KRAZATI- adagrasib tablet, coated
KRAZATI by
Drug Labeling and Warnings
KRAZATI by is a Prescription medication manufactured, distributed, or labeled by Mirati Therapeutics, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KRAZATI safely and effectively. See full prescribing information for KRAZATI.
KRAZATI® (adagrasib) tablets, for oral use
Initial U.S. Approval: 2022INDICATIONS AND USAGE
KRAZATI is an inhibitor of the RAS GTPase family indicated for:
Non-small cell lung cancer (NSCLC)*
- As a single agent, for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic NSCLC, as determined by an FDA-approved test, who have received at least one prior systemic therapy. (1.1)
Colorectal cancer (CRC)*
- In combination with cetuximab, for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic CRC, as determined by an FDA-approved test, who have received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy. (1.2)
*These indications are approved under accelerated approval based on objective response rate (ORR) and duration of response (DOR). Continued approval for these indications may be contingent upon verification and description of a clinical benefit in confirmatory trials. (1.1, 1.2)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Gastrointestinal Adverse Reactions: Monitor patients for diarrhea, nausea and vomiting and provide supportive care as needed. Withhold, reduce the dose or permanently discontinue based on severity. (2.3, 5.1)
- QTc Interval Prolongation: Avoid concomitant use of KRAZATI with other products with a known potential to prolong the QTc interval. Monitor ECG and electrolytes particularly potassium and magnesium, in patients at risk, and in patients taking medications known to prolong the QT interval. Correct electrolyte abnormalities. Withhold, reduce the dose, or permanently discontinue based on severity. (2.3, 5.2)
- Hepatotoxicity: Monitor liver laboratory tests prior to the start of KRAZATI and monthly for 3 months after and as clinically indicated. Reduce the dose, withhold, or permanently discontinue based on severity. (2.3, 5.3)
- Interstitial Lung Disease (ILD) / Pneumonitis: Monitor for new or worsening respiratory symptoms. Withhold KRAZATI for suspected ILD/pneumonitis and permanently discontinue if no other potential causes of ILD/pneumonitis are identified. (2.3, 5.4)
ADVERSE REACTIONS
- Single agent use in NCSLC: The most common adverse reactions (≥ 25%) were nausea, diarrhea, vomiting, fatigue, musculoskeletal pain, hepatotoxicity, renal impairment, edema, dyspnea, and decreased appetite. The most common (≥ 2%) Grade 3 or 4 laboratory abnormalities were decreased lymphocytes, decreased hemoglobin, increased alanine aminotransferase, increased aspartate aminotransferase, hypokalemia, hyponatremia, increased lipase, decreased leukocytes, decreased neutrophils and increased alkaline phosphatase. (6.1)
- In combination with cetuximab in CRC: The most common adverse reactions (≥ 25%) were rash, nausea, diarrhea, vomiting, fatigue, musculoskeletal pain, hepatotoxicity, headache, dry skin, abdominal pain, decreased appetite, edema, anemia, and cough. The most common (≥ 2%) Grade 3 or 4 laboratory abnormalities were decreased lymphocytes, decreased potassium, decreased magnesium, decreased hemoglobin, increased aspartate aminotransferase, increased lipase, decreased albumin, and increased alanine aminotransferase. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See full prescribing information for clinically significant drug interactions with KRAZATI. (7)
- Strong CYP3A4 Inducers: Avoid concomitant use. (7.1)
- Strong CYP3A4 Inhibitors: Avoid concomitant use until adagrasib concentrations have reached steady state. (7.1)
- Sensitive CYP3A4 Substrates: Avoid concomitant use with sensitive CYP3A4 substrates. (7.2)
- Sensitive CYP2C9 or CYP2D6 Substrates or P-gp Substrates: Avoid concomitant use with sensitive CYP2C9 or CYP2D6 substrates or P-gp substrates where minimal concentration changes may lead to serious adverse reactions. (7.2)
- Drugs That Prolong QT Interval: Avoid concomitant use with KRAZATI. (7.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 KRAS G12C-Mutated Locally Advanced or Metastatic Non-Small Cell Lung Cancer
1.2 KRAS G12C-Mutated Locally Advanced or Metastatic Colorectal Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Adverse Reactions
5.2 QTc Interval Prolongation
5.3 Hepatotoxicity
5.4 Interstitial Lung Disease / Pneumonitis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on KRAZATI
7.2 Effects of KRAZATI on Other Drugs
7.3 Drugs That Prolong QTc Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Non-Small Cell Lung Cancer
14.2 Colorectal Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 KRAS G12C-Mutated Locally Advanced or Metastatic Non-Small Cell Lung Cancer
KRAZATI, as a single-agent, is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test [see Dosage and Administration (2.1)], who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on objective response rate (ORR) and duration of response (DOR) [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of a clinical benefit in a confirmatory trial.
1.2 KRAS G12C-Mutated Locally Advanced or Metastatic Colorectal Cancer
KRAZATI in combination with cetuximab is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic colorectal cancer (CRC), as determined by an FDA-approved test [see Dosage and Administration (2.1)], who have received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy.
This indication is approved under accelerated approval based on ORR and DOR [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of a clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Non-Small Cell Lung Cancer
Select patients for treatment of locally advanced or metastatic NSCLC with KRAZATI based on the presence of KRAS G12C mutation in plasma or tumor specimens [see Clinical Studies (14.1)]. If no mutation is detected in a plasma specimen, test tumor tissue.
Colorectal Cancer
Select patients for treatment of locally advanced or metastatic CRC with KRAZATI based on the presence of KRAS G12C mutation in tumor specimens [see Clinical Studies (14.2)].
Information on FDA-approved tests for the detection of a KRAS G12C mutation is available at: https://www.fda.gov/CompanionDiagnostics
2.2 Recommended Dosage
The recommended dosage of KRAZATI as a single agent or in combination with cetuximab is 600 mg orally twice daily until disease progression or unacceptable toxicity.
Refer to the cetuximab prescribing information for cetuximab dosage information [see Clinical Studies (14.2)].
Take KRAZATI at the same time every day with or without food [see Clinical Pharmacology (12.3)]. Swallow tablets whole. Do not chew, crush or split tablets.
If vomiting occurs after taking KRAZATI, do not take an additional dose. Resume dosing at the next scheduled time.
If a dose is inadvertently missed, it should be skipped if greater than 4 hours have elapsed from the expected dosing time. Resume dosing at the next scheduled time.
2.3 Dosage Modifications for Adverse Reactions
Recommended dose reductions for adverse reactions for use of KRAZATI as a single agent or in combination with cetuximab are outlined in Table 1. If adverse reactions occur, a maximum of two dose reductions are permitted. Permanently discontinue KRAZATI in patients who are unable to tolerate 600 mg once daily.
Table 1: Recommended KRAZATI Dosage Reductions for Adverse Reactions Dose Reduction
Dosage
First dose reduction
400 mg twice daily
Second dose reduction
600 mg once daily
Refer to the cetuximab prescribing information for dose modifications for adverse reactions associated with cetuximab.
When KRAZATI is administered in combination with cetuximab, withhold or permanently discontinue cetuximab when KRAZATI is withheld or permanently discontinued.
Treatment with KRAZATI as a single agent may be continued if cetuximab is permanently discontinued. [see Clinical Pharmacology (12.1), Clinical Studies (14.2)].
The recommended dosage modifications for adverse reactions are provided in Table 2.
Table 2: Recommended KRAZATI Dosage Modifications for Adverse Reactions ALT = alanine aminotransferase; AST = aspartate aminotransferase; ILD = Interstitial Lung Disease; ULN = upper limit of normal - * Grading defined by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0.
- † When KRAZATI is administered in combination with cetuximab, withhold or permanently discontinue treatment with cetuximab when withholding or permanently discontinuing treatment with KRAZATI.
Adverse Reaction
Severity*
Dosage Modification†
Nausea or vomiting despite appropriate supportive care (including anti-emetic therapy)
[see Warnings and Precautions (5.1)]Grade 3 or 4
- Withhold KRAZATI until recovery to ≤ Grade 1 or return to baseline.
- Resume KRAZATI at the next lower dose level.
Diarrhea despite appropriate supportive care (including anti-diarrheal therapy)
[see Warnings and Precautions (5.1)]Grade 3 or 4
- Withhold KRAZATI until recovery to ≤ Grade 1 or return to baseline.
- Resume KRAZATI at the next lower dose level.
QTc Interval Prolongation
[see Warnings and Precautions (5.2)]QTc absolute value greater than 500 ms
or
Greater than an increase of 60 ms from baseline- Withhold KRAZATI until QTc interval less than 481 ms or return to baseline.
- Resume KRAZATI at the next lower dose level.
Torsade de pointes, polymorphic ventricular tachycardia or signs or symptoms of serious or life-threatening arrhythmia
- Permanently discontinue KRAZATI
Hepatotoxicity
[see Warnings and Precautions (5.3)]Grade 2
AST or ALT- Decrease KRAZATI to the next lower dose level.
Grade 3 or 4
AST or ALT- Withhold KRAZATI until recovery to ≤ Grade 1 or return to baseline.
- Resume KRAZATI at the next lower dose level.
AST or ALT > 3 × ULN with total bilirubin > 2 × ULN in the absence of alternative causes
- Permanently discontinue KRAZATI
Interstitial Lung Disease / Pneumonitis
[see Warnings and Precautions (5.4)]Any Grade
- Withhold KRAZATI if ILD/pneumonitis is suspected.
- Permanently discontinue KRAZATI if ILD/pneumonitis is confirmed
Other Adverse Reactions
[see Adverse Reactions (6.1)]Grade 3 or 4
- Withhold KRAZATI until ≤ Grade 1 or return to baseline.
- Resume KRAZATI at the next lower dose level.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Gastrointestinal Adverse Reactions
KRAZATI can cause severe gastrointestinal adverse reactions.
In the pooled safety population [see Adverse Reactions (6.1)], who received single-agent KRAZATI, serious gastrointestinal adverse reactions observed were gastrointestinal bleeding in 3.8% including 0.8% Grade 3 or 4, gastrointestinal obstruction in 1.6% including 1.4% Grade 3 or 4, colitis in 0.5% including 0.3% Grade 3, ileus in 0.5%, and stenosis in 0.3%. In addition, nausea, diarrhea, or vomiting occurred in 89% of 366 patients, including 9% Grade 3. Nausea, diarrhea, or vomiting led to dosage interruption or dose reduction in 29% of patients and permanent discontinuation of adagrasib in 0.3%.
In patients who received KRAZATI in combination with cetuximab [see Adverse Reactions (6.1)], serious gastrointestinal adverse reactions included gastrointestinal bleeding in 8.5% including 1.1% Grade 3 or 4, gastrointestinal obstruction in 5.3% including 5.3% Grade 3 or 4, colitis in 1.1% including 1.1% Grade 3 and ileus in 1.1%. In addition, nausea, diarrhea, or vomiting occurred in 92% of 94 patients, including 6% Grade 3. Nausea, diarrhea, or vomiting led to adagrasib dose interruption or dose reduction in 23% of patients.
Monitor and manage patients using supportive care, including antidiarrheals, antiemetics, or fluid replacement, as indicated. Withhold, reduce the dose, or permanently discontinue KRAZATI based on severity [see Dosage and Administration (2.3)].
5.2 QTc Interval Prolongation
KRAZATI can cause QTc interval prolongation, which can increase the risk for ventricular tachyarrhythmias (e.g., torsades de pointes) or sudden death.
In the pooled safety population [see Adverse Reactions (6.1)] who received single-agent KRAZATI, 6% of 366 patients with at least one post-baseline electrocardiogram (ECG) assessment had an average QTc ≥ 501 msec and 11% of patients had an increase from baseline of QTc > 60 msec. KRAZATI causes concentration-dependent increases in the QTc interval [see Clinical Pharmacology (12.2)].
In patients who received KRAZATI in combination with cetuximab [see Adverse Reactions (6.1)], 5% of 93 patients with at least one post-baseline electrocardiogram (ECG) assessment had an average QTc ≥ 501 msec and 16% of patients had an increase from baseline of QTc > 60 msec.
Avoid concomitant use of KRAZATI with other products with a known potential to prolong the QTc interval [see Drug Interactions (7.3) and Clinical Pharmacology (12.2)]. Avoid use of KRAZATI in patients with congenital long QT syndrome and in patients with concurrent QTc prolongation.
Monitor ECGs and electrolytes, particularly potassium and magnesium, prior to starting KRAZATI, during concomitant use, and as clinically indicated in patients with congestive heart failure, bradyarrhythmias, electrolyte abnormalities, and in patients who are unable to avoid concomitant medications that are known to prolong the QT interval. Correct electrolyte abnormalities. Withhold, reduce the dose, or permanently discontinue KRAZATI depending on severity [see Dosage and Administration (2.3)].
5.3 Hepatotoxicity
KRAZATI can cause hepatotoxicity, which may lead to drug-induced liver injury and hepatitis.
In the pooled safety population of 366 patients [see Adverse Reactions (6.1)] who received single-agent KRAZATI, drug-induced liver injury was reported in 0.3% of patients, including 0.3% Grade 3. A total of 32% of patients who received adagrasib had increased alanine aminotransferase (ALT)/increased aspartate aminotransferase (AST); 5% were Grade 3 and 0.5% were Grade 4. The median time to first onset of increased ALT/AST was 3 weeks (range: 0.1 to 48). Overall hepatotoxicity occurred in 37%, and 7% were Grade 3 or 4. Hepatotoxicity leading to dose interruption or reduction occurred in 12% of patients. Adagrasib was discontinued due to hepatotoxicity in 0.5% of patients.
In patients who received KRAZATI in combination with cetuximab [see Adverse Reactions (6.1)], 29% had increased alanine aminotransferase (ALT)/increased aspartate aminotransferase (AST); 5% were Grade 3 and 1.1% were Grade 4. The median time to first onset of increased ALT/AST was 4 weeks (range: 0.1 to 27). Overall hepatotoxicity occurred in 38%, and 10% were Grade 3 or 4. Hepatotoxicity leading to adagrasib dose interruption or reduction occurred in 12% of patients.
Monitor liver laboratory tests (AST, ALT, alkaline phosphatase and total bilirubin) prior to the start of KRAZATI and monthly for 3 months or as clinically indicated, with more frequent testing in patients who develop transaminase elevations. Reduce the dose, withhold, or permanently discontinue KRAZATI based on severity [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.4 Interstitial Lung Disease / Pneumonitis
KRAZATI can cause interstitial lung disease (ILD)/pneumonitis, which can be fatal.
In the pooled safety population [see Adverse Reactions (6.1)] who received single-agent KRAZATI, ILD/pneumonitis occurred in 4.1% of patients, 1.4% were Grade 3 or 4, and one case was fatal. The median time to first onset for ILD/pneumonitis was 12 weeks (range: 5 to 31 weeks). Adagrasib was discontinued due to ILD/pneumonitis in 0.8% of patients.
In patients who received KRAZATI in combination with cetuximab [see Adverse Reactions (6.1)], Grade 1 ILD/pneumonitis occurred in 1.1% of patients. The time to first onset for ILD/pneumonitis was 38 weeks.
Monitor patients for new or worsening respiratory symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever) during treatment with KRAZATI. Withhold KRAZATI in patients with suspected ILD/pneumonitis and permanently discontinue KRAZATI if no other potential causes of ILD/pneumonitis are identified [see Dosage and Administration (2.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.1)]
- QTc Interval Prolongation [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to adagrasib as a single agent at 600 mg orally twice daily in 366 patients with NSCLC and other solid tumors enrolled in KRYSTAL-1 and KRYSTAL-12 (NCT04685135), respectively. Among 366 patients who received adagrasib, 39% of patients were exposed for 6 months or longer and 12% were exposed for greater than one year. In this pooled safety population, the most common (≥ 25%) adverse reactions were nausea (70%), diarrhea (69%), vomiting (57%), fatigue (55%), musculoskeletal pain (38%), hepatotoxicity (37%), renal impairment (33%), edema (30%), dyspnea (26%), and decreased appetite (29%). In this pooled safety population, the most common Grade 3 or 4 (≥ 2%) laboratory abnormalities were decreased lymphocytes (20%), decreased hemoglobin (7%), increased alanine aminotransferase (4.5%), increased aspartate aminotransferase (4.2%), hypokalemia (3.6%), hyponatremia (3.4%), increased lipase (2.5%), decreased leukocytes (2.5%), decreased neutrophils (2.3%), and increased alkaline phosphatase (2.0%).
The data described in WARNINGS AND PRECAUTIONS and below also reflects exposure to adagrasib in combination with cetuximab in 94 patients with KRAS G12C-mutated, locally advanced or metastatic CRC in KRYSTAL-1.
Non-Small Cell Lung Cancer
The safety of adagrasib was evaluated in patients with KRAS G12C-mutated, locally advanced or metastatic NSCLC in KRYSTAL-1 [see Clinical Studies (14.1)]. Patients received adagrasib 600 mg orally twice daily (n = 116). Among patients who received adagrasib, 45% were exposed for 6 months or longer and 4% were exposed for greater than one year.
The median age of patients who received adagrasib was 64 years (range 25 to 89), 56% female, 84% White, 8% Black or African American, and 4.3% Asian.
Serious adverse reactions occurred in 57% of patients who received adagrasib. Serious adverse reactions in ≥ 2% of patients were pneumonia (17%), dyspnea (9%), renal impairment (8%), sepsis (5%), hypoxia (4.3%), pleural effusion (4.3%), respiratory failure (4.3%), anemia (3.4%), cardiac failure (3.4%), hyponatremia (3.4%), hypotension (3.4%), muscular weakness (3.4%), pyrexia (3.4%), dehydration (2.6%), diarrhea (2.6%), mental status changes (2.6%), pulmonary embolism (2.6%), and pulmonary hemorrhage (2.6%). Fatal adverse reactions occurred in 11% of patients who received adagrasib due to pneumonia (3.4%), respiratory failure (1.7%), sudden death (1.7%), cardiac failure (0.9%), cerebrovascular accident (0.9%), mental status change (0.9%), pulmonary embolism (0.9%), and pulmonary hemorrhage (0.9%).
Permanent discontinuation of adagrasib due to an adverse reaction occurred in 13% of patients. Adverse reactions which resulted in permanent discontinuation of adagrasib occurring in two patients each (1.7%) were pneumonia and pneumonitis and occurring in one patient each (0.9%) were cerebrovascular accident, dyspnea, decreased ejection fraction, encephalitis, gastrointestinal obstruction, hemorrhage, hepatotoxicity, hypotension, muscular weakness, pulmonary embolism, pyrexia, respiratory failure and sepsis.
Dose interruptions of adagrasib due to an adverse reaction occurred in 77% of patients. Adverse reactions requiring dosage interruption in ≥ 2% of patients who received adagrasib included nausea, hepatotoxicity, fatigue, vomiting, pneumonia, renal impairment, diarrhea, QTc interval prolongation, anemia, dyspnea, increased lipase, decreased appetite, dizziness, hyponatremia, muscular weakness, increased amylase, pneumonitis, sepsis and decreased weight.
Dose reductions of adagrasib due to an adverse reaction occurred in 28% of patients. Adverse reactions which required dose reductions in ≥ 2% of patients who received adagrasib included hepatotoxicity, fatigue, nausea, diarrhea, vomiting, and renal impairment.
The most common adverse reactions (≥ 20%) were diarrhea, nausea, fatigue, vomiting, musculoskeletal pain, hepatotoxicity, renal impairment, dyspnea, edema, decreased appetite, cough, pneumonia, dizziness, constipation, abdominal pain, and QTc interval prolongation. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, increased aspartate aminotransferase, decreased sodium, decreased hemoglobin, increased creatinine, decreased albumin, increased alanine aminotransferase, increased lipase, decreased platelets, decreased magnesium, and decreased potassium.
Table 3 summarizes the adverse reactions in KRYSTAL-1.
Table 3: Adverse Reactions (≥ 20%) in Patients with KRAS G12C-mutated NSCLC Who Received Adagrasib in KRYSTAL-1 - * Grouped term.
- † Hepatotoxicity includes mixed liver injury, blood alkaline phosphatase increased, alanine aminotransferase increased, aspartate aminotransferase increased, liver function test increased, blood bilirubin increased, and bilirubin conjugated increased.
- ‡ Renal impairment includes acute kidney injury and increased blood creatinine.
Adverse Reaction
Adagrasib
N = 116All Grades
(%)Grade 3 or 4
(%)Gastrointestinal Disorders
Diarrhea*
70
0.9
Nausea
69
4.3
Vomiting*
56
0.9
Constipation
22
0
Abdominal pain*
21
0
General Disorders and Administration Site Conditions
Fatigue*
59
7
Edema*
32
0
Musculoskeletal and Connective Tissue Disorders
Musculoskeletal pain*
41
7
Hepatobiliary Disorders
37
10
Renal and Urinary Disorders
36
6
Respiratory
Dyspnea*
35
10
Cough*
24
0.9
Metabolism and Nutrition Disorders
Decreased appetite
30
4.3
Infections and Infestations
Pneumonia*
24
17
Nervous System Disorders
Dizziness*
23
0.9
Cardiac Disorders
Electrocardiogram QT prolonged
20
6
Table 4 summarizes the laboratory abnormalities in KRYSTAL-1.
Table 4: Select Laboratory Abnormalities Occurring (≥ 25%) That Worsened from Baseline in Patients with KRAS G12C-mutated NSCLC Who Received Adagrasib in KRYSTAL-1 - * Denominator used to calculate the rate varied from 106 to 113 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality
Adagrasib*
All Grades
(%)Grade 3 or 4
(%)Hematology
Lymphocytes decreased
64
25
Hemoglobin decreased
51
8
Platelets decreased
27
0
Chemistry
Aspartate aminotransferase increased
52
6
Sodium decreased
52
8
Creatinine increased
50
0
Albumin decreased
50
0.9
Alanine aminotransferase increased
46
5
Lipase increased
35
1.8
Magnesium decreased
26
0
Potassium decreased
26
3.5
Colorectal Cancer
The safety of adagrasib combined with cetuximab was evaluated in 94 patients with KRAS G12C-mutated, locally advanced or metastatic CRC in KRYSTAL-1 [see Clinical Studies (14.2)]. Patients started treatment with adagrasib 600 mg twice daily in combination with cetuximab weekly (n = 17) or every two weeks (n = 77). Among patients who received adagrasib in combination with cetuximab, 60% were exposed for greater than 6 months and 12% were exposed for greater than 12 months.
Serious adverse reactions occurred in 30% of patients who received adagrasib in combination with cetuximab. The most common serious adverse reactions (≥ 2%) were pneumonia (4.3%), pleural effusion, pyrexia, acute kidney injury, dehydration, and small intestinal obstruction (2.1% each).
A fatal adverse reaction of pneumonia occurred in 1 patient who received adagrasib in combination with cetuximab.
Adverse reactions leading to discontinuation of adagrasib occurred in 2 patients. Adverse reactions which resulted in permanent discontinuation of adagrasib (1 patient each) included abdominal pain and prolonged QT interval.
Adverse reactions leading to dose interruptions of adagrasib occurred in 62% of patients. The most common adverse reactions or laboratory abnormalities leading to dose interruption in ≥ 2% of patients who received adagrasib included diarrhea, nausea, vomiting, abdominal pain, dizziness, headache, pneumonia, alanine aminotransferase increased, aspartate aminotransferase increased, dyspnea, fatigue, pleural effusion, rash, anemia, electrocardiogram QT prolongation, blood bilirubin increased, blood creatinine increased, decreased appetite, dehydration, hemorrhage, hypomagnesemia, lipase increased, muscular weakness, musculoskeletal pain, and pyrexia.
Adverse reactions leading to dose reductions of adagrasib occurred in 35% of patients. The most common adverse reactions or laboratory abnormalities leading to dose reductions in ≥ 2% of patients who received adagrasib included fatigue, increased aspartate aminotransferase, increased alanine aminotransferase, nausea, decreased appetite, electrocardiogram QT prolongation, dizziness, acute kidney injury, diarrhea, dysarthria, and vomiting.
The most common adverse reactions (≥ 20%) were rash, nausea, diarrhea, vomiting, fatigue, musculoskeletal pain, hepatotoxicity, headache, dry skin, abdominal pain, decreased appetite, edema, anemia, dizziness, cough, constipation, and peripheral neuropathy.
The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, decreased leukocytes, increased alanine aminotransferase, decreased magnesium, decreased albumin, increased lipase, decreased potassium, increased aspartate aminotransferase, increased creatinine, decreased sodium, decreased calcium, increased amylase, and increased alkaline phosphatase.
Table 5 summarizes the adverse reactions in patients with metastatic CRC in KRYSTAL-1.
Table 5: Adverse Reactions (≥ 20 %) in Patients with KRAS G12C-mutated CRC Received Adagrasib in Combination with Cetuximab in KRYSTAL-1 - * Graded per CTCAE version 5.0.
- † Grouped term; includes multiple related terms.
Adverse Reaction*
Adagrasib in Combination with Cetuximab
N = 94All Grades
(%)Grade 3 or 4
(%)Skin and subcutaneous tissue disorders
Rash†
84
4.3
Dry skin
36
0
Gastrointestinal Disorders
Nausea
68
2.1
Diarrhea†
65
5
Vomiting†
57
0
Abdominal pain†
30
4.3
Constipation
23
0
General Disorders and Administration Site Conditions
Fatigue†
57
3.2
Musculoskeletal pain†
47
4.3
Edema†
28
0
Hepatobiliary Disorders
Hepatotoxicity†
38
10
Nervous System Disorders
Headache
37
4.3
Dizziness†
24
2.1
Peripheral neuropathy†
20
1.1
Metabolism and Nutrition Disorders
Decreased appetite
30
0
Blood and lymphatic system disorders
Anemia
27
7
Respiratory
Cough†
25
0
Other clinically relevant adverse reactions observed in less than 20% of patients were infusion related reactions (15%).
Table 6 summarizes the laboratory abnormalities in patients with metastatic CRC in KRYSTAL-1.
Table 6: Selected Laboratory Abnormalities (≥ 25%) in Patients Who Received Adagrasib in Combination with Cetuximab in KRYSTAL-1 - * The denominator used to calculate the rate varied from 82 to 92 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality
Adagrasib in Combination with Cetuximab*
All Grades
(%)Grade 3 or 4
(%)Hematology
Lymphocytes decreased
63
17
Hemoglobin decreased
48
5
Leukocytes decreased
27
1.1
Chemistry
Alanine aminotransferase increased
51
2.2
Magnesium decreased
49
7
Albumin decreased
46
2.2
Lipase increased
41
3.3
Potassium decreased
40
9
Aspartate aminotransferase increased
39
4.3
Creatinine increased
30
1.1
Sodium decreased
30
0
Calcium decreased
29
1.1
Amylase increased
29
0
Alkaline phosphatase increased
29
1.1
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on KRAZATI
Strong CYP3A4 Inducers
Avoid concomitant use of KRAZATI with strong CYP3A inducers.
Adagrasib is a CYP3A4 substrate. Concomitant use of KRAZATI with a strong CYP3A inducer reduces adagrasib exposure [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of KRAZATI.
Strong CYP3A4 Inhibitors
Avoid concomitant use of KRAZATI with strong CYP3A inhibitors until adagrasib concentrations have reached steady state (after approximately 8 days).
Adagrasib is a CYP3A4 substrate. If adagrasib concentrations have not reached steady state, concomitant use of a strong CYP3A inhibitor will increase adagrasib concentrations, [see Clinical Pharmacology (12.3)], which may increase the risk of KRAZATI adverse reactions.
7.2 Effects of KRAZATI on Other Drugs
Sensitive CYP3A Substrates
Avoid concomitant use of KRAZATI with sensitive CYP3A substrates unless otherwise recommended in the Prescribing Information for these substrates.
Adagrasib is a CYP3A inhibitor. Concomitant use with KRAZATI increases exposure of CYP3A substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
Sensitive CYP2C9 Substrates
Avoid concomitant use of KRAZATI with sensitive CYP2C9 substrates where minimal concentration changes may lead to serious adverse reactions unless otherwise recommended in the Prescribing Information for these substrates.
Adagrasib is a CYP2C9 inhibitor. Concomitant use with KRAZATI increases exposure of CYP2C9 substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
Sensitive CYP2D6 Substrates
Avoid concomitant use of KRAZATI with sensitive CYP2D6 substrates where minimal concentration changes may lead to serious adverse reactions unless otherwise recommended in the Prescribing Information for these substrates.
Adagrasib is a CYP2D6 inhibitor. Concomitant use with KRAZATI increases exposure of CYP2D6 substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
P-gp Substrates
Avoid concomitant use of KRAZATI with P-gp substrates where minimal concentration changes may lead to serious adverse reactions unless otherwise recommended in the Prescribing Information for these substrates.
Adagrasib is a P-gp inhibitor. Concomitant use with KRAZATI increases exposure of P-gp substrates [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions related to these substrates.
7.3 Drugs That Prolong QTc Interval
Avoid concomitant use of KRAZATI with other product(s) with a known potential to prolong the QTc interval. If concomitant use cannot be avoided, monitor electrocardiogram and electrolytes prior to starting KRAZATI, during concomitant use, and as clinically indicated [see Warnings and Precautions (5.2)]. Withhold KRAZATI if the QTc interval is > 500 ms or the change from baseline is > 60 ms [see Dosage and Administration (2.3)].
Adagrasib causes QTc interval prolongation [see Clinical Pharmacology (12.2)]. Concomitant use of KRAZATI with other products that prolong the QTc interval may result in a greater increase in the QTc interval and adverse reactions associated with QTc interval prolongation, including Torsade de pointes, other serious arrythmias, and sudden death [see Warnings and Precautions (5.2)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of KRAZATI in pregnant women. In animal reproduction studies, oral administration of adagrasib to pregnant rats and rabbits during the period of organogenesis did not cause adverse development effects or embryo-fetal lethality at exposures below the human exposure at the recommended dose of 600 mg twice daily (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In a rat embryo-fetal development study, once daily oral administration of adagrasib to pregnant rats during the period of organogenesis resulted in maternal toxicity (reduced body weight and food intake, and adverse clinical signs leading to moribund condition and early termination) and lower fetal body weight at 270 mg/kg dose level (approximately 2 times the recommended dose of 600 mg twice daily based on body surface area [BSA]). Adagrasib induced skeletal malformations, such as bent limbs, and skeletal variations, such as bent scapula, wavy ribs, and supernumerary short cervical ribs at 270 mg/kg, which were secondary to maternal toxicity and reduced fetal body weight.
In a rabbit embryo-fetal development study, once daily oral administration of adagrasib during the period of organogenesis resulted in lower fetal body weight and increased litter frequency of unossified sternebra at 30 mg/kg (approximately 0.11 times the human exposure based on area under the curve [AUC] at the clinical dose of 600 mg twice daily). This skeletal variation was associated with maternal toxicities, including reduced mean body weight and decreased food consumption. Adagrasib exposure did not cause adverse developmental effects and did not affect embryo-fetal survival in rabbits at doses up to 30 mg/kg once daily.
8.2 Lactation
Risk Summary
There are no data on the presence of adagrasib or its metabolites in human milk, the effects on the breastfed child, or on milk production. Because of the potential for serious adverse reactions in breastfed children, advise women not to breastfeed during treatment with KRAZATI and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Infertility
Based on findings from animal studies, KRAZATI may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of KRAZATI has not been established in pediatric patients.
8.5 Geriatric Use
Of 116 patients with metastatic NSCLC who received adagrasib 600 mg orally twice daily in KRYSTAL-1, 49% (57 patients) were ≥ 65 years of age and 13% (15 patients) were ≥ 75 years of age. No overall differences in safety or effectiveness were observed between older and younger patients.
Of 94 patients with metastatic CRC who received adagrasib 600 mg orally twice daily in combination with cetuximab in KRISTAL-1, 33% (31 patients) were ≥ 65 years of age and 2.1% (2 patients) were ≥ 75 years of age. No overall differences in safety or effectiveness were observed between older and younger patients.
-
11 DESCRIPTION
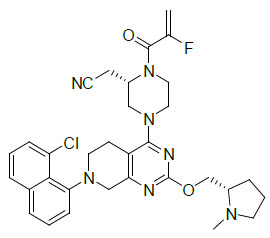
Adagrasib is an irreversible inhibitor of KRAS G12C and belongs to the RAS GTPase family. The molecular formula is C32H35ClFN7O2 and the molecular weight is 604.1 g/mol. The chemical name is {(2S)-4-[7-(8-chloronaphthalen-1-yl)-2-{[(2S)-1-methylpyrrolidin-2-yl]- methoxy}-5,6,7,8-tetrahydropyrido[3,4-d]pyrimidin-4-yl]-1-(2-fluoroprop-2-enoyl)piperazin-2-yl}acetonitrile. Adagrasib has the following chemical structure:
Adagrasib is a crystalline solid. The solubility of adagrasib in the aqueous media decreases over the range pH 1.2 to 7.4 from > 262 mg/mL to < 0.010 mg/mL.
KRAZATI (adagrasib) tablets for oral administration contain 200 mg of adagrasib. The following are inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate (vegetable sourced), mannitol, and microcrystalline cellulose. The tablet film coating contains hypromellose, maltodextrin, medium chain triglycerides (vegetable sourced), polydextrose, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Adagrasib is an irreversible inhibitor of KRAS G12C that covalently binds to the mutant cysteine in KRAS G12C and locks the mutant KRAS protein in its inactive state that prevents downstream signaling without affecting wild-type KRAS protein. Adagrasib inhibited tumor cell growth and viability in cells harboring KRAS G12C mutations and resulted in tumor regression in KRAS G12C-mutated tumor xenograft models with minimal off-target activity. Adagrasib in combination with cetuximab had increased antitumor activity in some cell line-derived and patient-derived KRAS G12C-mutant CRC tumor xenograft models compared to adagrasib or cetuximab alone.
12.2 Pharmacodynamics
Adagrasib exposure-response relationships and the time course of pharmacodynamic response are unknown.
Cardiac Electrophysiology
Adagrasib increased QTc in a concentration-dependent manner. Based on the concentration-QTcF relationship, the mean (90% CI) QTcF change from baseline (ΔQTcF) was 18 (15, 21) ms at the mean steady-state maximum concentration (Cmax,ss) in patients after administration of adagrasib 600 mg twice daily [see Warnings and Precautions (5.2)].
12.3 Pharmacokinetics
The pharmacokinetics of adagrasib were studied in healthy subjects and in patients with KRAS G12C-mutated NSCLC or CRC. Adagrasib pharmacokinetic data are presented as mean (percent coefficient of variation) unless otherwise specified.
Adagrasib AUC and Cmax increase dose proportionally over the dose range of 400 mg to 600 mg (0.67 to 1 times the approved recommended dose). Adagrasib steady-state was reached within 8 days following administration of the approved recommended dosage and accumulation was approximately 6-fold.
Distribution
The apparent volume of distribution of adagrasib is 942 L (57%). Human plasma protein binding of adagrasib is approximately 98% in vitro.
Elimination
The adagrasib terminal elimination half-life is 23 hours (16%) and the apparent oral clearance (CL/F) is 37 L/h (54%) in patients.
Specific Populations
No clinically significant differences in the pharmacokinetics of adagrasib based on age (19 to 89 years), sex, race (White, Black or African American, or Asian), body weight (36 to 146 kg), ECOG PS (0, 1), tumor type (NSCLC or CRC), or tumor burden. No clinically significant differences in the pharmacokinetics of adagrasib are expected in patients with mild to severe renal impairment (CLcr 15 to < 90 mL/min estimated by Cockcroft-Gault equation) or in patients with mild to severe hepatic impairment (Child-Pugh classes A to C).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
The following table describes the effect of other drugs on the pharmacokinetics of adagrasib.
Table 7: Effect of Other Drugs on Adagrasib Cmax = maximum plasma concentration; AUC = area under the plasma concentration-time curve - * Predicted changes in Cmax or AUC of adagrasib.
Concomitant Drug
Adagrasib Dosage
Changes in Cmax or AUC of Adagrasib
Cmax
% DecreaseAUC
% DecreaseRifampin
(a strong CYP3A inducer)600 mg single dose
88%
95%
600 mg multiple doses
> 61%*
> 66%*
Strong CYP3A Inhibitors: Adagrasib Cmax increased by 2.4-fold and AUC increased by 4-fold following concomitant use of a single dose of 200 mg (0.33 times the approved recommended dose) with itraconazole (a strong CYP3A inhibitor). No clinically significant differences in the pharmacokinetics of adagrasib at steady state were predicted when used concomitantly with itraconazole.
No clinically significant differences in the pharmacokinetics of adagrasib were predicted or observed when used concomitantly with efavirenz (a moderate CYP3A inducer), pantoprazole (a proton pump inhibitor), rosuvastatin (a BCRP/OATP substrate), or eltrombopag (a BCRP inhibitor).
The following table describes the effect of adagrasib on the pharmacokinetics of other drugs.
Table 8: Effect of Adagrasib on Other Drugs Cmax = maximum plasma concentration; AUC = area under the plasma concentration-time curve - * 0.66 times the approved recommended dosage.
- † Predicted changes in Cmax or AUC of concomitant drug.
Concomitant Drug
Adagrasib Dosage
Fold Increase of Concomitant Drug
Cmax
AUC
Midazolam
(a sensitive CYP3A substrate)400 mg* twice daily
4.8-fold
21-fold
600 mg twice daily
3.1-fold†
31-fold†
Warfarin
(a sensitive CYP2C9 substrate)600 mg twice daily
1.1-fold†
2.9-fold†
Dextromethorphan
(a sensitive CYP2D6 substrate)400 mg* twice daily
1.9-fold
1.8-fold
600 mg twice daily
1.7-fold†
2.4-fold†
Digoxin
(a P-gp substrate)600 mg twice daily
1.9-fold†
1.5-fold†
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with adagrasib.
Adagrasib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay and was not genotoxic in an in vitro chromosomal aberration assay or an in vivo micronucleus assay in rats.
Fertility studies were not conducted with adagrasib. In toxicology studies of up to 13-weeks in duration in rats, oral administration of adagrasib induced phospholipidosis which increased vacuolation in female reproductive organs, including vacuolation in ovaries (corpora lutea, macrophage or interstitial cells) and uterus (glandular epithelium), and atrophy with mucification of the vaginal mucosa at doses ≥ 150 mg/kg (approximately equal to or greater than the human exposure at the recommended dose based on area under the curve [AUC]). These findings reversed after cessation of dosing in the 28-day study but in the 13-week study, pigmented macrophage aggregates were observed in the ovaries of female rats after the recovery period. In a 28-day repeat-dose toxicology study, oral administration of adagrasib to male rats induced atrophy and epithelial vacuolation of the prostate gland and seminal vesicles at 300 mg/kg (approximately 1.6 times the human exposure at the recommended dose based on AUC). These findings resolved after cessation of treatment.
13.2 Animal Toxicology and/or Pharmacology
Phospholipidosis (vacuolation and/or presence of foamy macrophages) was observed in multiple organs (e.g., lung, trachea, heart, skeletal, ovaries, uterus, adrenal gland, kidney, liver, lymph nodes, spleen, thymus, and thyroid in rats; and heart and lung in dogs) after repeated oral administration of adagrasib in rats and dogs. In toxicology studies of up to 13-week duration in rats, phospholipidosis was observed at doses ≥ 150 mg/kg (approximately ≥ 2 times the human exposure at the recommended dose based on AUC). In a dog 28-day toxicity study, this effect was observed at 25 mg/kg (approximately equal to the human exposure at the recommended dose based on AUC). The extent of vacuolization and the presence of foamy macrophages were more prominent in the rat compared to dogs, and evidence of reversibility after cessation of treatment was noted for most organs. The significance of this finding in humans in unknown.
-
14 CLINICAL STUDIES
14.1 Non-Small Cell Lung Cancer
The efficacy of adagrasib was evaluated in KRYSTAL-1 (NCT03785249), a multicenter, single-arm, open-label expansion cohort study. Eligible patients were required to have locally advanced or metastatic KRAS G12C-mutated NSCLC who previously received treatment with a platinum-based regimen and an immune checkpoint inhibitor, an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, and at least one measurable lesion as defined by Response Evaluation criteria in Solid Tumors (RECIST v1.1). Identification of a KRAS G12C mutation was prospectively determined by local testing using tissue specimens. Patients received adagrasib 600 mg orally twice daily until unacceptable toxicity or disease progression. Tumor assessments were performed every 6 weeks. The major efficacy outcome measures were confirmed objective response rate (ORR) and duration of response (DOR) as evaluated by blinded independent central review (BICR) according to RECIST v1.1.
In the efficacy population, KRAS G12C mutation status was determined by prospective local testing using tumor tissue specimens. Of the 112 patients with KRAS G12C mutation, tissue samples from 88% (98/112) patients were tested retrospectively using the QIAGEN therascreen KRAS RGQ PCR Kit. While 89% (87/98) of patients were positive for KRAS G12C mutation, 11% (11/98) did not have a KRAS G12C mutation identified. In addition, plasma samples from 63% (71/112) patients were tested retrospectively using Agilent Resolution ctDx FIRST assay. While 66% (47/71) of patients were positive for KRAS G12C mutation, 34% (24/71) did not have a KRAS G12C mutation identified.
A total of 112 patients had at least one measurable lesion at baseline as assessed by BICR according to RECIST v1.1.
The baseline demographic and disease characteristics in the efficacy population were: median age 64 years (range: 25 to 89), 55% female, 83% White, 8% were Black or African American, 4% Asian, 4% race not reported, 0.9% American Indian or Alaska Native, 16% Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 and 83% ECOG PS 1. Tumor histology was 97% adenocarcinoma and 89% of patients had metastatic disease. Patients received a median of 2 prior systemic therapies (range 1 to 7); 43% received 1 prior line, 35% received 2 prior lines, 10% received 3 prior lines and 12% received 4 or more prior lines, 98% received both prior platinum and prior anti-PD-1/PD-L1 therapy. Sites of extra-thoracic disease included bone 42%, brain 30%, adrenals 21%, and liver 21%.
Efficacy results are summarized in Table 9.
Table 9: Efficacy Results for KRYSTAL-1 CI = Confidence Interval - * Assessed by BICR.
- † Estimate using Kaplan-Meier method.
- ‡ Observed proportion of patients with duration of response beyond landmark time.
Efficacy Parameter
Adagrasib
(n = 112)Objective Response Rate (95% CI)*
43 (34, 53)
Complete response rate, %
0.9
Partial response rate, %
42
Duration of Response*
Median† in months (95% CI)
8.5 (6.2, 13.8)
Patients with duration ≥ 6 months‡, %
58
14.2 Colorectal Cancer
The efficacy of adagrasib in combination with cetuximab was evaluated in KRYSTAL-1, a multicenter, single-arm, open-label expansion cohort study. Eligible patients were required to have locally advanced or metastatic KRAS G12C-mutated CRC and to have previously received therapy with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, a VEGF inhibitor if eligible, and an ECOG PS of 0 or 1.
Patients initiated treatment with adagrasib 600 mg orally twice daily in combination with cetuximab administered either biweekly (77 patients with 500 mg/m2 every two weeks) or weekly (17 patients with 400 mg/m2 initial dose followed by 250 mg/m2 weekly). Treatment continued until unacceptable toxicity or disease progression. Tumor assessments were performed every 6 weeks. Adagrasib discontinuation required cetuximab discontinuation, however patients could continue to receive adagrasib if cetuximab was discontinued [see Dosage and Administration (2.3)]. Six patients continued with adagrasib single agent therapy after discontinuing cetuximab. The length of time these 6 patients received adagrasib alone ranged from 43 days to 3 years. Patient treatment with adagrasib after disease progression continued if a patient was clinically stable and considered to be deriving clinical benefit by the investigator.
The major efficacy outcome measures were confirmed ORR and DOR according to RECIST v1.1 as assessed by BICR.
In the efficacy population, KRAS G12C mutation status was determined by prospective local testing using tumor tissue specimens. Of the 94 patients with KRAS G12C mutation, tissue samples from 79% (74/94) patients were tested retrospectively using the QIAGEN therascreen KRAS RGQ PCR Kit. Of the 74 tissue samples submitted, 81% (60/74) yielded a result with 93% (56/60) positive for KRAS G12C and 7% (4/60) without a KRAS G12C mutation identified.
The baseline demographic and disease characteristics in the efficacy population were: median age 57 years (range: 24 to 75 years), 53% female, 71% White, 14% were Black or African American, 5% Asian, 1.1% American Indian or Alaska Native, 9% reported as other; 51% ECOG PS 0 and 49% ECOG PS 1. Tumor histology was 100% adenocarcinoma and 99% of patients had metastatic disease. Patients received a median of 3 prior systemic therapies (range 1 to 9); 9% received 1 prior line, 36% received 2 prior lines, 31% received 3 prior lines and 25% received 4 or more prior lines. Sites of metastatic disease included lung (71%), liver (64%) and bone (14%).
Efficacy results are summarized in Table 10.
Table 10: Efficacy Results for KRYSTAL-1 CI = Confidence Interval - * Assessed by BICR.
- † Estimate using Kaplan-Meier method.
- ‡ Observed proportion of patients with duration of response beyond landmark time.
Efficacy Parameter
Adagrasib
(n = 94)Objective Response Rate (95% CI)*
34 (25, 45)
Complete response rate, %
0
Partial response rate, %
34.0
Duration of Response*
Median† in months (95% CI)
5.8 (4.2, 7.6)
Patients with duration ≥ 6 months‡, %
31
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
KRAZATI (adagrasib) tablets, 200 mg, oval shaped, white to off-white, immediate release, film coated tablets with "200" on one side and stylized "M" on the other side.
KRAZATI (adagrasib) tablets are packaged in high-density polyethylene, white opaque, square bottles with desiccant and polypropylene, white, child resistant closures with a tamper-proof heat induction seal.
- NDC: 80739-812-12: 200 mg, bottle containing 120 tablets.
- NDC: 80739-812-18: 200 mg, bottle containing 180 tablets.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Gastrointestinal Adverse Reactions
Advise patients that KRAZATI can cause severe gastrointestinal adverse reactions and to contact their healthcare provider for signs or symptoms of severe or persistent gastrointestinal adverse reactions [see Warnings and Precautions (5.1)].
QTc Interval Prolongation
Advise patients that KRAZATI can cause QTc interval prolongation and to contact their healthcare provider for signs or symptoms of arrhythmias [see Warnings and Precautions (5.2)].
Hepatotoxicity
Advise patients that KRAZATI can cause hepatotoxicity and to immediately contact their healthcare provider for signs or symptoms of liver dysfunction [see Warnings and Precautions (5.3)].
Interstitial Lung Disease (ILD)/Pneumonitis
Advise patients that KRAZATI can cause ILD / pneumonitis and to contact their healthcare provider immediately for new or worsening respiratory symptoms [see Warnings and Precautions (5.4)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7.1)].
Missed Dose
If a dose of KRAZATI is missed by greater than 4 hours, resume dosing at the next scheduled time [see Dosage and Administration (2.2)].
Lactation
Advise women not to breastfeed during treatment with KRAZATI and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Inform patients that KRAZATI may cause infertility [see Use in Specific Populations (8.3)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
KRAZATI® (krah zah tee)
(adagrasib)
TabletsWhat is KRAZATI?
KRAZATI is a prescription medicine used in adults:- o
alone to treat non-small cell lung cancer (NSCLC)
- o that has spread to other parts of the body or cannot be removed by surgery, and
- o whose tumor has an abnormal KRAS G12C gene, and
- o who have received at least one prior treatment.
- o
in combination with a medicine called cetuximab to treat colon or rectal cancer (CRC)
- o that has spread to other parts of the body or cannot be removed by surgery, and
- o whose tumor has an abnormal KRAS G12C gene, and
- o who have previously received certain chemotherapy medicines.
Your healthcare provider will perform a test to make sure that KRAZATI is right for you.
It is not known if KRAZATI is safe and effective in children.Before taking KRAZATI, tell your healthcare provider about all of your medical conditions, including if you:
- o have any heart problems, including heart failure and congenital long QT syndrome.
- o have liver problems.
- o are pregnant or plan to become pregnant. It is not known if KRAZATI can harm your unborn baby.
- o are breastfeeding or plan to breastfeed. It is not known if KRAZATI passes into your breastmilk. Do not breastfeed during treatment and for 1 week after your last dose of KRAZATI.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. KRAZATI can affect the way other medicines work, and other medicines can affect how KRAZATI works.
How should I take KRAZATI?
- o Take KRAZATI exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking KRAZATI unless your healthcare provider tells you to.
- o Your healthcare provider may change your dose, or temporarily or permanently stop treatment with KRAZATI if you develop certain side effects.
- o For colon or rectal cancer, you will also receive cetuximab through a vein in your arm (intravenously) given by your healthcare provider. Your healthcare provider will permanently or temporarily stop your treatment with cetuximab if your treatment with KRAZATI is permanently or temporarily stopped.
- o Take your prescribed dose of KRAZATI 2 times each day, at about the same time each day.
- o Take KRAZATI either with food or without food.
- o Swallow KRAZATI tablets whole. Do not chew, crush or split tablets.
- o If you vomit after taking a dose of KRAZATI, do not take an extra dose. Take your next dose at your next scheduled time.
- o If you miss a dose of KRAZATI, take the dose as soon as you remember. If it has been more than 4 hours, do not take the dose. Take your next dose of KRAZATI at your next scheduled time. Do not take 2 doses at the same time to make up for a missed dose.
What are possible side effects of KRAZATI?
KRAZATI can cause serious side effects, including:- o
Stomach and intestinal (gastrointestinal) problems. Stomach and intestinal side effects including nausea, diarrhea, or vomiting, are common with KRAZATI but can also sometimes be severe. KRAZATI can also cause serious stomach and intestinal side effects such as bleeding, obstruction, inflammation of the colon (colitis), and narrowing (stenosis).
- o Call your healthcare provider if you develop any of the signs or symptoms of stomach or intestinal problems listed above during treatment with KRAZATI.
- o Your healthcare provider may prescribe an antidiarrheal medicine or anti-nausea medicine, or other treatment, as needed.
- o
Changes in the electrical activity of your heart called QTc prolongation. Certain changes can occur in the electrical activity of your heart during treatment with KRAZATI and can be seen on a test called an electrocardiogram (ECG or EKG). QTc prolongation can increase your risk for irregular heartbeats that can be life-threatening, such as torsades de pointes, and can lead to sudden death.
- o
You should not take KRAZATI if you have congenital long QT syndrome or if you currently have QTc prolongation. See "Before taking KRAZATI, tell your healthcare provider about all of your medical conditions, including if you:"
- ▪ Your healthcare provider should monitor the electrical activity of your heart and the levels of body salts in your blood (electrolytes) especially potassium and magnesium before starting and during treatment with KRAZATI if you have heart failure, a slow heart rate, abnormal levels of electrolytes in your blood, or if you take a medicine that can prolong the QT interval of your heartbeat.
- ▪ Tell your healthcare provider if you feel dizzy, lightheaded, or faint, or if you get abnormal heartbeats during treatment with KRAZATI.
- o
You should not take KRAZATI if you have congenital long QT syndrome or if you currently have QTc prolongation. See "Before taking KRAZATI, tell your healthcare provider about all of your medical conditions, including if you:"
- o Liver problems. Abnormal liver blood test results are common with KRAZATI and can sometimes be severe. Your healthcare provider should do blood tests before starting and during treatment with KRAZATI to check your liver function. Tell your healthcare provider right away if you develop any signs or symptoms of liver problems, including:
- o your skin or the white part of your eyes turns yellow (jaundice)
- o dark or "tea-colored" urine
- o light-colored stools (bowel movements)
- o tiredness or weakness
- o nausea or vomiting
- o bleeding or bruising
- o loss of appetite
- o pain, aching or tenderness on the right side of your stomach area (abdomen)
- o Lung or breathing problems. KRAZATI may cause inflammation of the lungs that can lead to death. Tell your healthcare provider or get emergency medical help right away if you have new or worsening shortness of breath, cough, or fever.
The most common side effects of KRAZATI when used alone for NSCLC include:
- o nausea
- o diarrhea
- o vomiting
- o tiredness
- o muscle and bone pain
- o kidney problems
- o swelling
- o decreased appetite
- o trouble breathing
The most common side effects of KRAZATI when used in combination with cetuximab for CRC include:
- o skin rash
- o nausea
- o diarrhea
- o vomiting
- o tiredness
- o muscle and bone pain
- o headache
- o dry skin
- o stomach pain
- o decreased appetite
- o swelling
- o low red blood cell count
- o cough
- o dizziness
- o constipation
- o nerve damage in the arms and legs
Certain abnormal blood test results are common during treatment with KRAZATI, when used alone or in combination with cetuximab. Your healthcare provider will monitor you for abnormal blood tests and treat you if needed.
KRAZATI may cause fertility problems in males and females, which may affect your ability to have children. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of KRAZATI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store KRAZATI?
- o Store KRAZATI at room temperature between 68°F to 77°F (20°C to 25°C).
- o KRAZATI comes in a child-resistant container.
- o KRAZATI comes with a desiccant (drying agent) in the container to keep the medicine dry. Do not remove the desiccant from the container after opening. Do not eat or swallow the desiccant.
Keep KRAZATI and all medicines out of the reach of children.
General information about the safe and effective use of KRAZATI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use KRAZATI for a condition for which it was not prescribed. Do not give KRAZATI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about KRAZATI that is written for health professionals.What are the ingredients in KRAZATI?
Active ingredient: adagrasib
Inactive ingredients: colloidal silicon dioxide, crospovidone, magnesium stearate (vegetable sourced), mannitol, and microcrystalline cellulose. The tablet film coating contains hypromellose, maltodextrin, medium chain triglycerides (vegetable sourced), polydextrose, talc, and titanium dioxide.
Distributed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA
KRAZATI and the KRAZATI logo are trademarks of Mirati Therapeutics, Inc., a Bristol Myers Squibb company.
For more information, go to www.KRAZATI.com or call 1-866-4-KRAZATI (1-866-457-2928).This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 07/2024
- o
alone to treat non-small cell lung cancer (NSCLC)
- PRINCIPAL DISPLAY PANEL - 180 Tablet Bottle Carton
-
INGREDIENTS AND APPEARANCE
KRAZATI
adagrasib tablet, coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 80739-812 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ADAGRASIB (UNII: 8EOO6HQF8Y) (ADAGRASIB - UNII:8EOO6HQF8Y) ADAGRASIB 200 mg Inactive Ingredients Ingredient Name Strength CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) HYPROMELLOSE 2910 (15 MPA.S) (UNII: 36SFW2JZ0W) MAGNESIUM STEARATE (UNII: 70097M6I30) MALTODEXTRIN (UNII: 7CVR7L4A2D) MANNITOL (UNII: 3OWL53L36A) MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYDEXTROSE (UNII: VH2XOU12IE) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TALC (UNII: 7SEV7J4R1U) Product Characteristics Color WHITE Score no score Shape OVAL Size 16mm Flavor Imprint Code 200;M Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 80739-812-12 1 in 1 CARTON 12/12/2022 1 120 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 80739-812-18 1 in 1 CARTON 12/12/2022 2 180 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216340 12/12/2022 Labeler - Mirati Therapeutics, Inc (078870124)

Trademark Results [KRAZATI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 KRAZATI 97541729 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 97541713 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 97541695 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 97541672 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 97541653 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 97541596 not registered Live/Pending |
Mirati Therapeutics, Inc. 2022-08-09 |
 KRAZATI 90603915 not registered Live/Pending |
Mirati Therapeutics, Inc. 2021-03-25 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.