent upon first-line chemotherapy. Second-line treatment was administered until progressive disease or unacceptable toxicity. The main outcome measure was OS. A secondary outcome measure was ORR.
The median age was 63 years (21 to 84 years); 64% were male, 52% had an ECOG performance status of 1, 44% had an ECOG performance status of 0, 58% received irinotecan-based therapy as first-line treatment, 55% progressed on first-line treatment within 9 months, and 77% received their last dose of bevacizumab as first-line treatment within 42 days of being randomized. Second-line chemotherapy regimens were generally balanced between each arm.
The addition of bevacizumab to fluoropyrimidine-based chemotherapy resulted in a statistically significant prolongation of OS and PFS. There was no significant difference in ORR. Results are presented in Table 6 and Figure 2.
Table 6: Efficacy Results in Study ML18147 Efficacy Parameter Bevacizumab with
Chemotherapy
(N = 409)Chemotherapy
(N = 411)Overall Survivala Median, in months 11.2 9.8 Hazard ratio (95% CI) 0.81 (0.69, 0.94) Progression-Free Survivalb Median, in months 5.7 4.0 Hazard ratio (95% CI) 0.68 (0.59, 0.78) a p = 0.0057 by unstratified log-rank test.
b p-value < 0.0001 by unstratified log-rank test.
Figure 2: Kaplan-Meier Curves for Duration of Survival in Metastatic Colorectal Cancer in Study ML18147
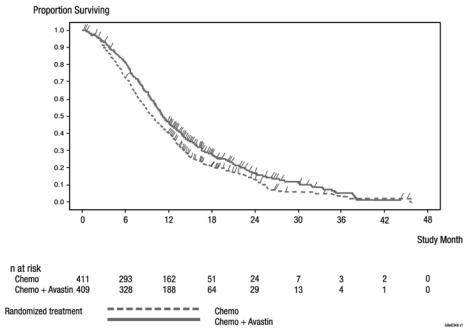
14.2 Lack of Efficacy in Adjuvant Treatment of Colon Cancer
Lack of efficacy of bevacizumab as an adjunct to standard chemotherapy for the adjuvant treatment of colon cancer was determined in two randomized, open-label, multicenter clinical studies. The first study [BO17920 (NCT00112918)] was conducted in 3451 patients with high-risk stage II and III colon cancer, who had undergone surgery for colon cancer with curative intent. Patients were randomized to receive bevacizumab at a dose equivalent to 2.5 mg/kg/week on either a 2-weekly schedule with FOLFOX4 (N = 1155), or on a 3-weekly schedule with XELOX (N = 1145) or FOLFOX4 alone (N = 1151). The main outcome measure was disease free survival (DFS) in patients with stage III colon cancer.
The median age was 58 years; 54% were male, 84% were White and 29% were ≥ 65 years. Eighty-three percent had stage III disease.
The addition of bevacizumab to chemotherapy did not improve DFS. As compared to FOLFOX4 alone, the proportion of stage III patients with disease recurrence or with death due to disease progression were numerically higher for patients receiving bevacizumab with FOLFOX4 or with XELOX. The hazard ratios for DFS were 1.17 (95% CI: 0.98, 1.39) for bevacizumab with FOLFOX4 versus FOLFOX4 alone and 1.07 (95% CI: 0.90, 1.28) for bevacizumab with XELOX versus FOLFOX4 alone. The hazard ratios for OS were 1.31 (95% CI = 1.03, 1.67) and 1.27 (95% CI = 1, 1.62) for the comparison of bevacizumab with FOLFOX4 versus FOLFOX4 alone and bevacizumab with XELOX versus FOLFOX4 alone, respectively. Similar lack of efficacy for DFS was observed in the bevacizumab-containing arms compared to FOLFOX4 alone in the high-risk stage II cohort.
In a second study [NSABP-C-08 (NCT00096278)], patients with stage II and III colon cancer who had undergone surgery with curative intent, were randomized to receive either bevacizumab administered at a dose equivalent to 2.5 mg/kg/week with mFOLFOX6 (N = 1354) or mFOLFOX6 alone (N = 1356). The median age was 57 years, 50% were male and 87% White. Seventy-five percent had stage III disease. The main outcome was DFS among stage III patients. The HR for DFS was 0.92 (95% CI: 0.77, 1.10). OS was not significantly improved with the addition of bevacizumab to mFOLFOX6 [HR 0.96 (95% CI: 0.75, 1.22)].
14.3 First-Line Non-Squamous Non-Small Cell Lung Cancer
Study E4599
The safety and efficacy of bevacizumab as first-line treatment of patients with locally advanced, metastatic, or recurrent non-squamous NSCLC was studied in a single, large, randomized, active-controlled, open-label, multicenter study [E4599 (NCT00021060)]. A total of 878 chemotherapy-naïve patients with locally advanced, metastatic or recurrent non-squamous NSCLC were randomized (1:1) to receive six 21-day cycles of paclitaxel (200 mg/m2) and carboplatin (AUC 6) with or without bevacizumab 15 mg/kg. After completing or discontinuing chemotherapy, patients randomized to receive bevacizumab continued to receive bevacizumab alone until disease progression or until unacceptable toxicity. The trial excluded patients with predominant squamous histology (mixed cell type tumors only), CNS metastasis, gross hemoptysis (1/2 teaspoon or more of red blood), unstable angina, or receiving therapeutic anticoagulation. The main outcome measure was duration of survival.
The median age was 63 years; 54% were male, 43% were ≥ 65 years, and 28% had ≥ 5% weight loss at study entry. Eleven percent had recurrent disease. Of the 89% with newly diagnosed NSCLC, 12% had Stage IIIB with malignant pleural effusion and 76% had Stage IV disease.
OS was statistically significantly longer for patients receiving bevacizumab with paclitaxel and carboplatin compared with those receiving chemotherapy alone. Median OS was 12.3 months vs. 10.3 months [HR 0.80 (95% CI: 0.68, 0.94), final p-value of 0.013, stratified log-rank test]. Based on investigator assessment which was not independently verified, patients were reported to have longer PFS with bevacizumab with paclitaxel and carboplatin compared to chemotherapy alone. Results are presented in Figure 3.
Figure 3: Kaplan-Meier Curves for Duration of Survival in First-Line Non-Squamous Non-Small Cell Lung Cancer in Study E4599
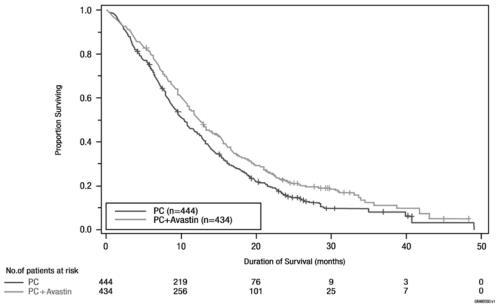
In an exploratory analysis across patient subgroups, the impact of bevacizumab on OS was less robust in the following subgroups: women [HR 0.99 (95% CI: 0.79, 1.25)], patients ≥ 65 years [HR 0.91 (95% CI: 0.72, 1.14)] and patients with ≥ 5% weight loss at study entry [HR 0.96 (95% CI: 0.73, 1.26)].
Study BO17704
The safety and efficacy of bevacizumab in patients with locally advanced, metastatic or recurrent non-squamous NSCLC, who had not received prior chemotherapy was studied in another randomized, double-blind, placebo-controlled study [BO17704 (NCT00806923)]. A total of 1043 patients were randomized (1:1:1) to receive cisplatin and gemcitabine with placebo, bevacizumab 7.5 mg/kg or bevacizumab 15 mg/kg. The main outcome measure was PFS. Secondary outcome measure was OS.
The median age was 58 years; 36% were female and 29% were ≥ 65 years. Eight percent had recurrent disease and 77% had Stage IV disease.
PFS was significantly higher in both bevacizumab-containing arms compared to the placebo arm [HR 0.75 (95% CI: 0.62, 0.91), p-value of 0.0026 for bevacizumab 7.5 mg/kg and HR 0.82 (95% CI: 0.68, 0.98), p-value of 0.0301 for bevacizumab 15 mg/kg]. The addition of bevacizumab to cisplatin and gemcitabine failed to demonstrate an improvement in the duration of OS [HR 0.93 (95% CI: 0.78, 1.11), p-value of 0.420 for bevacizumab 7.5 mg/kg and HR 1.03 (95% CI: 0.86, 1.23), p-value of 0.761 for bevacizumab 15 mg/kg].
14.4 Recurrent Glioblastoma
Study EORTC 26101
The safety and efficacy of bevacizumab were evaluated in a multicenter, randomized (2:1), open-label study in patients with recurrent GBM (EORTC 26101, NCT01290939). Patients with first progression following radiotherapy and temozolomide were randomized (2:1) to receive bevacizumab (10 mg/kg every 2 weeks) with lomustine (90 mg/m2 every 6 weeks) or lomustine (110 mg/m2 every 6 weeks) alone until disease progression or unacceptable toxicity. Randomization was stratified by World Health Organization performance status (0 vs. > 0), steroid use (yes vs. no), largest tumor diameter (≤ 40 vs. > 40 mm), and institution. The main outcome measure was OS. Secondary outcome measures were investigator-assessed PFS and ORR per the modified Response Assessment in Neuro-oncology (RANO) criteria, health related quality of life (HRQoL), cognitive function, and corticosteroid use.
A total of 432 patients were randomized to receive lomustine alone (N = 149) or bevacizumab with lomustine (N = 283). The median age was 57 years; 24.8% of patients were ≥ 65 years. The majority of patients with were male (61%); 66% had a WHO performance status score > 0; and in 56% the largest tumor diameter was ≤ 40 mm. Approximately 33% of patients randomized to receive lomustine received bevacizumab following documented progression.
No difference in OS (HR 0.91, p-value of 0.4578) was observed between arms; therefore, all secondary outcome measures are descriptive only. PFS was longer in the bevacizumab with lomustine arm [HR 0.52 (95% CI: 0.41, 0.64)] with a median PFS of 4.2 months in the bevacizumab with lomustine arm and 1.5 months in the lomustine arm. Among the 50% of patients receiving corticosteroids at the time of randomization, a higher percentage of patients in the bevacizumab with lomustine arm discontinued corticosteroids (23% vs. 12%).
Study AVF3708g and Study NCI 06-C-0064E
The efficacy and safety of bevacizumab 10 mg/kg every 2 weeks in patients with previously treated GBM were evaluated in one single arm single center study (NCI 06-C-0064E) and a randomized noncomparative multicenter study [AVF3708g (NCT00345163)]. Response rates in both studies were evaluated based on modified WHO criteria that considered corticosteroid use. In AVF3708g, the response rate was 25.9% (95% CI: 17%, 36.1%) with a median duration of response of 4.2 months (95% CI: 3, 5.7). In Study NCI 06-C-0064E, the response rate was 19.6% (95% CI: 10.9%, 31.3%) with a median duration of response of 3.9 months (95% CI: 2.4, 17.4).
14.5 Metastatic Renal Cell Carcinoma
Study BO17705
The safety and efficacy of Avastin were evaluated in patients with treatment-naïve mRCC in a multicenter, randomized, double-blind, international study [BO17705 (NCT00738530)] comparing interferon-alfa and bevacizumab versus interferon alfa and placebo. A total of 649 patients who had undergone a nephrectomy were randomized (1:1) to receive either bevacizumab (10 mg/kg every 2 weeks; N = 327) or placebo (every 2 weeks; N = 322) with interferon-alfa (9 MIU subcutaneously three times weekly, for a maximum of 52 weeks). Patients were treated until disease progression or unacceptable toxicity. The main outcome measure was investigator-assessed PFS. Secondary outcome measures were ORR and OS.
The median age was 60 years (18 to 82 years); 70% were male and 96% were White. The study population was characterized by Motzer scores as follows: 28% favorable (0), 56% intermediate (1-2), 8% poor (3-5), and 7% missing.
PFS was statistically significantly prolonged among patients receiving bevacizumab compared to placebo; median PFS was 10.2 months vs. 5.4 months [HR 0.60 (95% CI: 0.49, 0.72), p-value < 0.0001, stratified log-rank test]. Among the 595 patients with measurable disease, ORR was also significantly higher (30% vs. 12%, p-value < 0.0001, stratified CMH test). There was no improvement in OS based on the final analysis conducted after 444 deaths, with a median OS of 23 months in the patients receiving bevacizumab with interferon-alfa and 21 months in patients receiving interferon alone [HR 0.86, (95% CI: 0.72, 1.04)]. Results are presented in Figure 4.
Figure 4: Kaplan-Meier Curves for Progression-Free Survival in Metastatic Renal Cell Carcinoma in Study BO17705 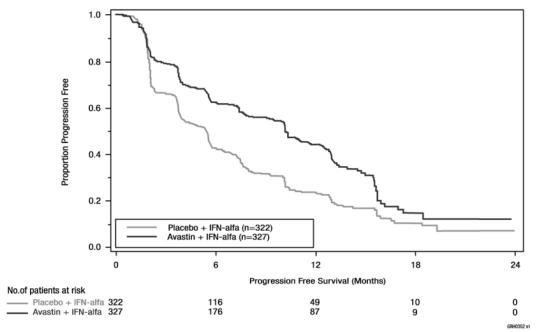
14.6 Persistent, Recurrent, or Metastatic Cervical Cancer
Study GOG-0240
The safety and efficacy of Avastin were evaluated in patients with persistent, recurrent, or metastatic cervical cancer in a randomized, four-arm, multicenter study comparing bevacizumab with chemotherapy versus chemotherapy alone [GOG-0240 (NCT00803062)]. A total of 452 patients were randomized (1:1:1:1) to receive paclitaxel and cisplatin with or without bevacizumab, or paclitaxel and topotecan with or without bevacizumab.
The dosing regimens for bevacizumab, paclitaxel, cisplatin and topotecan were as follows:
- Day 1: Paclitaxel 135 mg/m2 over 24 hours, Day 2: cisplatin 50 mg/m2 with bevacizumab;
- Day 1: Paclitaxel 175 mg/m2 over 3 hours, Day 2: cisplatin 50 mg/m2 with bevacizumab;
- Day 1: Paclitaxel 175 mg/m2 over 3 hours with cisplatin 50 mg/m2 with bevacizumab;
- Day 1: Paclitaxel 175 mg/m2 over 3 hours with bevacizumab, Days 1-3: topotecan 0.75 mg/m2 over 30 minutes.
Patients were treated until disease progression or unacceptable adverse reactions. The main outcome measure was OS. Secondary outcome measures included ORR.
The median age was 48 years (20 to 85 years). Of the 452 patients randomized at baseline, 78% of patients were White, 80% had received prior radiation, 74% had received prior chemotherapy concurrent with radiation, and 32% had a platinum-free interval of less than 6 months. Patients had a GOG performance status of 0 (58%) or 1 (42%). Demographic and disease characteristics were balanced across arms.
Results are presented in Figure 5 and Table 7.
Figure 5: Kaplan-Meier Curves for Overall Survival in Persistent, Recurrent, or Metastatic Cervical Cancer in Study GOG-0240 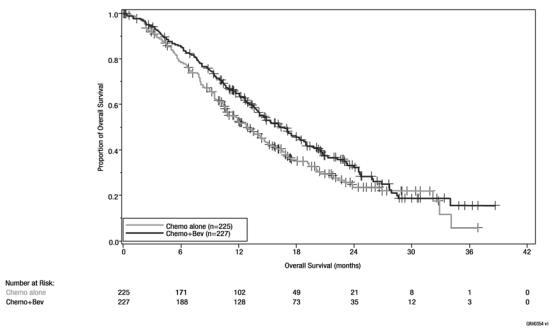
Table 7: Efficacy Results in Study GOG-0240 Efficacy Parameter Bevacizumab with Chemotherapy
(N = 227)Chemotherapy
(N = 225)Overall Survival Median, in monthsa 16.8 12.9 Hazard ratio (95% CI) 0.74 (0.58;0.94) p-valueb 0.0132 a Kaplan-Meier estimates.
b log-rank test (stratified).
The ORR was higher in patients who received bevacizumab with chemotherapy [45% (95% CI: 39, 52)] compared to patients who received chemotherapy alone [34% (95% CI: 28, 40)].
Table 8: Efficacy Results in Study GOG-0240 Efficacy Parameter Topotecan and Paclitaxel with or without
Bevacizumab
(N = 223)Cisplatin and Paclitaxel with or without
Bevacizumab
(N = 229)Overall Survival Median, in monthsa 13.3 15.5 Hazard ratio (95% CI)] 1.15 (0.91, 1.46) p-value 0.23 a Kaplan-Meier estimates.
The HR for OS with bevacizumab with cisplatin and paclitaxel as compared to cisplatin and paclitaxel alone was 0.72 (95% CI: 0.51, 1.02). The HR for OS with bevacizumab with topotecan and paclitaxel as compared to topotecan and paclitaxel alone was 0.76 (95% CI: 0.55, 1.06).
- Day 1: Paclitaxel 135 mg/m2 over 24 hours, Day 2: cisplatin 50 mg/m2 with bevacizumab;
-
16
HOW SUPPLIED/STORAGE AND HANDLING
MVASI (bevacizumab-awwb) injection is a clear to slightly opalescent, colorless to pale yellow, sterile solution for intravenous infusion supplied as single-dose vials in the following strengths: 100 mg/4 mL (NDC 55513-206-01) and 400 mg/16 mL (NDC 55513-207-01). Each carton contains one vial.
Store refrigerated at 2°C to 8ºC (36°F to 46ºF) in the original carton until time of use to protect from light. Do not freeze or shake the vial or carton.
-
17
PATIENT COUNSELING INFORMATION
Gastrointestinal Perforations and Fistulae
Bevacizumab products may increase the risk of developing gastrointestinal perforations and fistulae. Advise patients to immediately contact their health care provider for high fever, rigors, persistent or severe abdominal pain, severe constipation, or vomiting [see Warnings and Precautions (5.1)].
Surgery and Wound Healing Complications
Bevacizumab products can increase the risk of wound healing complications. Advise patients that MVASI should not be used for at least 28 days before or after surgery and until surgical wounds are fully healed [see Warnings and Precautions (5.2)].
Hemorrhage
Bevacizumab products can increase the risk of hemorrhage. Advise patients to immediately contact their health care provider for signs and symptoms of serious or unusual bleeding including coughing or spitting blood [see Warnings and Precautions (5.3)].
Arterial and Venous Thromboembolic Events
Bevacizumab products increase the risk of arterial and venous thromboembolic events. Advise patients to immediately contact their health care provider for signs and symptoms of arterial or venous thromboembolism [see Warnings and Precautions (5.4, 5.5)].
Hypertension
Bevacizumab products can increase blood pressure. Advise patients that they will undergo routine blood pressure monitoring and to contact their healthcare provider if they experience changes in blood pressure [see Warnings and Precautions (5.6)].
Posterior Reversible Leukoencephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES) has been associated with bevacizumab products treatment. Advise patients to immediately contact their health care provider for new onset or worsening neurological function [see Warnings and Precautions (5.7)].
Renal Injury and Proteinuria
Bevacizumab products increase the risk of proteinuria and renal injury, including nephrotic syndrome. Advise patients that treatment with MVASI requires regular monitoring of renal function and to contact their health care provider for proteinuria or signs and symptoms of nephrotic syndrome [see Warnings and Precautions (5.8)].
Infusion-Related Reactions
Bevacizumab products can cause infusion-related reactions. Advise patients to contact their healthcare provider immediately for signs or symptoms of infusion-related reactions [see Warnings and Precautions (5.9)].
Congestive Heart Failure
Bevacizumab products can increase the risk of developing congestive heart failure. Advise patients to contact their healthcare provider immediately for signs and symptoms of CHF [see Warnings and Precautions (5.12)].
Embryo-Fetal Toxicity
Advise female patients that bevacizumab products may cause fetal harm and to inform their healthcare provider with a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with MVASI and for 6 months after the last dose of MVASI [see Use in Specific Populations (8.3)].
Ovarian Failure
Bevacizumab products may lead to ovarian failure. Advise patients of potential options for preservation of ova prior to starting treatment [see Warnings and Precautions (5.11)].
Lactation
Advise women not to breastfeed during treatment with MVASI and for 6 months after the last dose [see Use in Specific Populations (8.2)].
MVASITM (bevacizumab-awwb)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
US License No. 1080Marketed by Amgen Inc.
AMGEN® and MVASITM (bevacizumab-awwb) are trademarks owned or licensed by Amgen Inc., its subsidiaries, or affiliates.
AVASTIN® (bevacizumab) is a registered trademark owned or licensed by Genentech Inc., its subsidiaries, or affiliates.
© 2017-2019 Amgen Inc. All rights reserved.
1xxxxxx- v2
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
100 mg/4 mL
MVASI™
(bevacizumab-awwb)
Injection
NDC: 55513-206-01
Single dose vial
Discard unused portion
100 mg/4 mL (25 mg/mL)
Contains 1 single dose vial
For Intravenous Infusion After Dilution
Store refrigerated at 2°C to 8°C (36°F to 46°F).
Protect from light. Do not Freeze or Shake
Sterile Solution – No Preservative
Rx Only
AMGEN®

-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
400 mg/16 mL
MVASI™
(bevacizumab-awwb)
Injection
NDC: 55513-207-01
Single dose vial
Discard unused portion
400 mg/16 mL (25 mg/mL)
Contains 1 single dose vial
For Intravenous Infusion After Dilution
Store refrigerated at 2°C to 8°C (36°F to 46°F). Protect from light. Do not Freeze or Shake
Sterile Solution – No Preservative
Rx Only
AMGEN®

-
INGREDIENTS AND APPEARANCE
MVASI
bevacizumab-awwb injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-206 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BEVACIZUMAB-AWWB (UNII: 2S9ZZM9Q9V) (BEVACIZUMAB-AWWB - UNII:2S9ZZM9Q9V) BEVACIZUMAB-AWWB 100 mg in 4 mL Inactive Ingredients Ingredient Name Strength TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 240 mg in 4 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 1.6 mg in 4 mL SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 4.8 mg in 4 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 23.2 mg in 4 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-206-01 1 in 1 CARTON 06/01/2018 1 4.0 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761028 06/01/2018 MVASI
bevacizumab-awwb injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-207 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BEVACIZUMAB-AWWB (UNII: 2S9ZZM9Q9V) (BEVACIZUMAB-AWWB - UNII:2S9ZZM9Q9V) BEVACIZUMAB-AWWB 400 mg in 16 mL Inactive Ingredients Ingredient Name Strength TREHALOSE DIHYDRATE (UNII: 7YIN7J07X4) 960 mg in 16 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 6.4 mg in 16 mL SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 19.2 mg in 16 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 92.8 mg in 16 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-207-01 1 in 1 CARTON 06/01/2018 1 16 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761028 06/01/2018 Labeler - Amgen Inc (039976196)
Trademark Results [MVASI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MVASI 87194207 5897547 Live/Registered |
Amgen Inc. 2016-10-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.