IMBRUVICA- ibrutinib capsule IMBRUVICA- ibrutinib tablet, film coated
Imbruvica by
Drug Labeling and Warnings
Imbruvica by is a Prescription medication manufactured, distributed, or labeled by Pharmacyclics LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IMBRUVICA safely and effectively. See full prescribing information for IMBRUVICA.
IMBRUVICA® (ibrutinib) capsules, for oral use
IMBRUVICA® (ibrutinib) tablets, for oral use
Initial U.S. Approval: 2013RECENT MAJOR CHANGES
INDICATIONS AND USAGE
IMBRUVICA is a kinase inhibitor indicated for the treatment of adult patients with:
- Mantle cell lymphoma (MCL) who have received at least one prior therapy (1.1).
Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. - Chronic lymphocytic leukemia (CLL)/Small lymphocytic lymphoma (SLL) (1.2).
- Chronic lymphocytic leukemia (CLL)/Small lymphocytic lymphoma (SLL) with 17p deletion (1.3).
- Waldenström's macroglobulinemia (WM) (1.4).
- Marginal zone lymphoma (MZL) who require systemic therapy and have received at least one prior anti-CD20-based therapy (1.5).
Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial. - Chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy (1.6).
DOSAGE AND ADMINISTRATION
- MCL and MZL: 560 mg taken orally once daily (2.2).
- CLL/SLL, WM, and cGVHD: 420 mg taken orally once daily (2.2).
Dose should be taken orally with a glass of water. Do not open, break, or chew the capsules. Do not cut, crush, or chew the tablets (2.1).
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hemorrhage: Monitor for bleeding and manage (5.1).
- Infections: Monitor patients for fever and infections, evaluate promptly, and treat (5.2).
- Cytopenias: Check complete blood counts monthly (5.3).
- Cardiac arrhythmias: Monitor for symptoms of arrhythmias and manage (5.4).
- Hypertension: Monitor blood pressure and treat (5.5).
- Second Primary Malignancies: Other malignancies have occurred in patients, including skin cancers, and other carcinomas (5.6).
- Tumor Lysis Syndrome (TLS): Assess baseline risk and take precautions. Monitor and treat for TLS (5.7).
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise women of the potential risk to a fetus and to avoid pregnancy while taking the drug and for 1 month after cessation of therapy. Advise men to avoid fathering a child during the same time period (5.8, 8.3).
ADVERSE REACTIONS
The most common adverse reactions (≥20%) in patients with B-cell malignancies (MCL, CLL/SLL, WM and MZL) were thrombocytopenia, diarrhea, anemia, neutropenia, musculoskeletal pain, rash, bruising, nausea, fatigue, hemorrhage, and pyrexia (6).
The most common adverse reactions (≥20%) in patients with cGVHD were fatigue, bruising, diarrhea, thrombocytopenia, muscle spasms, stomatitis, nausea, hemorrhage, anemia, and pneumonia (6).
To report SUSPECTED ADVERSE REACTIONS, contact Pharmacyclics at 1-877-877-3536 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Hepatic Impairment (based on Child-Pugh criteria): Avoid use of IMBRUVICA in patients with severe baseline hepatic impairment. In patients with mild or moderate impairment, reduce IMBRUVICA dose (2.5, 8.6).
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2019
- Mantle cell lymphoma (MCL) who have received at least one prior therapy (1.1).
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Mantle Cell Lymphoma
1.2 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
1.3 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma with 17p deletion
1.4 Waldenström's Macroglobulinemia
1.5 Marginal Zone Lymphoma
1.6 Chronic Graft versus Host Disease
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Guidelines
2.2 Recommended Dosage
2.3 Dose Modifications for Adverse Reactions
2.4 Dose Modifications for Use with CYP3A Inhibitors
2.5 Dose Modifications for Use in Hepatic Impairment
2.6 Missed Dose
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
5.2 Infections
5.3 Cytopenias
5.4 Cardiac Arrhythmias
5.5 Hypertension
5.6 Second Primary Malignancies
5.7 Tumor Lysis Syndrome
5.8 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of CYP3A Inhibitors on Ibrutinib
7.2 Effect of CYP3A Inducers on Ibrutinib
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Plasmapheresis
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Mantle Cell Lymphoma
14.2 Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma
14.3 Waldenström's Macroglobulinemia
14.4 Marginal Zone Lymphoma
14.5 Chronic Graft versus Host Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Mantle Cell Lymphoma
IMBRUVICA is indicated for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
Accelerated approval was granted for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial [see Clinical Studies (14.1)].
1.2 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
IMBRUVICA is indicated for the treatment of adult patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL).
1.3 Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma with 17p deletion
IMBRUVICA is indicated for the treatment of adult patients with chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) with 17p deletion.
1.4 Waldenström's Macroglobulinemia
IMBRUVICA is indicated for the treatment of adult patients with Waldenström's macroglobulinemia (WM).
1.5 Marginal Zone Lymphoma
IMBRUVICA is indicated for the treatment of adult patients with marginal zone lymphoma (MZL) who require systemic therapy and have received at least one prior anti-CD20-based therapy.
Accelerated approval was granted for this indication based on overall response rate [see Clinical Studies (14.4)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Guidelines
Administer IMBRUVICA orally once daily at approximately the same time each day. The dose should be taken orally with a glass of water. Do not open, break, or chew the capsules. Do not cut, crush, or chew the tablets.
2.2 Recommended Dosage
Mantle Cell Lymphoma and Marginal Zone Lymphoma
The recommended dose of IMBRUVICA for MCL and MZL is 560 mg orally once daily until disease progression or unacceptable toxicity.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma and Waldenström's Macroglobulinemia
The recommended dose of IMBRUVICA for CLL/SLL and WM as a single agent, in combination with rituximab for WM, or in combination with bendamustine and rituximab or with obinutuzumab for CLL/SLL is 420 mg orally once daily until disease progression or unacceptable toxicity.
When administering IMBRUVICA in combination with rituximab or obinutuzumab, consider administering IMBRUVICA prior to rituximab or obinutuzumab when given on the same day.
Chronic Graft versus Host Disease
The recommended dose of IMBRUVICA for cGVHD is 420 mg orally once daily until cGVHD progression, recurrence of an underlying malignancy, or unacceptable toxicity. When a patient no longer requires therapy for the treatment of cGVHD, IMBRUVICA should be discontinued considering the medical assessment of the individual patient.
2.3 Dose Modifications for Adverse Reactions
Interrupt IMBRUVICA therapy for any Grade 3 or greater non-hematological toxicities, Grade 3 or greater neutropenia with infection or fever, or Grade 4 hematological toxicities. Once the symptoms of the toxicity have resolved to Grade 1 or baseline (recovery), IMBRUVICA therapy may be reinitiated at the starting dose. If the toxicity reoccurs, reduce dose by 140 mg per day. A second reduction of dose by 140 mg may be considered as needed. If these toxicities persist or recur following two dose reductions, discontinue IMBRUVICA.
Recommended dose modifications are described below:
Toxicity Occurrence Dose Modification for MCL and MZL After Recovery
Starting Dose = 560 mgDose Modification for CLL/SLL, WM, and cGVHD After Recovery
Starting Dose = 420 mgFirst Restart at 560 mg daily Restart at 420 mg daily Second Restart at 420 mg daily Restart at 280 mg daily Third Restart at 280 mg daily Restart at 140 mg daily Fourth Discontinue IMBRUVICA Discontinue IMBRUVICA 2.4 Dose Modifications for Use with CYP3A Inhibitors
Recommended dose modifications are described below [see Drug Interactions (7.1)] :
Patient Population Coadministered Drug Recommended IMBRUVICA Dose B-Cell Malignancies - Moderate CYP3A inhibitor
280 mg once daily
Modify dose as recommended [see Dosage and Administration (2.3)].- Voriconazole 200 mg twice daily
- Posaconazole suspension 100 mg once daily, 100 mg twice daily, or 200 mg twice daily
140 mg once daily
Modify dose as recommended [see Dosage and Administration (2.3)].- Posaconazole suspension 200 mg three times daily or 400 mg twice daily
- Posaconazole IV injection 300 mg once daily
- Posaconazole delayed-release tablets 300 mg once daily
70 mg once daily
Interrupt dose as recommended [see Dosage and Administration (2.3)].- Other strong CYP3A inhibitors
Avoid concomitant use.
If these inhibitors will be used short-term (such as anti-infectives for seven days or less), interrupt IMBRUVICA.
Chronic Graft versus Host Disease - Moderate CYP3A inhibitor
420 mg once daily
Modify dose as recommended [see Dosage and Administration (2.3)].- Voriconazole 200 mg twice daily
- Posaconazole suspension 100 mg once daily, 100 mg twice daily, or 200 mg twice daily
280 mg once daily
Modify dose as recommended [see Dosage and Administration (2.3)].- Posaconazole suspension 200 mg three times daily or 400 mg twice daily
- Posaconazole IV injection 300 mg once daily
- Posaconazole delayed-release tablets 300 mg once daily
140 mg once daily
Interrupt dose as recommended [see Dosage and Administration (2.3)].- Other strong CYP3A inhibitors
Avoid concomitant use.
If these inhibitors will be used short-term (such as anti-infectives for seven days or less), interrupt IMBRUVICA.After discontinuation of a CYP3A inhibitor, resume previous dose of IMBRUVICA [see Dosage and Administration (2.2) and Drug Interactions (7.1)].
2.5 Dose Modifications for Use in Hepatic Impairment
The recommended dose is 140 mg daily for patients with mild hepatic impairment (Child-Pugh class A).
The recommended dose is 70 mg daily for patients with moderate hepatic impairment (Child-Pugh class B).
Avoid the use of IMBRUVICA in patients with severe hepatic impairment (Child-Pugh class C) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
Capsules:
Each 70 mg capsule is a yellow, opaque capsule marked with "ibr 70 mg" in black ink.
Each 140 mg capsule is a white, opaque capsule marked with "ibr 140 mg" in black ink.
Tablets:
Each 140 mg tablet is a yellow green to green round tablet debossed with "ibr" on one side and "140" on the other side.
Each 280 mg tablet is a purple oblong tablet debossed with "ibr" on one side and "280" on the other side.
Each 420 mg tablet is a yellow green to green oblong tablet debossed with "ibr" on one side and "420" on the other side.
Each 560 mg tablet is a yellow to orange oblong tablet debossed with "ibr" on one side and "560" on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemorrhage
Fatal bleeding events have occurred in patients treated with IMBRUVICA. Major hemorrhage (≥ Grade 3, serious, or any central nervous system events; e.g., intracranial hemorrhage [including subdural hematoma], gastrointestinal bleeding, hematuria, and post procedural hemorrhage) have occurred in 4% of patients, with fatalities occurring in 0.4% of 2,838 patients exposed to IMBRUVICA in 27 clinical trials. Bleeding events of any grade, including bruising and petechiae, occurred in 39% of patients treated with IMBRUVICA.
The mechanism for the bleeding events is not well understood.
Use of either anticoagulant or antiplatelet agents concomitantly with IMBRUVICA increases the risk of major hemorrhage. In IMBRUVICA clinical trials, 3.1% of patients taking IMBRUVICA without antiplatelet or anticoagulant therapy experienced major hemorrhage. The addition of antiplatelet therapy with or without anticoagulant therapy increased this percentage to 4.4%, and the addition of anticoagulant therapy with or without antiplatelet therapy increased this percentage to 6.1%. Consider the risks and benefits of anticoagulant or antiplatelet therapy when co-administered with IMBRUVICA. Monitor for signs and symptoms of bleeding.
Consider the benefit-risk of withholding IMBRUVICA for at least 3 to 7 days pre- and post-surgery depending upon the type of surgery and the risk of bleeding [see Clinical Studies (14)].
5.2 Infections
Fatal and non-fatal infections (including bacterial, viral, or fungal) have occurred with IMBRUVICA therapy. Grade 3 or greater infections occurred in 24% of 1,124 patients exposed to IMBRUVICA in clinical trials [see Adverse Reactions (6.1, 6.2)]. Cases of progressive multifocal leukoencephalopathy (PML) and Pneumocystis jirovecii pneumonia (PJP) have occurred in patients treated with IMBRUVICA. Consider prophylaxis according to standard of care in patients who are at increased risk for opportunistic infections. Monitor and evaluate patients for fever and infections and treat appropriately.
5.3 Cytopenias
Treatment-emergent Grade 3 or 4 cytopenias including neutropenia (23%), thrombocytopenia (8%), and anemia (3%) based on laboratory measurements occurred in patients with B-cell malignancies treated with single agent IMBRUVICA.
Monitor complete blood counts monthly.
5.4 Cardiac Arrhythmias
Fatal and serious cardiac arrhythmias have occurred with IMBRUVICA therapy. Grade 3 or greater ventricular tachyarrhythmias occurred in 0.2% of patients, and Grade 3 or greater atrial fibrillation and atrial flutter occurred in 4% of 1,124 patients exposed to IMBRUVICA in clinical trials. These events have occurred particularly in patients with cardiac risk factors, hypertension, acute infections, and a previous history of cardiac arrhythmias. See Additional Important Adverse Reactions (6.1) .
Periodically monitor patients clinically for cardiac arrhythmias. Obtain an ECG for patients who develop arrhythmic symptoms (e.g., palpitations, lightheadedness, syncope, chest pain) or new onset dyspnea. Manage cardiac arrhythmias appropriately, and if it persists, consider the risks and benefits of IMBRUVICA treatment and follow dose modification guidelines [see Dosage and Administration (2.3)].
5.5 Hypertension
Hypertension of any grade occurred in 12% of 1,124 patients treated with IMBRUVICA in clinical trials. Grade 3 or greater hypertension occurred in 5% of patients with a median time to onset of 5.9 months (range, 0.03 to 24 months).
Monitor blood pressure in patients treated with IMBRUVICA and initiate or adjust anti-hypertensive medication throughout treatment with IMBRUVICA as appropriate.
5.6 Second Primary Malignancies
Other malignancies (10%) including non-skin carcinomas (4%) have occurred in 1,124 patients treated with IMBRUVICA in clinical trials. The most frequent second primary malignancy was non-melanoma skin cancer (6%).
5.7 Tumor Lysis Syndrome
Tumor lysis syndrome has been infrequently reported with IMBRUVICA therapy. Assess the baseline risk (e.g., high tumor burden) and take appropriate precautions. Monitor patients closely and treat as appropriate.
5.8 Embryo-Fetal Toxicity
Based on findings in animals, IMBRUVICA can cause fetal harm when administered to a pregnant woman. Administration of ibrutinib to pregnant rats and rabbits during the period of organogenesis caused embryo-fetal toxicity including malformations at exposures that were 2-20 times higher than those reported in patients with hematologic malignancies. Advise women to avoid becoming pregnant while taking IMBRUVICA and for 1 month after cessation of therapy. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in more detail in other sections of the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Cytopenias [see Warnings and Precautions (5.3)]
- Cardiac Arrhythmias [see Warnings and Precautions (5.4)]
- Hypertension [see Warnings and Precautions (5.5)]
- Second Primary Malignancies [see Warnings and Precautions (5.6)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely variable conditions, adverse event rates observed in clinical trials of a drug cannot be directly compared with rates of clinical trials of another drug and may not reflect the rates observed in practice.
Mantle Cell Lymphoma
The data described below reflect exposure to IMBRUVICA in a clinical trial (Study 1104) that included 111 patients with previously treated MCL treated with 560 mg daily with a median treatment duration of 8.3 months.
The most commonly occurring adverse reactions (≥ 20%) were thrombocytopenia, diarrhea, neutropenia, anemia, fatigue, musculoskeletal pain, peripheral edema, upper respiratory tract infection, nausea, bruising, dyspnea, constipation, rash, abdominal pain, vomiting and decreased appetite (see Tables 1 and 2).
The most common Grade 3 or 4 non-hematological adverse reactions (≥ 5%) were pneumonia, abdominal pain, atrial fibrillation, diarrhea, fatigue, and skin infections.
Fatal and serious cases of renal failure have occurred with IMBRUVICA therapy. Increases in creatinine 1.5 to 3 times the upper limit of normal occurred in 9% of patients.
Adverse reactions from the MCL trial (N=111) using single agent IMBRUVICA 560 mg daily occurring at a rate of ≥ 10% are presented in Table 1.
Table 1: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with MCL (N=111) Body System Adverse Reaction All Grades (%) Grade 3 or Higher (%) - * Includes one event with a fatal outcome.
Gastrointestinal disorders Diarrhea 51 5 Nausea 31 0 Constipation 25 0 Abdominal pain 24 5 Vomiting 23 0 Stomatitis 17 1 Dyspepsia 11 0 Infections and infestations Upper respiratory tract infection 34 0 Urinary tract infection 14 3 Pneumonia 14 8* Skin infections 14 5 Sinusitis 13 1 General disorders and administration site conditions Fatigue 41 5 Peripheral edema 35 3 Pyrexia 18 1 Asthenia 14 3 Skin and subcutaneous tissue disorders Bruising 30 0 Rash 25 3 Petechiae 11 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain 37 1 Muscle spasms 14 0 Arthralgia 11 0 Respiratory, thoracic and mediastinal disorders Dyspnea 27 5* Cough 19 0 Epistaxis 11 0 Metabolism and nutrition disorders Decreased appetite 21 2 Dehydration 12 4 Nervous system disorders Dizziness 14 0 Headache 13 0 Table 2: Treatment-Emergent* Hematologic Laboratory Abnormalities in Patients with MCL (N=111) Percent of Patients (N=111) All Grades (%) Grade 3 or 4 (%) Treatment-emergent Grade 4 thrombocytopenia (6%) and neutropenia (13%) occurred in patients. - * Based on laboratory measurements and adverse reactions
Platelets Decreased 57 17 Neutrophils Decreased 47 29 Hemoglobin Decreased 41 9 Ten patients (9%) discontinued treatment due to adverse reactions in the trial (N=111). The most frequent adverse reaction leading to treatment discontinuation was subdural hematoma (1.8%). Adverse reactions leading to dose reduction occurred in 14% of patients.
Patients with MCL who develop lymphocytosis greater than 400,000/mcL have developed intracranial hemorrhage, lethargy, gait instability, and headache. However, some of these cases were in the setting of disease progression.
Forty percent of patients had elevated uric acid levels on study including 13% with values above 10 mg/dL. Adverse reaction of hyperuricemia was reported for 15% of patients.
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
The data described below reflect exposure in one single-arm, open-label clinical trial (Study 1102) and four randomized controlled clinical trials (RESONATE, RESONATE-2, and HELIOS, and iLLUMINATE) in patients with CLL/SLL (n=1,506 total and n=781 patients exposed to IMBRUVICA). Patients with creatinine clearance (CrCl) ≤ 30 mL/min, AST or ALT ≥ 2.5 × ULN (upper limit of normal), or total bilirubin ≥ 1.5× ULN (unless of non-hepatic origin) were excluded from these trials. Study 1102 included 51 patients with previously treated CLL/SLL, RESONATE included 386 randomized patients with previously treated CLL or SLL who received single agent IMBRUVICA or ofatumumab, RESONATE-2 included 267 randomized patients with treatment naïve-CLL or SLL who were 65 years or older and received single agent IMBRUVICA or chlorambucil, HELIOS included 574 randomized patients with previously treated CLL or SLL who received IMBRUVICA in combination with bendamustine and rituximab or placebo in combination with bendamustine and rituximab, and iLLUMINATE included 228 randomized patients with treatment naïve CLL who were 65 years or older or with coexisting medical conditions and received IMBRUVICA in combination with obinutuzumab or chlorambucil in combination with obinutuzumab.
The most commonly occurring adverse reactions in patients with CLL/SLL receiving IMBRUVICA (≥ 20%) were neutropenia, thrombocytopenia, anemia, diarrhea, rash, musculoskeletal pain, bruising, nausea, fatigue, pyrexia, hemorrhage, and cough.
Four to 10 percent of patients with CLL/SLL receiving IMBRUVICA discontinued treatment due to adverse reactions. These included pneumonia, hemorrhage, atrial fibrillation, rash and neutropenia. Adverse reactions leading to dose reduction occurred in approximately 7% of patients.
Study 1102
Adverse reactions and laboratory abnormalities from the CLL/SLL trial (N=51) using single agent IMBRUVICA 420 mg daily in patients with previously treated CLL/SLL occurring at a rate of ≥ 10% with a median duration of treatment of 15.6 months are presented in Tables 3 and 4.
Table 3: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with CLL/SLL (N=51) in Study 1102 Body System Adverse Reaction All Grades (%) Grade 3 or Higher (%) - * One patient death due to histiocytic sarcoma.
Gastrointestinal disorders Diarrhea 59 4 Constipation 22 2 Nausea 20 2 Stomatitis 20 0 Vomiting 18 2 Abdominal pain 14 0 Dyspepsia 12 0 Infections and infestations Upper respiratory tract infection 47 2 Sinusitis 22 6 Skin infection 16 6 Pneumonia 12 10 Urinary tract infection 12 2 General disorders and administration site conditions Fatigue 33 6 Pyrexia 24 2 Peripheral edema 22 0 Asthenia 14 6 Chills 12 0 Skin and subcutaneous tissue disorders Bruising 51 2 Rash 25 0 Petechiae 16 0 Respiratory, thoracic and mediastinal disorders Cough 22 0 Oropharyngeal pain 14 0 Dyspnea 12 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain 25 6 Arthralgia 24 0 Muscle spasms 18 2 Nervous system disorders Dizziness 20 0 Headache 18 2 Metabolism and nutrition disorders Decreased appetite 16 2 Neoplasms benign, malignant, unspecified Second malignancies 10 2* Vascular disorders Hypertension 16 8 Table 4: Treatment-Emergent* Hematologic Laboratory Abnormalities in Patients with CLL/SLL (N=51) in Study 1102 Percent of Patients (N=51) All Grades (%) Grade 3 or 4 (%) Treatment-emergent Grade 4 thrombocytopenia (8%) and neutropenia (12%) occurred in patients. - * Based on laboratory measurements per IWCLL criteria and adverse reactions.
Platelets Decreased 69 12 Neutrophils Decreased 53 26 Hemoglobin Decreased 43 0 RESONATE
Adverse reactions and laboratory abnormalities described below in Tables 5 and 6 reflect exposure to IMBRUVICA with a median duration of 8.6 months and exposure to ofatumumab with a median of 5.3 months in RESONATE in patients with previously treated CLL/SLL.
Table 5: Adverse Reactions Reported in ≥ 10% of Patients in the IMBRUVICA Treated Arm in Patients with CLL/SLL in RESONATE Body System
Adverse ReactionIMBRUVICA
(N=195)Ofatumumab
(N=191)All Grades
(%)Grade 3 or Higher (%) All Grades
(%)Grade 3 or Higher (%) The body system and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm. - * Includes multiple ADR terms
- † Includes 3 events of pneumonia with fatal outcome in each arm, and 1 event of pyrexia and upper respiratory tract infection with a fatal outcome in the ofatumumab arm.
Gastrointestinal disorders Diarrhea 48 4 18 2 Nausea 26 2 18 0 Stomatitis* 17 1 6 1 Constipation 15 0 9 0 Vomiting 14 0 6 1 General disorders and administration site conditions Pyrexia 24 2 15 2† Infections and infestations Upper respiratory tract infection 16 1 11 2† Pneumonia* 15 12† 13 10† Sinusitis* 11 1 6 0 Urinary tract infection 10 4 5 1 Skin and subcutaneous tissue disorders Rash* 24 3 13 0 Petechiae 14 0 1 0 Bruising* 12 0 1 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 28 2 18 1 Arthralgia 17 1 7 0 Muscle spasms 13 0 8 0 Respiratory, thoracic and mediastinal disorders Cough 19 0 23 1 Dyspnea 12 2 10 1 Nervous system disorders Headache 14 1 6 0 Dizziness 11 0 5 0 Injury, poisoning and procedural complications Contusion 11 0 3 0 Eye disorders Vision blurred 10 0 3 0 Table 6: Treatment-Emergent Hematologic Laboratory Abnormalities in Patients with CLL/SLL in RESONATE IMBRUVICA
(N=195)Ofatumumab
(N=191)All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Treatment-emergent Grade 4 thrombocytopenia (2% in the IMBRUVICA arm vs 3% in the ofatumumab arm) and neutropenia (8% in the IMBRUVICA arm vs 8% in the ofatumumab arm) occurred in patients. Neutrophils Decreased 51 23 57 26 Platelets Decreased 52 5 45 10 Hemoglobin Decreased 36 0 21 0 RESONATE-2
Adverse reactions and laboratory abnormalities described below in Tables 7 and 8 reflect exposure to IMBRUVICA with a median duration of 17.4 months. The median exposure to chlorambucil was 7.1 months in RESONATE-2.
Table 7: Adverse Reactions Reported in ≥ 10% of Patients in the IMBRUVICA Treated Arm in Patients with CLL/SLL in RESONATE-2 Body System
Adverse ReactionIMBRUVICA
(N=135)Chlorambucil
(N=132)All Grades
(%)Grade 3 or Higher (%) All Grades
(%)Grade 3 or Higher (%) Subjects with multiple events for a given ADR term are counted once only for each ADR term.
The body system and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm.- * Includes multiple ADR terms
Gastrointestinal disorders Diarrhea 42 4 17 0 Nausea 22 1 39 1 Constipation 16 1 16 0 Stomatitis* 14 1 4 1 Vomiting 13 0 20 1 Abdominal pain 13 3 11 1 Dyspepsia 11 0 2 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 36 4 20 0 Arthralgia 16 1 7 1 Muscle spasms 11 0 5 0 Eye disorders Dry eye 17 0 5 0 Lacrimation increased 13 0 6 0 Vision blurred 13 0 8 0 Visual acuity reduced 11 0 2 0 Skin and subcutaneous tissue disorders Rash* 21 4 12 2 Bruising* 19 0 7 0 Infections and infestations Upper respiratory tract infection 17 2 17 2 Skin infection* 15 2 3 1 Pneumonia* 14 8 7 4 Urinary tract infections 10 1 8 1 Respiratory, thoracic and mediastinal disorders Cough 22 0 15 0 Dyspnea 10 1 10 0 General disorders and administration site conditions Fatigue 30 1 38 5 Peripheral edema 19 1 9 0 Pyrexia 17 0 14 2 Vascular disorders Hypertension* 14 4 1 0 Nervous system disorders Headache 12 1 10 2 Dizziness 11 0 12 1 Investigations Weight decreased 10 0 12 0 Table 8: Treatment-Emergent Hematologic Laboratory Abnormalities in Patients with CLL/SLL in RESONATE-2 IMBRUVICA
(N=135)Chlorambucil
(N=132)All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)Treatment-emergent Grade 4 thrombocytopenia (1% in the IMBRUVICA arm vs 3% in the chlorambucil arm) and neutropenia (11% in the IMBRUVICA arm vs 12% in the chlorambucil arm) occurred in patients. Neutrophils Decreased 55 28 67 31 Platelets Decreased 47 7 58 14 Hemoglobin Decreased 36 0 39 2 HELIOS
Adverse reactions described below in Table 9 reflect exposure to IMBRUVICA + BR with a median duration of 14.7 months and exposure to placebo + BR with a median of 12.8 months in HELIOS in patients with previously treated CLL/SLL.
Table 9: Adverse Reactions Reported in at Least 10% of Patients and at Least 2% Greater in the IMBRUVICA Arm in Patients with CLL/SLL in HELIOS Body System
Adverse ReactionIbrutinib + BR
(N=287)Placebo + BR
(N=287)All Grades
(%)Grade 3 or Higher (%) All Grades
(%)Grade 3 or Higher (%) The body system and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm.
<1 used for frequency above 0 and below 0.5%- * Includes multiple ADR terms
- † Includes 2 events of hemorrhage with fatal outcome in the IMBRUVICA arm and 1 event of neutropenia with a fatal outcome in the placebo + BR arm.
Blood and lymphatic system disorders Neutropenia* 66 61 60 56† Thrombocytopenia* 34 16 26 16 Skin and subcutaneous tissue disorders Rash * 32 4 25 1 Bruising * 20 <1 8 <1 Gastrointestinal disorders Diarrhea 36 2 23 1 Abdominal pain 12 1 8 <1 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 29 2 20 0 Muscle spasms 12 <1 5 0 General disorders and administration site conditions Pyrexia 25 4 22 2 Vascular disorders Hemorrhage* 19 2† 9 1 Hypertension * 11 5 5 2 Infections and infestations Bronchitis 13 2 10 3 Skin infection* 10 3 6 2 Metabolism and nutrition disorders Hyperuricemia 10 2 6 0 Atrial fibrillation of any grade occurred in 7% of patients treated with IMBRUVICA + BR and 2% of patients treated with placebo + BR. The frequency of Grade 3 and 4 atrial fibrillation was 3% in patients treated with IMBRUVICA + BR and 1% in patients treated with placebo + BR.
iLLUMINATE
Adverse reactions described below in Table 10 reflect exposure to IMBRUVICA + obinutuzumab with a median duration of 29.3 months and exposure to chlorambucil + obinutuzumab with a median of 5.1 months in iLLUMINATE in patients with previously untreated CLL/SLL.
Table 10: Adverse Reactions Reported in at Least 10% of Patients in the IMBRUVICA Arm in Patients with CLL/SLL in iLLUMINATE Body System
Adverse ReactionIMBRUVICA + Obinutuzumab
(N=113)Chlorambucil + Obinutuzumab
(N=115)All Grades
(%)Grade 3 or Higher (%) All Grades
(%)Grade 3 or Higher (%) The body system and individual ADR terms are sorted in descending frequency order in the IMBRUVICA arm. - * Includes multiple ADR terms
- † Includes one event with a fatal outcome.
Blood and lymphatic system disorders Neutropenia* 48 39 64 48 Thrombocytopenia* 36 19 28 11 Anemia 17 4 25 8 Skin and subcutaneous tissue disorders Rash* 36 3 11 0 Bruising* 32 3 3 0 Gastrointestinal Disorders Diarrhea 34 3 10 0 Constipation 16 0 12 1 Nausea 12 0 30 0 Musculoskeletal and Connective Tissue Disorders Musculoskeletal Pain* 33 1 23 3 Arthralgia 22 1 10 0 Muscle spasms 13 0 6 0 Respiratory, Thoracic and Mediastinal Disorders Cough 27 1 12 0 Injury, Poisoning and Procedural Complications Infusion related reaction 25 2 58 8 Vascular disorders Hemorrhage* 25 1 9 0 Hypertension* 17 4 4 3 Infections and Infestations Pneumonia* 16 9 9 4† Upper Respiratory Tract Infection 14 1 6 0 Skin infection* 13 1 3 0 Urinary tract infection 12 3 7 1 Nasopharyngitis 12 0 3 0 Conjunctivitis 11 0 2 0 Metabolism and Nutrition Disorders Hyperuricemia 13 1 0 0 Cardiac Disorders Atrial Fibrillation 12 5 0 0 General Disorders and Administration Site Conditions Pyrexia 19 2 26 1 Fatigue 18 0 17 2 Peripheral edema 12 0 7 0 Psychiatric disorders Insomnia 12 0 4 0 Waldenström's Macroglobulinemia and Marginal Zone Lymphoma
The data described below reflect exposure to IMBRUVICA in three single-arm open-label clinical trials (Study 1118, Study 1121, and INNOVATE monotherapy arm) and one randomized controlled trial (INNOVATE) in patients with WM or MZL, including a total n=307 patients overall and n=232 patients exposed to IMBRUVICA. Study 1118 included 63 patients with previously treated WM who received single agent IMBRUVICA. Study 1121 included 63 patients with previously treated MZL who received single agent IMBRUVICA. INNOVATE included 150 patients with treatment naïve or previously treated WM who received IMBRUVICA or placebo in combination with rituximab. The INNOVATE monotherapy arm included 31 patients with previously treated WM who failed prior rituximab-containing therapy and received IMBRUVICA.
The most commonly occurring adverse reactions in Studies 1118, 1121, and INNOVATE (≥ 20%) were thrombocytopenia, diarrhea, bruising, neutropenia, musculoskeletal pain, hemorrhage, anemia, rash, fatigue, and nausea.
Seven percent of patients receiving IMBRUVICA across Studies 1118, 1121, and INNOVATE discontinued treatment due to adverse reactions. The most common adverse reactions leading to discontinuation were atrial fibrillation, interstitial lung disease, diarrhea and rash. Adverse reactions leading to dose reduction occurred in 13% of patients.
Study 1118 and INNOVATE Monotherapy Arm
Adverse reactions and laboratory abnormalities described below in Tables 11 and 12 reflect exposure to IMBRUVICA with a median duration of 11.7 months in Study 1118 and 33 months in the INNOVATE Monotherapy Arm.
Table 11: Non-Hematologic Adverse Reactions in ≥ 10% in Patients with WM in Study 1118 and the INNOVATE Monotherapy Arm (N=94) Body System Adverse Reaction All Grades
(%)Grade 3 or Higher (%) The body system and individual ADR preferred terms are sorted in descending frequency order. - * Includes multiple ADR terms.
Gastrointestinal disorders Diarrhea 38 2 Nausea 21 0 Stomatitis* 15 0 Constipation 12 1 Gastroesophageal reflux disease 12 0 Skin and subcutaneous tissue disorders Bruising* 28 1 Rash* 21 1 Vascular disorders Hemorrhage* 28 0 Hypertension* 14 4 General disorders and administrative site conditions Fatigue 18 2 Pyrexia 12 2 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 21 0 Muscle spasms 19 0 Infections and infestations Upper respiratory tract infection 19 0 Skin infection* 18 3 Sinusitis* 16 0 Pneumonia* 13 5 Nervous system disorders Headache 14 0 Dizziness 13 0 Respiratory, thoracic and mediastinal disorders Cough 13 0 Table 12: Treatment-Emergent Hematologic Laboratory Abnormalities in Patients with WM in Study 1118 and the INNOVATE Monotherapy Arm (N=94) Percent of Patients (N=94) All Grades (%) Grade 3 or 4 (%) Treatment-emergent Grade 4 thrombocytopenia (4%) and neutropenia (7%) occurred in patients. Platelets Decreased 38 11 Neutrophils Decreased 43 16 Hemoglobin Decreased 21 6 INNOVATE
Adverse reactions described below in Table 13 reflect exposure to IMBRUVICA + R with a median duration of 25.8 months and exposure to placebo + R with a median duration of 15.5 months in patients with treatment naïve or previously treated WM in INNOVATE.
Table 13: Adverse Reactions Reported in at Least 10% of Patients and at Least 2% Greater in the IMBRUVICA Arm in Patients with WM in INNOVATE Body System
Adverse ReactionIMBRUVICA + R
(N=75)Placebo + R
(N=75)All Grades
(%)Grade 3 or Higher
(%)All Grades
(%)Grade 3 or Higher
(%)The body system and individual ADR preferred terms are sorted in descending frequency order. - * Includes multiple ADR terms.
- † Includes one event with a fatal outcome.
Skin and subcutaneous tissue disorders Bruising* 37 1 5 0 Rash* 24 1 11 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 35 4 21 3 Arthralgia 24 3 11 1 Muscle spasms 17 0 12 1 Vascular disorders Hemorrhage* 32 3 17 4† Hypertension* 20 13 5 4 Gastrointestinal disorders Diarrhea 28 0 15 1 Nausea 21 0 12 0 Dyspepsia 16 0 1 0 Constipation 13 1 11 1 Infections and infestations Pneumonia* 19 13 5 3 Skin infection* 17 3 3 0 Urinary tract infection 13 0 0 0 Bronchitis 12 3 7 0 Influenza 12 0 7 1 Viral upper respiratory tract infection 11 0 7 0 General disorders and administration site conditions Peripheral edema 17 0 12 1 Respiratory, thoracic, and mediastinal disorders Cough 17 0 11 0 Blood and Lymphatic System Disorders Neutropenia* 16 12 11 4 Cardiac Disorders Atrial fibrillation 15 12 3 1 Nervous system disorders Dizziness 11 0 7 0 Psychiatric disorders Insomnia 11 0 4 0 Metabolism and nutrition disorders Hypokalemia 11 0 1 1 Grade 3 or 4 infusion related reactions were observed in 1% of patients treated with IMBRUVICA + R.
Study 1121
Adverse reactions and laboratory abnormalities described below in Tables 14 and 15 reflect exposure to IMBRUVICA with a median duration of 11.6 months in Study 1121.
Table 14: Non-Hematologic Adverse Reactions in ≥ 10% in Patients with MZL in Study 1121 (N=63) Body System Adverse Reaction All Grades
(%)Grade 3 or Higher (%) The body system and individual ADR preferred terms are sorted in descending frequency order. - * Includes multiple ADR terms.
- † Includes one event with a fatal outcome.
Gastrointestinal disorders Diarrhea 43 5 Nausea 25 0 Dyspepsia 19 0 Stomatitis* 17 2 Abdominal pain 16 2 Constipation 14 0 Abdominal pain upper 13 0 Vomiting 11 2 General disorders and administrative site conditions Fatigue 44 6 Peripheral edema 24 2 Pyrexia 17 2 Skin and subcutaneous tissue disorders Bruising * 41 0 Rash* 29 5 Pruritus 14 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain* 40 3 Arthralgia 24 2 Muscle spasms 19 3 Infections and infestations Upper respiratory tract infection 21 0 Sinusitis* 19 0 Bronchitis 11 0 Pneumonia* 11 10 Metabolism and nutrition disorders Decreased appetite 16 2 Hyperuricemia 16 0 Hypoalbuminemia 14 0 Hypokalemia 13 0 Vascular disorders Hemorrhage* 30 2† Hypertension* 14 5 Respiratory, thoracic and mediastinal disorders Cough 22 2 Dyspnea 21 2 Nervous system disorders Dizziness 19 0 Headache 13 0 Psychiatric disorders Anxiety 16 2 Table 15: Treatment-Emergent Hematologic Laboratory Abnormalities in Patients with MZL in Study 1121 (N=63) Percent of Patients (N=63) All Grades (%) Grade 3 or 4 (%) Treatment-emergent Grade 4 thrombocytopenia (3%) and neutropenia (6%) occurred in patients. Platelets Decreased 49 6 Hemoglobin Decreased 43 13 Neutrophils Decreased 22 13 Chronic Graft versus Host Disease
The data described below reflect exposure to IMBRUVICA in an open-label clinical trial (Study 1129) that included 42 patients with cGVHD after failure of first line corticosteroid therapy and required additional therapy.
The most commonly occurring adverse reactions in the cGVHD trial (≥ 20%) were fatigue, bruising, diarrhea, thrombocytopenia, stomatitis, muscle spasms, nausea, hemorrhage, anemia, and pneumonia. Atrial fibrillation occurred in one patient (2%) which was Grade 3.
Twenty-four percent of patients receiving IMBRUVICA in the cGVHD trial discontinued treatment due to adverse reactions. The most common adverse reactions leading to discontinuation were fatigue and pneumonia. Adverse reactions leading to dose reduction occurred in 26% of patients.
Adverse reactions and laboratory abnormalities described below in Tables 16 and 17 reflect exposure to IMBRUVICA with a median duration of 4.4 months in the cGVHD trial.
Table 16: Non-Hematologic Adverse Reactions in ≥ 10% of Patients with cGVHD (N=42) Body System Adverse Reaction All Grades
(%)Grade 3 or Higher (%) The system organ class and individual ADR preferred terms are sorted in descending frequency order. - * Includes multiple ADR terms.
- † Includes 2 events with a fatal outcome.
General disorders and administration site conditions Fatigue 57 12 Pyrexia 17 5 Edema peripheral 12 0 Skin and subcutaneous tissue disorders Bruising* 40 0 Rash* 12 0 Gastrointestinal disorders Diarrhea 36 10 Stomatitis* 29 2 Nausea 26 0 Constipation 12 0 Musculoskeletal and connective tissue disorders Muscle spasms 29 2 Musculoskeletal pain* 14 5 Vascular disorders Hemorrhage* 26 0 Infections and infestations Pneumonia* 21 14† Upper respiratory tract infection 19 0 Sepsis* 10 10 Nervous system disorders Headache 17 5 Injury, poisoning and procedural complications Fall 17 0 Respiratory, thoracic and mediastinal disorders Cough 14 0 Dyspnea 12 2 Metabolism and nutrition disorders Hypokalemia 12 7 Table 17: Treatment-Emergent Hematologic Laboratory Abnormalities in Patients with cGVHD (N=42) Percent of Patients (N=42) All Grades (%) Grade 3 or 4 (%) Treatment-emergent Grade 4 neutropenia occurred in 2% of patients. Platelets Decreased 33 0 Neutrophils Decreased 10 10 Hemoglobin Decreased 24 2 Additional Important Adverse Reactions
Cardiac Arrhythmias
In randomized controlled trials (n=1605; median treatment duration of 14.8 months for 805 patients treated with IMBRUVICA and 5.6 months for 800 patients in the control arm), the incidence of ventricular tachyarrhythmias (ventricular extrasystoles, ventricular arrhythmias, ventricular fibrillation, ventricular flutter, and ventricular tachycardia) of any grade was 1.0% versus 0.5% and of Grade 3 or greater was 0.2% versus 0% in patients treated with IMBRUVICA compared to patients in the control arm. In addition, the incidence of atrial fibrillation and atrial flutter of any grade was 9% versus 1.4% and for Grade 3 or greater was 4.1% versus 0.4% in patients treated with IMBRUVICA compared to patients in the control arm.
Diarrhea
In randomized controlled trials (n=1605; median treatment duration of 14.8 months for 805 patients treated with IMBRUVICA and 5.6 months for 800 patients in the control arm), diarrhea of any grade occurred at a rate of 39% of patients treated with IMBRUVICA compared to 18% of patients in the control arm. Grade 3 diarrhea occurred in 3% versus 1% of IMBRUVICA-treated patients compared to the control arm, respectively. The median time to first onset was 21 days (range, 0 to 708) versus 46 days (range, 0 to 492) for any grade diarrhea and 117 days (range, 3 to 414) versus 194 days (range, 11 to 325) for Grade 3 diarrhea in IMBRUVICA-treated patients compared to the control arm, respectively. Of the patients who reported diarrhea, 85% versus 89% had complete resolution, and 15% versus 11% had not reported resolution at time of analysis in IMBRUVICA-treated patients compared to the control arm, respectively. The median time from onset to resolution in IMBRUVICA-treated subjects was 7 days (range, 1 to 655) versus 4 days (range, 1 to 367) for any grade diarrhea and 7 days (range, 1 to 78) versus 19 days (range, 1 to 56) for Grade 3 diarrhea in IMBRUVICA-treated subjects compared to the control arm, respectively. Less than 1% of subjects discontinued IMBRUVICA due to diarrhea compared with 0% in the control arm.
Visual Disturbance
In randomized controlled trials (n=1605; median treatment duration of 14.8 months for 805 patients treated with IMBRUVICA and 5.6 months for 800 patients in the control arm), blurred vision and decreased visual acuity of any grade occurred in 11% of patients treated with IMBRUVICA (10% Grade 1, 2% Grade 2, no Grade 3 or higher) compared to 6% in the control arm (6% Grade 1 and <1% Grade 2 and 3). The median time to first onset was 91 days (range, 0 to 617) versus 100 days (range, 2 to 477) in IMBRUVICA-treated patients compared to the control arm, respectively. Of the patients who reported visual disturbances, 60% versus 71% had complete resolution and 40% versus 29% had not reported resolution at the time of analysis in IMBRUVICA-treated patients compared to the control arm, respectively. The median time from onset to resolution was 37 days (range, 1 to 457) versus 26 days (range, 1 to 721) in IMBRUVICA-treated subjects compared to the control arm, respectively.
Long-Term Safety
The safety data from long-term follow-up over 5 years of 1,178 patients (treatment-naïve CLL/SLL n=162, relapsed/refractory CLL/SLL n=646, and relapsed/refractory MCL n=370) treated with IMBRUVICA were analyzed. The median treatment duration for CLL/SLL was 51 months (range, 0.2 to 98 months). The median treatment duration for MCL was 11 months (range, 0 to 87 months). The cumulative rate of hypertension increased over time with prolonged IMBRUVICA treatment. The prevalence for Grade 3 or greater hypertension was 4% (year 0-1), 6% (year 1-2), 8% (year 2-3), 9% (year 3-4), and 9% (year 4-5). The incidence for the 5-year period was 11%.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of IMBRUVICA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Hepatobiliary disorders: hepatic failure including acute and/or fatal events, hepatic cirrhosis
- Respiratory disorders: interstitial lung disease
- Metabolic and nutrition disorders: tumor lysis syndrome [see Warnings & Precautions (5.7)]
- Immune system disorders: anaphylactic shock, angioedema, urticaria
- Skin and subcutaneous tissue disorders: Stevens-Johnson Syndrome (SJS), onychoclasis, panniculitis
- Infections: hepatitis B reactivation
- Nervous system disorders: peripheral neuropathy
-
7 DRUG INTERACTIONS
7.1 Effect of CYP3A Inhibitors on Ibrutinib
The coadministration of IMBRUVICA with a strong or moderate CYP3A inhibitor may increase ibrutinib plasma concentrations [see Clinical Pharmacology (12.3)]. Increased ibrutinib concentrations may increase the risk of drug-related toxicity.
Dose modifications of IMBRUVICA are recommended when used concomitantly with posaconazole, voriconazole and moderate CYP3A inhibitors [see Dosage and Administration (2.4)].
Avoid concomitant use of other strong CYP3A inhibitors. Interrupt IMBRUVICA if these inhibitors will be used short-term (such as anti-infectives for seven days or less) [see Dosage and Administration (2.4)].
Avoid grapefruit and Seville oranges during IMBRUVICA treatment, as these contain strong or moderate inhibitors of CYP3A.
7.2 Effect of CYP3A Inducers on Ibrutinib
The coadministration of IMBRUVICA with strong CYP3A inducers may decrease ibrutinib concentrations. Avoid coadministration with strong CYP3A inducers [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
IMBRUVICA, a kinase inhibitor, can cause fetal harm based on findings from animal studies. There are no available data on IMBRUVICA use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. In animal reproduction studies, administration of ibrutinib to pregnant rats and rabbits during the period of organogenesis at exposures up to 2-20 times the clinical doses of 420-560 mg daily produced embryofetal toxicity including structural abnormalities (see Data). If IMBRUVICA is used during pregnancy or if the patient becomes pregnant while taking IMBRUVICA, the patient should be apprised of the potential hazard to the fetus.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Ibrutinib was administered orally to pregnant rats during the period of organogenesis at doses of 10, 40 and 80 mg/kg/day. Ibrutinib at a dose of 80 mg/kg/day was associated with visceral malformations (heart and major vessels) and increased resorptions and post-implantation loss. The dose of 80 mg/kg/day in rats is approximately 14 times the exposure (AUC) in patients with MCL or MZL and 20 times the exposure in patients with CLL/SLL or WM administered the dose of 560 mg daily and 420 mg daily, respectively. Ibrutinib at doses of 40 mg/kg/day or greater was associated with decreased fetal weights. The dose of 40 mg/kg/day in rats is approximately 6 times the exposure (AUC) in patients with MCL administered the dose of 560 mg daily.
Ibrutinib was also administered orally to pregnant rabbits during the period of organogenesis at doses of 5, 15, and 45 mg/kg/day. Ibrutinib at a dose of 15 mg/kg/day or greater was associated with skeletal variations (fused sternebrae) and ibrutinib at a dose of 45 mg/kg/day was associated with increased resorptions and post-implantation loss. The dose of 15 mg/kg/day in rabbits is approximately 2.0 times the exposure (AUC) in patients with MCL and 2.8 times the exposure in patients with CLL/SLL or WM administered the dose of 560 and 420 mg daily, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of ibrutinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production.
The development and health benefits of breastfeeding should be considered along with the mother's clinical need for IMBRUVICA and any potential adverse effects on the breastfed child from IMBRUVICA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential prior to initiating IMBRUVICA therapy.
Contraception
Females
Advise females of reproductive potential to avoid pregnancy while taking IMBRUVICA and for up to 1 month after ending treatment. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be informed of the potential hazard to a fetus.
8.4 Pediatric Use
The safety and effectiveness of IMBRUVICA in pediatric patients has not been established.
8.5 Geriatric Use
Of the 1,124 patients in clinical studies of IMBRUVICA, 64% were ≥ 65 years of age, while 23% were ≥75 years of age. No overall differences in effectiveness were observed between younger and older patients. Anemia (all grades), pneumonia (Grade 3 or higher), thrombocytopenia, hypertension, and atrial fibrillation occurred more frequently among older patients treated with IMBRUVICA.
8.6 Hepatic Impairment
Avoid use of IMBRUVICA in patients with severe hepatic impairment (Child-Pugh class C). The safety of IMBRUVICA has not been evaluated in patients with mild to severe hepatic impairment by Child-Pugh criteria.
Dose modifications of IMBRUVICA are recommended in patients with mild or moderate hepatic impairment (Child-Pugh class A and B). Monitor patients for adverse reactions of IMBRUVICA closely [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no specific experience in the management of ibrutinib overdose in patients. One healthy subject experienced reversible Grade 4 hepatic enzyme increases (AST and ALT) after a dose of 1680 mg. Closely monitor patients who ingest more than the recommended dosage and provide appropriate supportive treatment.
-
11 DESCRIPTION
Ibrutinib is an inhibitor of Bruton's tyrosine kinase (BTK). It is a white to off-white solid with the empirical formula C25H24N6O2 and a molecular weight 440.50. Ibrutinib is freely soluble in dimethyl sulfoxide, soluble in methanol and practically insoluble in water.
The chemical name for ibrutinib is 1-[(3R)-3-[4-amino-3-(4-phenoxyphenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl]-1-piperidinyl]-2-propen-1-one and has the following structure:
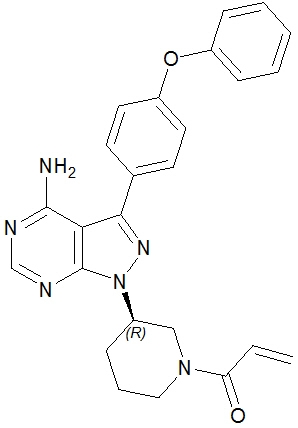
IMBRUVICA (ibrutinib) is available as immediate-release oral capsules and immediate-release oral tablets.
IMBRUVICA (ibrutinib) capsules for oral administration are available in the following dosage strengths: 70 mg and 140 mg. Each capsule contains ibrutinib (active ingredient) and the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate. The capsule shell contains gelatin, titanium dioxide, yellow iron oxide (70 mg capsule only), and black ink.
IMBRUVICA (ibrutinib) tablets for oral administration are available in the following dosage strengths: 140 mg, 280 mg, 420 mg, and 560 mg. Each tablet contains ibrutinib (active ingredient) and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The film coating for each tablet contains ferrosoferric oxide (140 mg, 280 mg, and 420 mg tablets), polyvinyl alcohol, polyethylene glycol, red iron oxide (280 mg and 560 mg tablets), talc, titanium dioxide, and yellow iron oxide (140 mg, 420 mg, and 560 mg tablets).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ibrutinib is a small-molecule inhibitor of BTK. Ibrutinib forms a covalent bond with a cysteine residue in the BTK active site, leading to inhibition of BTK enzymatic activity. BTK is a signaling molecule of the B-cell antigen receptor (BCR) and cytokine receptor pathways. BTK's role in signaling through the B-cell surface receptors results in activation of pathways necessary for B-cell trafficking, chemotaxis, and adhesion. Nonclinical studies show that ibrutinib inhibits malignant B-cell proliferation and survival in vivo as well as cell migration and substrate adhesion in vitro.
12.2 Pharmacodynamics
In patients with recurrent B-cell lymphoma > 90% occupancy of the BTK active site in peripheral blood mononuclear cells was observed up to 24 hours after ibrutinib doses of ≥ 2.5 mg/kg/day (≥ 175 mg/day for average weight of 70 kg).
In vitro Platelet Aggregation
Ibrutinib demonstrated inhibition of collagen-induced platelet aggregation, with IC50 values at 4.6 μM (2026 ng/mL), 0.8 μM (352 ng/mL), and 3 μM (1321 ng/mL) in blood samples from healthy donors, donors taking warfarin, and donors with severe renal dysfunction, respectively. Ibrutinib did not show meaningful inhibition of platelet aggregation for ADP, arachidonic acid, ristocetin, and TRAP-6.
Cardiac Electrophysiology
At a single dose 3 times the maximum recommended dose (1680 mg), IMBRUVICA did not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Ibrutinib exposure increases with doses up to 840 mg (1.5 times the maximum approved recommended dosage) in patients with B-cell malignancies. The mean steady-state AUC (% coefficient of variation) observed in patients at 560 mg with MCL is 865 (69%) ng∙h/mL and with MZL is 978 (82%) ng∙h/mL, and in patients at 420 mg with CLL/SLL is 708 (71%) ng∙h/mL, with WM is 707 (72%) ng∙h/mL, and with cGVHD is 1159 (50%) ng∙h/mL. Steady-state concentrations of ibrutinib without CYP3A inhibitors were achieved with an accumulation ratio of 1 to 1.6 after 1 week of multiple daily doses of 420 mg or 560 mg.
Absorption
Absolute bioavailability of ibrutinib in fasted condition was 2.9% (90% CI: 2.1, 3.9) in healthy subjects. Ibrutinib is absorbed after oral administration with a median Tmax of 1 hour to 2 hours.
Effect of Food
The administration of IMBRUVICA with a high-fat and high-calorie meal (800 calories to 1,000 calories with approximately 50% of total caloric content of the meal from fat) increased ibrutinib Cmax by 2- to 4-fold and AUC by approximately 2-fold, compared with administration of ibrutinib after overnight fasting.
In vitro studies suggest that ibrutinib is not a substrate of p-glycoprotein (P-gp) or breast cancer resistance protein (BCRP).
Distribution
Reversible binding of ibrutinib to human plasma protein in vitro was 97.3% with no concentration dependence in the range of 50 ng/mL to 1000 ng/mL. The volume of distribution (Vd) was 683 L, and the apparent volume of distribution at steady state (Vd,ss/F) was approximately 10,000 L.
Elimination
Intravenous clearance was 62 L/h in fasted conditions and 76 L/h in fed conditions. In line with the high first-pass effect, the apparent oral clearance is 2000 L/h in fasted conditions and 1000 L/h in fed conditions. The half-life of ibrutinib is 4 hours to 6 hours.
Metabolism
Metabolism is the main route of elimination for ibrutinib. It is metabolized to several metabolites primarily by cytochrome P450 (CYP) 3A and to a minor extent by CYP2D6. The active metabolite, PCI-45227, is a dihydrodiol metabolite with inhibitory activity towards BTK approximately 15 times lower than that of ibrutinib. The range of the mean metabolite to parent ratio for PCI-45227 at steady-state is 1 to 2.8.
Excretion
Ibrutinib, mainly in the form of metabolites, is eliminated primarily via feces. After a single oral administration of radiolabeled ibrutinib, 90% of radioactivity was excreted within 168 hours, with 80% excreted in the feces and less than 10% eliminated in urine. Unchanged ibrutinib accounted for 1% of the radiolabeled excreted dose in feces and none in urine, with the remainder of the excreted dose being metabolites.
Patients with Renal Impairment
Mild and moderate renal impairment (creatinine clearance [CLcr] > 25 mL/min as estimated by Cockcroft-Gault equation) had no influence on the exposure of ibrutinib. No data is available in patients with severe renal impairment (CLcr < 25 mL/min) or in patients on dialysis.
Patients with Hepatic Impairment
The AUC of ibrutinib increased 2.7-fold in subjects with mild hepatic impairment (Child-Pugh class A), 8.2-fold in subjects with moderate hepatic impairment (Child-Pugh class B) and 9.8-fold in subjects with severe hepatic impairment (Child-Pugh class C) relative to subjects with normal liver function. The Cmax of ibrutinib increased 5.2-fold in mild hepatic impairment, 8.8-fold in moderate hepatic impairment and 7-fold in severe hepatic impairment relative to subjects with normal liver function [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Effect of CYP3A Inhibitors on Ibrutinib
The coadministration of multiple doses of ketoconazole (strong CYP3A inhibitor) increased the Cmax of ibrutinib by 29-fold and AUC by 24-fold. The coadministration of multiple doses of voriconazole (strong CYP3A inhibitor) increased steady state Cmax of ibrutinib by 6.7-fold and AUC by 5.7-fold. Simulations under fed conditions suggest that posaconazole (strong CYP3A inhibitor) may increase the AUC of ibrutinib 3-fold to 10-fold.
The coadministration of multiple doses of erythromycin (moderate CYP3A inhibitor) increased steady state Cmax of ibrutinib by 3.4-fold and AUC by 3-fold.
Effect of CYP3A Inducers on Ibrutinib
The coadministration of rifampin (strong CYP3A inducer) decreased the Cmax of ibrutinib by more than 13-fold and AUC by more than 10-fold. Simulations suggest that efavirenz (moderate CYP3A inducer) may decrease the AUC of ibrutinib by 3-fold.
Effect of Ibrutinib on CYP Substrates
In vitro studies suggest that ibrutinib and PCI-45227 are unlikely to inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 or 3A at clinical doses. Both ibrutinib and PCI-45227 are unlikely to induce CYP1A2, CYP2B6 or CYP3A at clinical doses.
Effect of Ibrutinib on Substrates of Transporters
In vitro studies suggest that ibrutinib may inhibit BCRP and P-gp transport at clinical doses. The coadministration of oral P-gp or BCRP substrates with a narrow therapeutic index (e.g., digoxin, methotrexate) with IMBRUVICA may increase their concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Ibrutinib was not carcinogenic in a 6-month rasH2 mouse study at oral doses up to 2000 mg/kg/day resulting in exposures approximately 23 (males) to 37 (females) times higher than the exposure in humans at a dose of 560 mg daily [see Warnings and Precautions (5.6)].
Ibrutinib was not mutagenic in a bacterial mutagenicity (Ames) assay, was not clastogenic in a chromosome aberration assay in mammalian (CHO) cells, nor was it clastogenic in an in vivo bone marrow micronucleus assay in mice at doses up to 2000 mg/kg.
Rats were administered oral daily doses of ibrutinib for 4 weeks prior to pairing and during pairing in males and 2 weeks prior to pairing and during pairing in females. Treatment of female rats continued following pregnancy up to gestation day (GD) 7, and treatment of male rats continued until end of study. No effects on fertility or reproductive capacities were observed in male or female rats up to the maximum dose tested, 100 mg/kg/day (Human Equivalent Dose [HED] 16 mg/kg).
-
14 CLINICAL STUDIES
14.1 Mantle Cell Lymphoma
The safety and efficacy of IMBRUVICA in patients with MCL who have received at least one prior therapy were evaluated in Study PCYC-1104-CA (referred to as Study 1104) (NCT01236391), an open-label, multi-center, single-arm trial of 111 previously treated patients. The median age was 68 years (range, 40 to 84 years), 77% were male, and 92% were Caucasian. At baseline, 89% of patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 42 months, and median number of prior treatments was 3 (range, 1 to 5 treatments), including 11% with prior stem cell transplantation. At baseline, 39% of subjects had at least one tumor ≥ 5 cm, 49% had bone marrow involvement, and 54% had extranodal involvement at screening.
IMBRUVICA was administered orally at 560 mg once daily until disease progression or unacceptable toxicity. Tumor response was assessed according to the revised International Working Group (IWG) for non-Hodgkin's lymphoma (NHL) criteria. The primary endpoint in this study was investigator-assessed overall response rate (ORR). Responses to IMBRUVICA are shown in Table 18.
Table 18: Overall Response Rate (ORR) and Duration of Response (DOR) Based on Investigator Assessment in Patients with MCL in Study 1104 Total (N=111) CI = confidence interval; CR = complete response; PR = partial response; NE = not evaluable ORR (%) 65.8 95% CI (%) (56.2, 74.5) CR (%) 17.1 PR (%) 48.6 Median DOR months (95% CI) 17.5 (15.8, NE) An Independent Review Committee (IRC) performed independent reading and interpretation of imaging scans. The IRC review demonstrated an ORR of 69%.
The median time to response was 1.9 months.
Lymphocytosis
Upon initiation of IMBRUVICA, a temporary increase in lymphocyte counts (i.e., ≥ 50% increase from baseline and above absolute lymphocyte count of 5,000/mcL) occurred in 33% of patients in the MCL study. The onset of isolated lymphocytosis occurs during the first few weeks of IMBRUVICA therapy and resolves by a median of 8 weeks.
14.2 Chronic Lymphocytic Leukemia / Small Lymphocytic Lymphoma
The safety and efficacy of IMBRUVICA in patients with CLL/SLL were demonstrated in one uncontrolled trial and four randomized, controlled trials.
Study 1102
Study PCYC-1102-CA (referred to as Study 1102) (NCT01105247), an open-label, multi-center trial, was conducted in 48 previously treated CLL patients. The median age was 67 years (range, 37 to 82 years), 71% were male, and 94% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 80 months and the median number of prior treatments was 4 (range, 1 to 12 treatments). At baseline, 46% of subjects had at least one tumor ≥ 5 cm.
IMBRUVICA was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The ORR and DOR were assessed using a modified version of the International Workshop on CLL Criteria by an Independent Review Committee. The ORR was 58.3% (95% CI: 43.2%, 72.4%), all partial responses. None of the patients achieved a complete response. The DOR ranged from 5.6 to 24.2+ months. The median DOR was not reached.
RESONATE
The RESONATE study (A Randomized, Multicenter, Open-label, Phase 3 Study of the Bruton's Tyrosine Kinase (BTK) Inhibitor Ibrutinib versus Ofatumumab in Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma) (NCT01578707) was conducted in patients with previously treated CLL or SLL. Patients (n=391) were randomized 1:1 to receive either IMBRUVICA 420 mg daily until disease progression, or unacceptable toxicity or ofatumumab at an initial dose of 300 mg, followed one week later by a dose of 2000 mg weekly for 7 doses and then every 4 weeks for 4 additional doses. Fifty-seven patients randomized to ofatumumab crossed over following progression to receive IMBRUVICA. The median age was 67 years (range, 30 to 88 years), 68% were male, and 90% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The trial enrolled 373 patients with CLL and 18 patients with SLL. The median time since diagnosis was 91 months and the median number of prior treatments was 2 (range, 1 to 13 treatments). At baseline, 58% of patients had at least one tumor ≥ 5 cm. Thirty-two percent of patients had 17p deletion.
Efficacy results for RESONATE are shown in Table 19 and the Kaplan-Meier curves for PFS, assessed by an IRC according to IWCLL criteria, and OS are shown in Figures 1 and 2, respectively.
Table 19: Efficacy Results in Patients with CLL/SLL in RESONATE Endpoint IMBRUVICA
N=195Ofatumumab
N=196a Median OS not evaluable for either arm
b IRC evaluated. All partial responses achieved; none of the patients achieved a complete response.
CI = confidence interval; HR = hazard ratio; NE = not evaluableProgression Free Survivalb Number of events (%) 35 (17.9) 111 (56.6) Disease progression 26 93 Death events 9 18 Median (95% CI), months NE 8.1 (7.2, 8.3) HR (95% CI) 0.22 (0.15, 0.32) Overall Survivala Number of deaths (%) 16 (8.2) 33 (16.8) HR (95% CI) 0.43 (0.24, 0.79) Overall Response Rateb 42.6% 4.1% Figure 1: Kaplan-Meier Curve of Progression Free Survival (ITT Population) in Patients with CLL/SLL in RESONATE
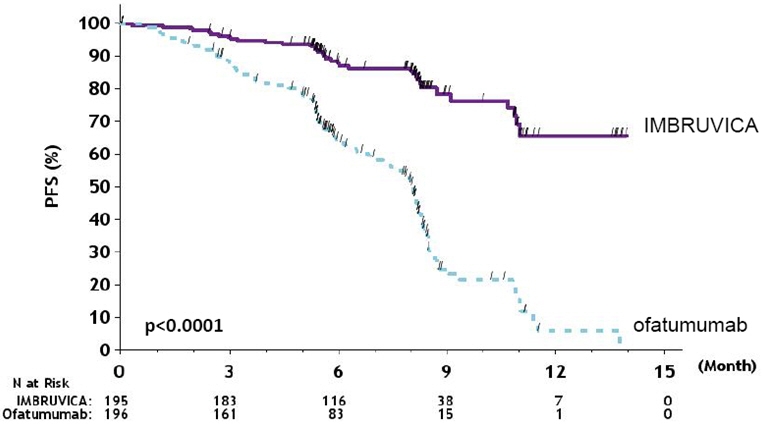
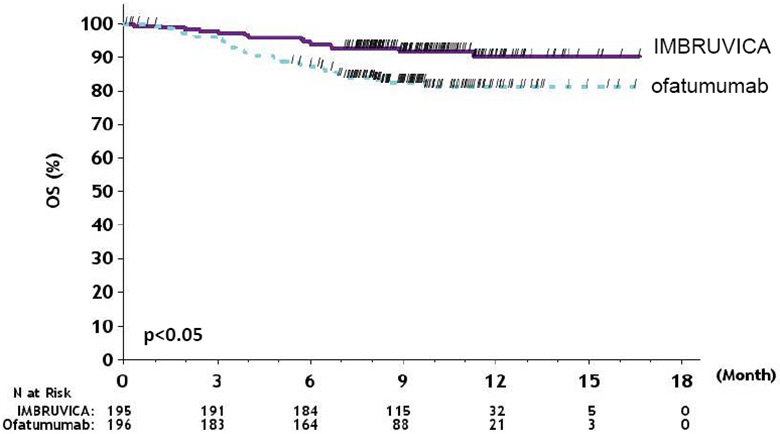
Figure 2: Kaplan-Meier Curve of Overall Survival (ITT Population) in Patients with CLL/SLL in RESONATE
63-Month Follow-Up
With an overall follow-up of 63 months, the median investigator-assessed PFS per IWCLL criteria was 44.1 months [95% CI (38.5, 56.9)] in the IMBRUVICA arm and 8.1 months [95% CI (7.8, 8.3)] in the ofatumumab arm, respectively. Overall response rate as assessed by investigators was 87.2% in the IMBRUVICA arm versus 22.4% in the ofatumumab arm.
CLL/SLL with 17p deletion (del 17p CLL/SLL) in RESONATE
RESONATE included 127 patients with del 17p CLL/SLL. The median age was 67 years (range, 30 to 84 years), 62% were male, and 88% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. PFS and ORR were assessed by an IRC. Efficacy results for del 17p CLL/SLL are shown in Table 20.
Table 20: Efficacy Results in Patients with del 17p CLL/SLL in RESONATE Endpoint IMBRUVICA
N=63Ofatumumab
N=64a IRC evaluated. All partial responses achieved; none of the patients achieved a complete response.
CI = confidence interval; HR = hazard ratio; NE = not evaluableProgression Free Survivala Number of events (%) 16 (25.4) 38 (59.4) Disease progression 12 31 Death events 4 7 Median (95% CI), months NE 5.8 (5.3, 7.9) HR (95% CI) 0.25 (0.14, 0.45) Overall Response Ratea 47.6% 4.7% 63-Month Follow-Up
With an overall follow-up of 63 months, the median investigator-assessed PFS in patients with del 17p per IWCLL criteria was 40.6 months [95% CI (25.4, 44.6)] in the IMBRUVICA arm and 6.2 months [95% CI (4.6, 8.1)] in the ofatumumab arm, respectively. Overall response rate as assessed by investigators in patients with del 17p was 88.9% in the IMBRUVICA arm versus 18.8% in the ofatumumab arm.
RESONATE-2
The RESONATE-2 study (A Randomized, Multicenter, Open-label, Phase 3 Study of the Bruton's Tyrosine Kinase Inhibitor PCI-32765 versus Chlorambucil in Patients 65 Years or Older with Treatment-naive Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma) (NCT01722487) was conducted in patients with treatment naïve CLL or SLL who were 65 years of age or older. Patients (n = 269) were randomized 1:1 to receive either IMBRUVICA 420 mg daily until disease progression or unacceptable toxicity, or chlorambucil at a starting dose of 0.5 mg/kg on Days 1 and 15 of each 28-day cycle for a maximum of 12 cycles, with an allowance for intrapatient dose increases up to 0.8 mg/kg based on tolerability.
The median age was 73 years (range, 65 to 90 years), 63% were male, and 91% were Caucasian. Ninety one percent of patients had a baseline ECOG performance status of 0 or 1 and 9% had an ECOG performance status of 2. The trial enrolled 249 patients with CLL and 20 patients with SLL. At baseline, 20% of patients had 11q deletion. The most common reasons for initiating CLL therapy include: progressive marrow failure demonstrated by anemia and/or thrombocytopenia (38%), progressive or symptomatic lymphadenopathy (37%), progressive or symptomatic splenomegaly (30%), fatigue (27%) and night sweats (25%).
With a median follow-up of 28.1 months, there were 32 observed death events [11 (8.1%) and 21 (15.8%) in IMBRUVICA and chlorambucil treatment arms, respectively]. With 41% of patients switching from chlorambucil to IMBRUVICA, the overall survival analysis in the ITT patient population resulted in a statistically significant HR of 0.44 [95% CI (0.21, 0.92)] and 2-year survival rate estimates of 94.7% [95% CI (89.1, 97.4)] and 84.3% [95% CI (76.7, 89.6)] in the IMBRUVICA and chlorambucil arms, respectively.
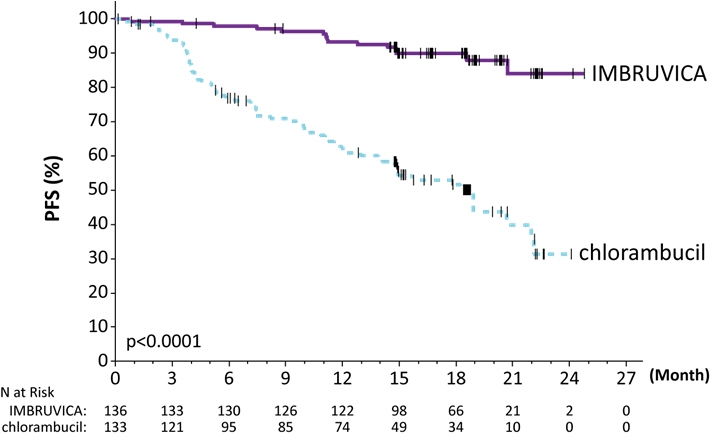
Efficacy results for RESONATE-2 are shown in Table 21 and the Kaplan-Meier curve for PFS, assessed by an IRC according to IWCLL criteria is shown in Figure 3.
Table 21: Efficacy Results in Patients with CLL/SLL in RESONATE-2 Endpoint IMBRUVICA
N=136Chlorambucil
N=133a IRC evaluated; Five subjects (3.7%) in the IMBRUVICA arm and two subjects (1.5%) in the Chlorambucil arm achieved complete response
b HR = hazard ratio; NE = not evaluableProgression Free Survivala Number of events (%) 15 (11.0) 64 (48.1) Disease progression 12 57 Death events 3 7 Median (95% CI), months NE 18.9 (14.1, 22.0) HRb (95% CI) 0.16 (0.09, 0.28) Overall Response Ratea (CR + PR) 82.4% 35.3% P-value <0.0001 Figure 3: Kaplan-Meier Curve of Progression-Free Survival (ITT Population) in Patients with CLL/SLL in RESONATE-2
HELIOS
The HELIOS study (Randomized, Double-blind, Placebo-controlled Phase 3 Study of Ibrutinib, a Bruton's Tyrosine Kinase (BTK) Inhibitor, in Combination with Bendamustine and Rituximab (BR) in Subjects with Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma) (NCT01611090) was conducted in patients with previously treated CLL or SLL. Patients (n = 578) were randomized 1:1 to receive either IMBRUVICA 420 mg daily or placebo in combination with BR until disease progression, or unacceptable toxicity. All patients received BR for a maximum of six 28-day cycles. Bendamustine was dosed at 70 mg/m2 infused IV over 30 minutes on Cycle 1, Days 2 and 3, and on Cycles 2-6, Days 1 and 2 for up to 6 cycles, and all patients had a CrCl ≥ 40 mL/min at baseline. Rituximab was administered at a dose of 375 mg/m2 in the first cycle, Day 1, and 500 mg/m2 Cycles 2 through 6, Day 1.
The median age was 64 years (range, 31 to 86 years), 66% were male, and 91% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 5.9 years and the median number of prior treatments was 2 (range, 1 to 11 treatments). At baseline, 56% of patients had at least one tumor ≥ 5 cm and 26% presented with del11q.
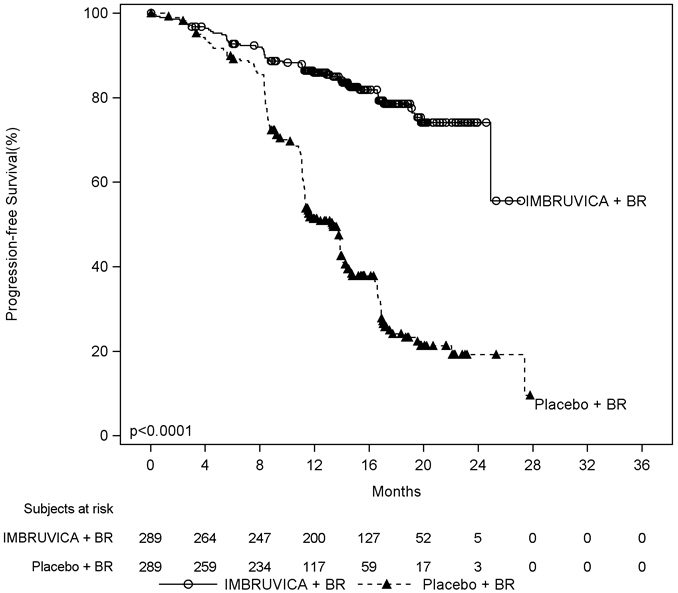
Efficacy results for HELIOS are shown in Table 22 and the Kaplan-Meier curves for PFS are shown in Figure 4.
Table 22: Efficacy Results in Patients with CLL/SLL in HELIOS Endpoint IMBRUVICA + BR
N=289Placebo + BR
N=289a IRC evaluated, twenty-four subjects (8.3%) in the IMBRUVICA + BR arm and six subjects (2.1%) in the placebo + BR arm achieved complete response
BR = bendamustine and rituximab; CI = confidence interval; HR = hazard ratio; NE = not evaluableProgression Free Survivala Number of events (%) 56 (19.4) 183 (63.3) Median (95% CI), months NE 13.3 (11.3, 13.9) HR (95% CI) 0.20 (0.15, 0.28) Overall Response Ratea 82.7% 67.8% Figure 4: Kaplan-Meier Curve of Progression-Free Survival (ITT Population) in Patients with CLL/SLL in HELIOS
iLLUMINATE
The iLLUMINATE study (a multi-center study of ibrutinib in combination with obinutuzumab versus chlorambucil in combination with obinutuzumab) (NCT02264574) was conducted in patients with treatment naïve CLL or SLL. Patients were 65 years of age or older or < 65 years of age with coexisting medical conditions, reduced renal function as measured by creatinine clearance < 70 mL/min, or presence of del 17p/TP53 mutation. Patients (n = 229) were randomized 1:1 to receive either IMBRUVICA 420 mg daily until disease progression or unacceptable toxicity or chlorambucil at a dose of 0.5 mg/kg on Days 1 and 15 of each 28-day cycle for 6 cycles. In both arms, patients received 1,000 mg of obinutuzumab on Days 1, 8, and 15 of the first cycle, followed by treatment on the first day of 5 subsequent cycles (total of 6 cycles, 28 days each). The first dose of obinutuzumab was divided between Day 1 (100 mg) and Day 2 (900 mg).
The median age was 71 years (range, 40 to 87 years), 64% were male, and 96% were Caucasian. All patients had a baseline ECOG performance status of 0 (48%) or 1-2 (52%). The trial enrolled 214 patients with CLL and 15 patients with SLL. At baseline, 65% of patients presented with CLL/SLL with high risk factors (del 17p/TP53 mutation [18%], del 11q [15%], or unmutated immunoglobulin heavy-chain variable region (unmutated IGHV) [54%]). The most common reasons for initiating CLL therapy included: lymphadenopathy (38%), night sweats (34%), progressive marrow failure (31%), fatigue (29%), splenomegaly (25%), and progressive lymphocytosis (21%).
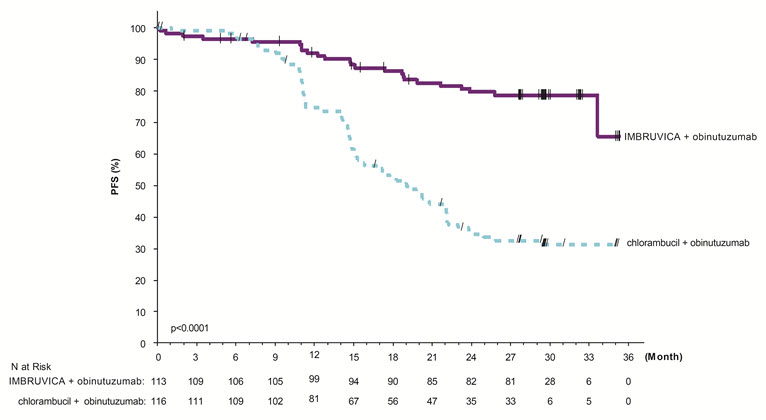
With a median follow-up time on study of 31 months, efficacy results for iLLUMINATE assessed by an IRC according to IWCLL criteria are shown in Table 23, and the Kaplan-Meier curve for PFS is shown in Figure 5.
Table 23: Efficacy Results in Patients with CLL/SLL in iLLUMINATE Endpoint IMBRUVICA + Obinutuzumab
N=113Chlorambucil + Obinutuzumab
N=116HR = hazard ratio; NE = not evaluable - * IRC-evaluated
- † P-value is from unstratified log-rank test
- ‡ Includes 1 patient in the IMBRUVICA + obinutuzumab arm with a complete response with incomplete marrow recovery (CRi)
- § PR = nPR +PR
Progression Free Survival* Number of events (%) 24 (21) 74 (64) Disease progression 11 64 Death events 13 10 Median (95% CI), months NE 19.0 (15.1, 22.1) HR (95% CI) 0.23 (0.15, 0.37) P-value† <0.0001 Overall Response Rate (%)* 88.5 73.3 CR‡ (%) 19.5 7.8 PR§ (%) 69.0 65.5 Figure 5: Kaplan-Meier Curve of Progression-Free Survival (ITT Population) in Patients with CLL/SLL in iLLUMINATE
In the high risk CLL/SLL population (del 17p/TP53 mutation, del 11q, or unmutated IGHV), the PFS HR was 0.15 [95% CI (0.09, 0.27)].
Lymphocytosis
Upon initiation of single-agent IMBRUVICA, an increase in lymphocyte counts (i.e., ≥ 50% increase from baseline and above absolute lymphocyte count of 5,000/mcL) occurred in 66% of patients in the CLL studies. The onset of isolated lymphocytosis occurs during the first month of IMBRUVICA therapy and resolves by a median of 14 weeks (range, 0.1 to 104 weeks). When IMBRUVICA was administered in combination, lymphocytosis was 7% with IMBRUVICA + BR versus 6% with placebo + BR and 7% with IMBRUVICA + obinutuzumab versus 1% with chlorambucil + obinutuzumab.
14.3 Waldenström's Macroglobulinemia
The safety and efficacy of IMBRUVICA in patients with WM were demonstrated in two single-arm trials and one randomized, controlled trial.
Study 1118 and INNOVATE Monotherapy Arm
The safety and efficacy of IMBRUVICA in WM were evaluated in Study PCYC-1118E (referred to as Study 1118) (NCT01614821), an open-label, multi-center, single-arm trial of 63 previously treated patients. The median age was 63 years (range, 44 to 86 years), 76% were male, and 95% were Caucasian. All patients had a baseline ECOG performance status of 0 or 1. The median time since diagnosis was 74 months, and the median number of prior treatments was 2 (range, 1 to 11 treatments). At baseline, the median serum IgM value was 3.5 g/dL (range, 0.7 to 8.4 g/dL).
IMBRUVICA was administered orally at 420 mg once daily until disease progression or unacceptable toxicity. The responses were assessed by investigators and an IRC using criteria adopted from the International Workshop of Waldenström's Macroglobulinemia. Responses, defined as partial response or better, per IRC are shown in Table 24.
Table 24: Response Rate and Duration of Response (DOR) Based on IRC Assessment in Patients with WM in Study 1118 Total (N=63) CI = confidence interval; NE = not evaluable Response rate (CR+VGPR+PR), (%) 61.9 95% CI (%) (48.8, 73.9) Complete Response (CR) 0 Very Good Partial Response (VGPR), (%) 11.1 Partial Response (PR), (%) 50.8 Median duration of response, months (range) NE (2.8+, 18.8+) The median time to response was 1.2 months (range, 0.7-13.4 months).
The INNOVATE monotherapy arm included 31 patients with previously treated WM who failed prior rituximab-containing therapy and received single-agent IMBRUVICA. The median age was 67 years (range, 47 to 90 years). Eighty-one percent of patients had a baseline ECOG performance status of 0 or 1, and 19% had a baseline ECOG performance status of 2. The median number of prior treatments was 4 (range, 1 to 7 treatments). The response rate observed in the INNOVATE monotherapy arm was 71% (0% CR, 29% VGPR, 42% PR). With a median follow-up time on study of 34 months (range, 8.6+ to 37.7 months), the median duration of response has not been reached.
INNOVATE
The INNOVATE study (A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Study of Ibrutinib or Placebo in Combination with Rituximab in Subjects with Waldenström's Macroglobulinemia) (NCT02165397) was conducted in treatment naïve or previously treated patients with WM. Patients (n = 150) were randomized 1:1 to receive either IMBRUVICA 420 mg daily or placebo in combination with rituximab until disease progression or unacceptable toxicity. Rituximab was administered weekly at a dose of 375 mg/m2 for 4 consecutive weeks (weeks 1-4) followed by a second course of weekly rituximab for 4 consecutive weeks (weeks 17-20). The major efficacy outcome measure is progression-free survival (PFS) assessed by an IRC with additional efficacy measure of response rate.
The median age was 69 years (range, 36 to 89 years), 66% were male, and 79% were Caucasian. Ninety-three percent of patients had a baseline ECOG performance status of 0 or 1, and 7% of patients had a baseline ECOG performance status of 2. Forty-five percent of patients were treatment naïve, and 55% of patients were previously treated. Among previously treated patients, the median number of prior treatments was 2 (range, 1 to 6 treatments). At baseline, the median serum IgM value was 3.2 g/dL (range, 0.6 to 8.3 g/dL), and MYD88 L265P mutations were present in 77% of patients, absent in 13% of patients, and 9% of patients were not evaluable for mutation status.
Efficacy results for INNOVATE as assessed by an IRC are shown in Table 25, and the Kaplan-Meier curves for PFS are shown in Figure 6.
Table 25: Efficacy Results in Patients with WM in INNOVATE Endpoint IMBRUVICA + R
N=75Placebo + R
N=75CI = confidence interval; HR = hazard ratio; NE = not evaluable; R = rituximab - * P-value is from log-rank test stratified by WM IPSS (low, med, high) and number of prior systemic treatment regimens (0, ≥1)
- † P-value associated with response rate was <0.0001.
Median follow-up time on study = 26.5 monthsProgression Free Survival Number of events (%) 14 (19) 42 (56) Median (95% CI), months NE 20.3 (13.7, 27.6) HR (95% CI) 0.20 (0.11, 0.38) P-value* <0.0001 Response Rate (CR+VGPR+PR)† 72% 32% 95% CI (0.62, 0.82) (0.21, 0.43) Complete Response (CR) 3% 1% Very Good Partial Response (VGPR) 23% 4% Partial Response (PR) 47% 27% Median duration of response, months (range) NE (1.9+, 36.4+) 21.2 (4.6, 25.8) Figure 6: Kaplan-Meier Curve of Progression-Free Survival (ITT Population) in Patients with WM in INNOVATE
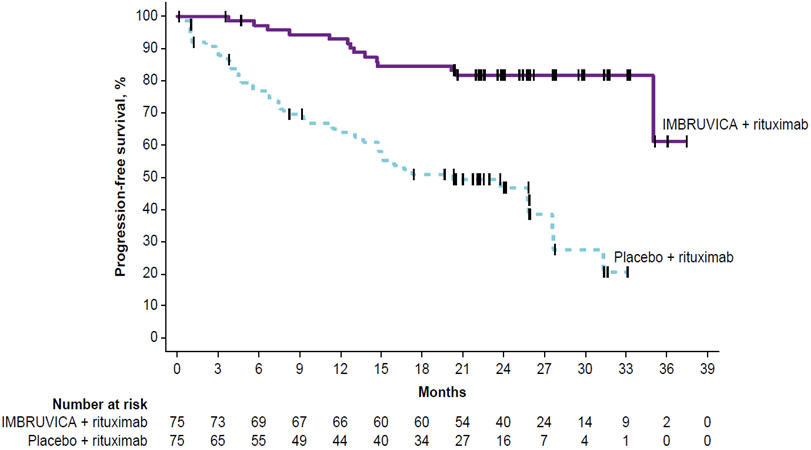
An exploratory analysis demonstrated a sustained hemoglobin improvement (defined as increase of ≥ 2 g/dL over baseline for at least 8 weeks without blood transfusions or growth factor support) in 65% of patients in the IMBRUVICA + R group and 39% of patients in the placebo + R group.
14.4 Marginal Zone Lymphoma
The safety and efficacy of IMBRUVICA in MZL were evaluated in Study PCYC-1121-CA (referred to as Study 1121) (NCT01980628), an open-label, multi-center, single-arm trial of patients who received at least one prior therapy. The efficacy analysis included 63 patients with 3 sub-types of MZL: mucosa-associated lymphoid tissue (MALT; N=32), nodal (N=17), and splenic (N=14). The median age was 66 years (range, 30 to 92 years), 59% were female, and 84% were Caucasian. Ninety two percent of patients had a baseline ECOG performance status of 0 or 1 and 8% had ECOG performance status 2. The median time since diagnosis was 3.8 years, and the median number of prior treatments was 2 (range, 1 to 9 treatments).
IMBRUVICA was administered orally at 560 mg once daily until disease progression or unacceptable toxicity. The responses were assessed by investigators and an IRC using criteria adopted from the International Working Group criteria for malignant lymphoma. Responses per IRC are shown in Table 26.
Table 26: Overall Response Rate (ORR) and Duration of Response (DOR) Based on IRC Assessment in Patients with MZL in Study 1121 Total (N=63) CI = confidence interval; NE = not evaluable
Median follow-up time on study = 19.4 monthsResponse rate (CR + PR), (%) 46.0% 95% CI (%) (33.4, 59.1) Complete Response (CR), (%) 3.2 Partial Response (PR), (%) 42.9 Median duration of response, months (range) NE (16.7, NE) The median time to response was 4.5 months (range, 2.3 to 16.4 months). Overall response rates were 46.9%, 41.2%, and 50.0% for the 3 MZL sub-types (MALT, nodal, splenic), respectively.
14.5 Chronic Graft versus Host Disease
The safety and efficacy of IMBRUVICA in cGVHD were evaluated in Study PCYC-1129-CA (referred to as Study 1129) (NCT02195869), an open-label, multi-center, single-arm trial of 42 patients with cGVHD after failure of first line corticosteroid therapy and requiring additional therapy. The median age was 56 years (range, 19 to 74 years), 52% were male, and 93% were Caucasian. The most common underlying malignancies leading to transplantation were acute lymphocytic leukemia, acute myeloid leukemia, and CLL. The median time since cGVHD diagnosis was 14 months, the median number of prior cGVHD treatments was 2 (range, 1 to 3 treatments), and 60% of patients had a Karnofsky performance score of ≤ 80. The majority of patients (88 %) had at least 2 organs involved at baseline, with the most commonly involved organs being mouth (86%), skin (81%), and gastrointestinal tract (33%). The median daily corticosteroid dose (prednisone or prednisone equivalent) at baseline was 0.3 mg/kg/day, and 52% of patients were receiving ongoing immunosuppressants in addition to systemic corticosteroids at baseline. Prophylaxis for infections were managed per institutional guidelines with 79% of patients receiving combinations of sulfonamides and trimethoprim and 64% receiving triazole derivatives.
IMBRUVICA was administered orally at 420 mg once daily. The responses were assessed by investigators using the 2005 National Institute of Health (NIH) Consensus Panel Response Criteria with two modifications to align with the updated 2014 NIH Consensus Panel Response Criteria. Efficacy results are shown in Table 27.
Table 27: Best Overall Response Rate (ORR) and Sustained Response Rate Based on Investigator Assessmenta in Patients with cGVHD in Study 1129 Total (N=42) CI = confidence interval
a Investigator assessment based on the 2005 NIH Response Criteria with two modifications (added "not evaluable" for organs with non-cGVHD abnormalities, and organ score change from 0 to 1 was not considered disease progression)
b Sustained response rate is defined as the proportion of patients who achieved a CR or PR that was sustained for at least 20 weeks.ORR 28 (67%) 95% CI (51%, 80%) Complete Response (CR) 9 (21%) Partial Response (PR) 19 (45%) Sustained response rateb 20 (48%) The median time to response coinciding with the first scheduled response assessment was 12.3 weeks (range, 4.1 to 42.1 weeks). Responses were seen across all organs involved for cGVHD (skin, mouth, gastrointestinal tract, and liver).
ORR results were supported by exploratory analyses of patient-reported symptom bother which showed at least a 7-point decrease in Lee Symptom Scale overall summary score in 24% (10/42) of patients on at least 2 consecutive visits.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
The 70 mg capsules are supplied as yellow opaque capsules, marked with "ibr 70 mg" in black ink, and are available in white HDPE bottles with a child-resistant closure:
- 28 capsules per bottle: NDC: 57962-070-28
The 140 mg capsules are supplied as white opaque capsules, marked with "ibr 140 mg" in black ink, and are available in white HDPE bottles with a child-resistant closure:
- 90 capsules per bottle: NDC: 57962-140-09
- 120 capsules per bottle: NDC: 57962-140-12
Store bottles at room temperature 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F to 86°F). Retain in original package until dispensing.
The IMBRUVICA (ibrutinib) tablets are supplied in 4 strengths in the following packaging configurations:
- 140 mg tablets: Yellow green to green round tablets debossed with "ibr" on one side and "140" on the other side. Carton of one folded blister card containing two 14-count blister strips for a total of 28 tablets: NDC: 57962-014-28
- 280 mg tablets: Purple oblong tablets debossed with "ibr" on one side and "280" on the other side. Carton of one folded blister card containing two 14-count blister strips for a total of 28 tablets: NDC: 57962-280-28
- 420 mg tablets: Yellow green to green oblong tablets debossed with "ibr" on one side and "420" on the other side. Carton of one folded blister card containing two 14-count blister strips for a total of 28 tablets: NDC: 57962-420-28
- 560 mg tablets: Yellow to orange oblong tablets debossed with "ibr" on one side and "560" on the other side. Carton of one folded blister card containing two 14-count blister strips for a total of 28 tablets: NDC: 57962-560-28
Store tablets in original packaging at room temperature 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C and 30°C (59°F to 86°F).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
-
Hemorrhage:
Inform patients of the possibility of bleeding, and to report any signs or symptoms (severe headache, blood in stools or urine, prolonged or uncontrolled bleeding). Inform the patient that IMBRUVICA may need to be interrupted for medical or dental procedures [see Warnings and Precautions (5.1)]. -
Infections:
Inform patients of the possibility of serious infection, and to report any signs or symptoms (fever, chills, weakness, confusion) suggestive of infection [see Warnings and Precautions (5.2)]. -
Cardiac Arrhythmias:
Counsel patients to report any signs of palpitations, lightheadedness, dizziness, fainting, shortness of breath, and chest discomfort [see Warnings and Precautions (5.4)]. -
Hypertension:
Inform patients that high blood pressure has occurred in patients taking IMBRUVICA, which may require treatment with anti-hypertensive therapy [see Warnings and Precautions (5.5)]. -
Second primary malignancies:
Inform patients that other malignancies have occurred in patients who have been treated with IMBRUVICA, including skin cancers and other carcinomas [see Warnings and Precautions (5.6)]. -
Tumor lysis syndrome:
Inform patients of the potential risk of tumor lysis syndrome and to report any signs and symptoms associated with this event to their healthcare provider for evaluation [see Warnings and Precautions (5.7)]. -
Embryo-fetal toxicity:
Advise women of the potential hazard to a fetus and to avoid becoming pregnant during treatment and for 1 month after the last dose of IMBRUVICA [see Warnings and Precautions (5.8)]. - Inform patients to take IMBRUVICA orally once daily according to their physician's instructions and that the oral dosage (capsules or tablets) should be swallowed whole with a glass of water without opening, breaking or chewing the capsules or cutting, crushing or chewing the tablets approximately the same time each day [see Dosage and Administration (2.1)].
- Advise patients that in the event of a missed daily dose of IMBRUVICA, it should be taken as soon as possible on the same day with a return to the normal schedule the following day. Patients should not take extra doses to make up the missed dose [see Dosage and Administration (2.6)].
- Advise patients of the common side effects associated with IMBRUVICA [see Adverse Reactions (6)]. Direct the patient to a complete list of adverse drug reactions in PATIENT INFORMATION.
- Advise patients to inform their health care providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
- Advise patients that they may experience loose stools or diarrhea and should contact their doctor if their diarrhea persists. Advise patients to maintain adequate hydration [see Adverse Reactions (6.1)].
-
Hemorrhage:
-
SPL UNCLASSIFIED SECTION
Active ingredient made in China.
Distributed and Marketed by:
Pharmacyclics LLC
Sunnyvale, CA USA 94085
and
Marketed by:
Janssen Biotech, Inc.
Horsham, PA USA 19044Patent http://www.imbruvica.com
IMBRUVICA® is a registered trademark owned by Pharmacyclics LLC© Pharmacyclics LLC 2019
© Janssen Biotech, Inc. 2019 -
PATIENT PACKAGE INSERT
PATIENT INFORMATION IMBRUVICA (im-BRU-vih-kuh)
(ibrutinib)
capsulesIMBRUVICA (im-BRU-vih-kuh)
(ibrutinib)
tabletsWhat is IMBRUVICA?
IMBRUVICA is a prescription medicine used to treat adults with:- Mantle cell lymphoma (MCL) who have received at least one prior treatment
- Chronic lymphocytic leukemia (CLL)/Small lymphocytic lymphoma (SLL)
- Chronic lymphocytic leukemia (CLL)/Small lymphocytic lymphoma (SLL) with 17p deletion
- Waldenström's macroglobulinemia (WM)
- Marginal zone lymphoma (MZL) who require a medicine by mouth or injection (systemic therapy) and have received a certain type of prior treatment
- Chronic graft versus host disease (cGVHD) after failure of 1 or more lines of systemic therapy
Before taking IMBRUVICA, tell your healthcare provider about all of your medical conditions, including if you: - have had recent surgery or plan to have surgery. Your healthcare provider may stop IMBRUVICA for any planned medical, surgical, or dental procedure.
- have bleeding problems
- have or had heart rhythm problems, smoke, or have a medical condition that increases your risk of heart disease, such as high blood pressure, high cholesterol, or diabetes
- have an infection
- have liver problems
- are pregnant or plan to become pregnant. IMBRUVICA can harm your unborn baby. If you are able to become pregnant, your healthcare provider will do a pregnancy test before starting treatment with IMBRUVICA.
- Females should not become pregnant during treatment and for 1 month after the last dose of IMBRUVICA.
- Males should avoid getting female partners pregnant during treatment and for 1 month after the last dose of IMBRUVICA.
- are breastfeeding or plan to breastfeed. You and your healthcare provider should decide if you will take IMBRUVICA or breastfeed.
How should I take IMBRUVICA? - Take IMBRUVICA exactly as your healthcare provider tells you to take it.
- Take IMBRUVICA 1 time a day.
- Swallow IMBRUVICA capsules and tablets whole with a glass of water.
- Do not open, break, or chew IMBRUVICA capsules.
- Do not cut, crush, or chew IMBRUVICA tablets.
- Take IMBRUVICA at about the same time each day.
- If you miss a dose of IMBRUVICA take it as soon as you remember on the same day. Take your next dose of IMBRUVICA at your regular time on the next day. Do not take extra doses of IMBRUVICA to make up for a missed dose.
- If you take too much IMBRUVICA call your healthcare provider or go to the nearest hospital emergency room right away.
What should I avoid while taking IMBRUVICA?
You should not drink grapefruit juice, eat grapefruit, or eat Seville oranges (often used in marmalades) during treatment with IMBRUVICA. These products may increase the amount of IMBRUVICA in your blood.What are the possible side effects of IMBRUVICA?
IMBRUVICA may cause serious side effects, including:- Bleeding problems (hemorrhage) are common during treatment with IMBRUVICA, and can also be serious and may lead to death. Your risk of bleeding may increase if you are also taking a blood thinner medicine. Tell your healthcare provider if you have any signs of bleeding, including:
- blood in your stools or black stools (looks like tar)
- pink or brown urine
- unexpected bleeding, or bleeding that is severe or
that you cannot control - vomit blood or vomit looks like coffee grounds
- cough up blood or blood clots
- increased bruising
- dizziness
- weakness
- confusion
- change in your speech
- headache that lasts a long time
- Infections can happen during treatment with IMBRUVICA. These infections can be serious and may lead to death. Tell your healthcare provider right away if you have fever, chills, weakness, confusion, or other signs or symptoms of an infection during treatment with IMBRUVICA.
- Decrease in blood cell counts. Decreased blood counts (white blood cells, platelets, and red blood cells) are common with IMBRUVICA, but can also be severe. Your healthcare provider should do monthly blood tests to check your blood counts.
- Heart rhythm problems (ventricular arrhythmias, atrial fibrillation and atrial flutter). Serious heart rhythm problems and death have happened in people treated with IMBRUVICA, especially in people who have an increased risk for heart disease, have an infection, or who have had heart rhythm problems in the past. Tell your healthcare provider if you get any symptoms of heart rhythm problems, such as feeling as if your heart is beating fast and irregular, lightheadedness, dizziness, shortness of breath, chest discomfort, or you faint. If you develop any of these symptoms, your healthcare provider may do a test to check your heart (ECG) and may change your IMBRUVICA dose.
- High blood pressure (hypertension). New or worsening high blood pressure has happened in people treated with IMBRUVICA. Your healthcare provider may start you on blood pressure medicine or change current medicines to treat your blood pressure.
- Second primary cancers. New cancers have happened during treatment with IMBRUVICA, including cancers of the skin or other organs.
- Tumor lysis syndrome (TLS). TLS is caused by the fast breakdown of cancer cells. TLS can cause kidney failure and the need for dialysis treatment, abnormal heart rhythm, seizure, and sometimes death. Your healthcare provider may do blood tests to check you for TLS.
- diarrhea
- muscle and bone pain
- rash
- bruising
- nausea
- tiredness
- fever
The most common side effects of IMBRUVICA in adults with cGVHD include: - tiredness
- bruising
- diarrhea
- mouth sores (stomatitis)
- muscle spasms
- nausea
- pneumonia
Diarrhea is a common side effect in people who take IMBRUVICA. Drink plenty of fluids during treatment with IMBRUVICA to help reduce your risk of losing too much fluid (dehydration) due to diarrhea. Tell your healthcare provider if you have diarrhea that does not go away.
These are not all the possible side effects of IMBRUVICA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store IMBRUVICA? - Store IMBRUVICA capsules and tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep IMBRUVICA capsules in the original container with the lid tightly closed.
- Keep IMBRUVICA tablets in the original carton.
General information about the safe and effective use of IMBRUVICA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use IMBRUVICA for a condition for which it was not prescribed. Do not give IMBRUVICA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about IMBRUVICA that is written for health professionals.What are the ingredients in IMBRUVICA?
Active ingredient: ibrutinib
Inactive ingredients:
IMBRUVICA capsules: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The 70 mg capsule shell contains gelatin, titanium dioxide, yellow iron oxide, and black ink. The 140 mg capsule shell contains gelatin, titanium dioxide, and black ink.
IMBRUVICA tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The film coating for each tablet contains ferrosoferric oxide (140 mg, 280 mg, and 420 mg tablets), polyvinyl alcohol, polyethylene glycol, red iron oxide (280 mg and 560 mg tablets), talc, titanium dioxide, and yellow iron oxide (140 mg, 420 mg, and 560 mg tablets).
Distributed and Marketed by: Pharmacyclics LLC Sunnyvale, CA USA 94085
Marketed by: Janssen Biotech, Inc. Horsham, PA USA 19044.
© Pharmacyclics LLC 2019
© Janssen Biotech, Inc. 2019
For more information, go to www.imbruvica.com or call 1-877-877-3536.This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 01/2019 -
PRINCIPAL DISPLAY PANEL - 28 Capsule Bottle Carton
NDC: 57962-070-28
Imbruvica®
(ibrutinib) capsules70 mg
Each capsule contains: ibrutinib 70 mg
Swallow capsules whole with water28 Capsules
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.

-
PRINCIPAL DISPLAY PANEL - 28 Capsule Bottle Label
NDC: 57962-070-28
Imbruvica®
(ibrutinib) capsules70 mg
Each capsule contains: ibrutinib 70 mg
Swallow capsules whole with water28 Capsules
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.

-
PRINCIPAL DISPLAY PANEL - 90 Capsule Bottle Carton
NDC: 57962-140-09
Imbruvica®
(ibrutinib) capsules140 mg
Each capsule contains: ibrutinib 140 mg
Swallow capsules whole with water90 Capsules
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
-
PRINCIPAL DISPLAY PANEL - 120 Capsule Bottle Carton
NDC: 57962-140-12
Imbruvica®
(ibrutinib) capsules140 mg
Each capsule contains: ibrutinib 140 mg
Swallow capsules whole with water120 Capsules
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
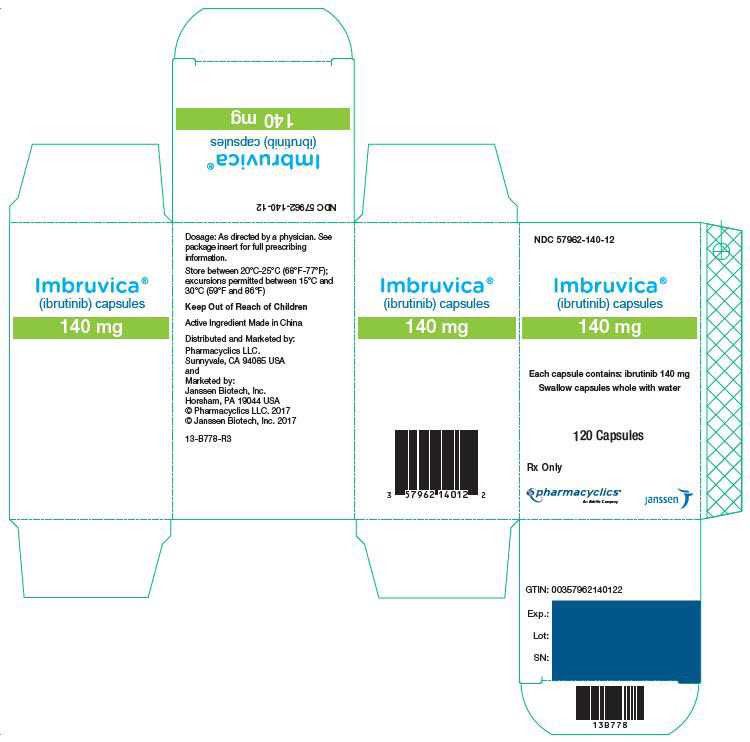
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Carton
NDC: 57962-014-28
Imbruvica®
(ibrutinib) tablets140 mg
Each tablet contains: ibrutinib 140 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
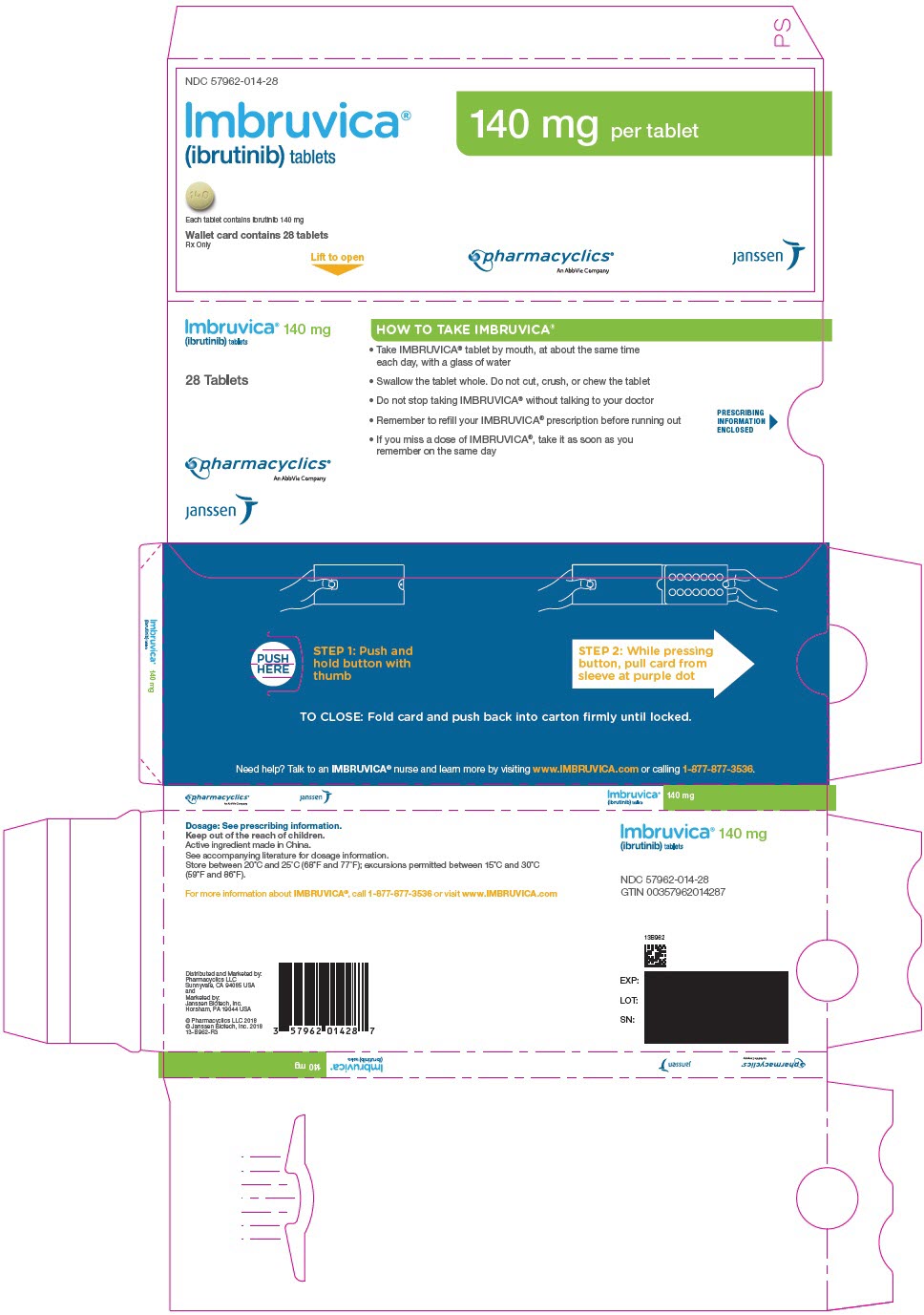
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Front)
NDC: 57962-014-28
Imbruvica®
(ibrutinib) tablets140 mg
Each tablet contains: ibrutinib 140 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
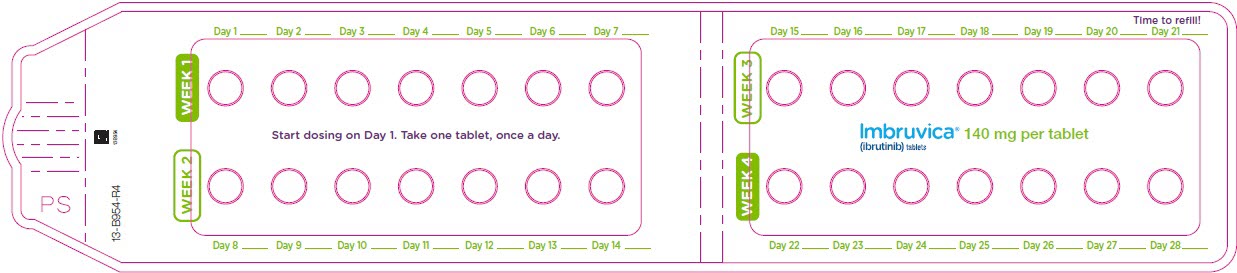
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Back)
NDC: 57962-014-28
Imbruvica®
(ibrutinib) tablets140 mg
Each tablet contains: ibrutinib 140 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
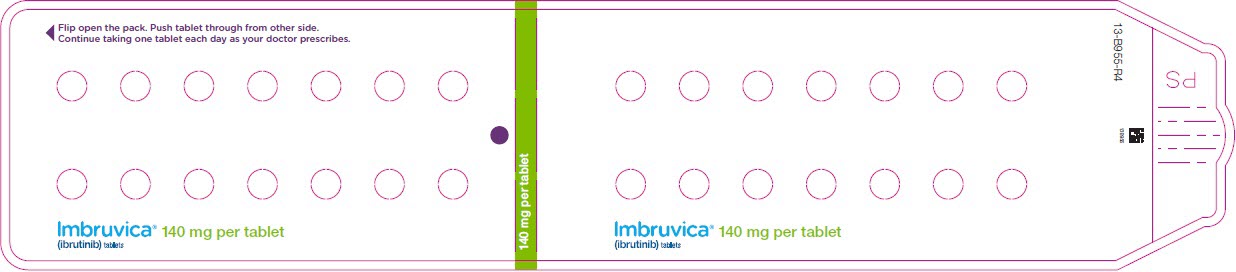
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Carton
NDC: 57962-280-28
Imbruvica®
(ibrutinib) tablets280 mg
Each tablet contains: ibrutinib 280 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.

-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Front)
NDC: 57962-280-28
Imbruvica®
(ibrutinib) tablets280 mg
Each tablet contains: ibrutinib 280 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
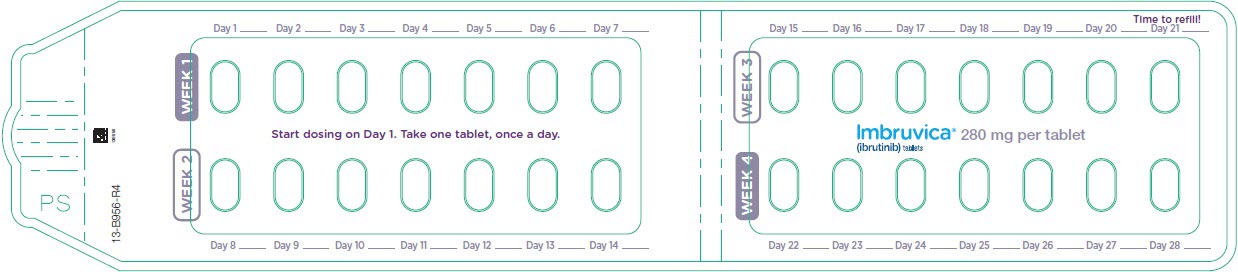
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Back)
NDC: 57962-280-28
Imbruvica®
(ibrutinib) tablets280 mg
Each tablet contains: ibrutinib 280 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Carton
NDC: 57962-420-28
Imbruvica®
(ibrutinib) tablets420 mg
Each tablet contains: ibrutinib 420 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.

-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Front)
NDC: 57962-420-28
Imbruvica®
(ibrutinib) tablets420 mg
Each tablet contains: ibrutinib 420 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
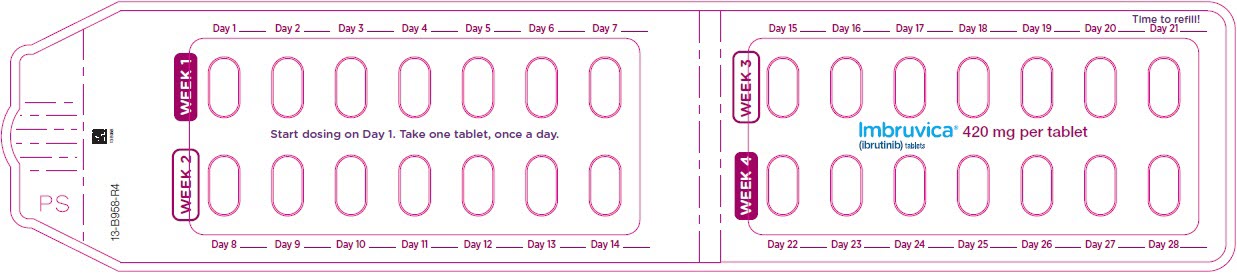
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Back)
NDC: 57962-420-28
Imbruvica®
(ibrutinib) tablets420 mg
Each tablet contains: ibrutinib 420 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Carton
NDC: 57962-560-28
Imbruvica®
(ibrutinib) tablets560 mg
Each tablet contains: ibrutinib 560 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Front)
NDC: 57962-560-28
Imbruvica®
(ibrutinib) tablets560 mg
Each tablet contains: ibrutinib 560 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
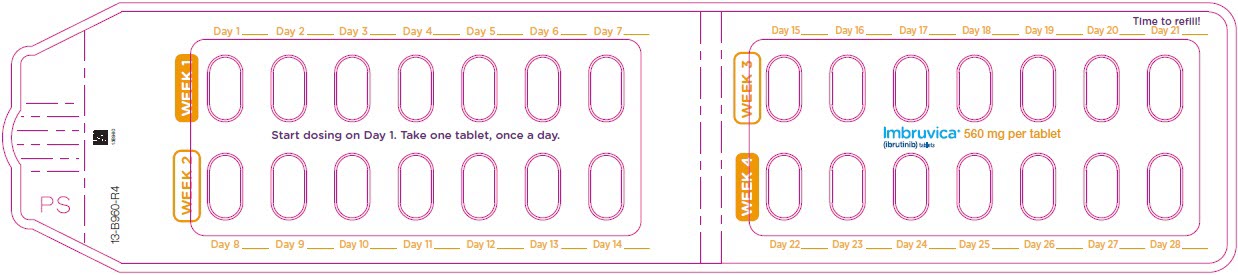
-
PRINCIPAL DISPLAY PANEL - 28 Tablet Blister Strip (Back)
NDC: 57962-560-28
Imbruvica®
(ibrutinib) tablets560 mg
Each tablet contains: ibrutinib 560 mg
Swallow tablet whole with water28 Tablets
Rx Only
Pharmacyclics LLC
An AbbVie CompanyJanssen Biotech, Inc.
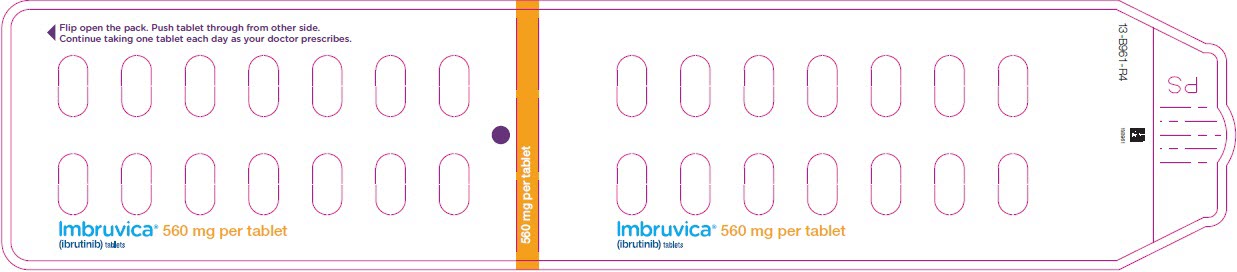
-
INGREDIENTS AND APPEARANCE
IMBRUVICA
ibrutinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-140 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 140 mg Product Characteristics Color WHITE Score no score Shape CAPSULE Size 22mm Flavor Imprint Code ibr;140;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-140-09 90 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 11/13/2013 2 NDC: 57962-140-12 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 11/13/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205552 11/13/2013 IMBRUVICA
ibrutinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-070 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 70 mg Product Characteristics Color YELLOW Score no score Shape CAPSULE Size 18mm Flavor Imprint Code ibr;70;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-070-28 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/20/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA205552 12/20/2017 IMBRUVICA
ibrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-014 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 140 mg Product Characteristics Color GREEN (Yellow-green to green) Score no score Shape ROUND Size 9mm Flavor Imprint Code ibr;140 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-014-28 1 in 1 CARTON 02/16/2018 1 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210563 02/16/2018 IMBRUVICA
ibrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-280 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 280 mg Product Characteristics Color PURPLE Score no score Shape OVAL (Oblong) Size 15mm Flavor Imprint Code ibr;280 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-280-28 1 in 1 CARTON 02/16/2018 1 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210563 02/16/2018 IMBRUVICA
ibrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-420 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 420 mg Product Characteristics Color GREEN (Yellow-green to green) Score no score Shape OVAL (Oblong) Size 18mm Flavor Imprint Code ibr;420 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-420-28 1 in 1 CARTON 02/16/2018 1 28 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 57962-420-71 1 in 1 CARTON 02/06/2019 2 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210563 02/16/2018 IMBRUVICA
ibrutinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57962-560 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRUTINIB (UNII: 1X70OSD4VX) (IBRUTINIB - UNII:1X70OSD4VX) IBRUTINIB 560 mg Product Characteristics Color YELLOW (Yellow to orange) Score no score Shape OVAL (Oblong) Size 19mm Flavor Imprint Code ibr;560 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57962-560-28 1 in 1 CARTON 02/16/2018 1 28 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 57962-560-71 1 in 1 CARTON 02/06/2019 2 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210563 02/16/2018 Labeler - Pharmacyclics LLC (079857498) Establishment Name Address ID/FEI Business Operations Pharmacyclics LLC 079857498 ANALYSIS(57962-140, 57962-070, 57962-014, 57962-280, 57962-420, 57962-560)
Trademark Results [Imbruvica]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 IMBRUVICA 86012748 not registered Dead/Abandoned |
Pharmacyclics, Inc. 2013-07-17 |
 IMBRUVICA 86007097 4523481 Live/Registered |
Pharmacyclics, Inc. 2013-07-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.