AQNEURSA- levacetylleucine granule, for suspension
AQNEURSA by
Drug Labeling and Warnings
AQNEURSA by is a Prescription medication manufactured, distributed, or labeled by IntraBio Inc, AndersonBrecon Inc., Eurofins Amatsi Analytics, SGS France, Patheon Austria GmbH & CoKG, AndersonBrecon (UK) Limited, Patheon France S.A.S, American Health Packaging. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AQNEURSA safely and effectively. See full prescribing information for AQNEURSA.
AQNEURSA™ (levacetylleucine) for oral suspension
Initial U.S. Approval: 2024INDICATIONS AND USAGE
AQNEURSA is indicated for the treatment of neurological manifestations of Niemann-Pick disease type C (NPC) in adults and pediatric patients weighing ≥15 kg. (1)
DOSAGE AND ADMINISTRATION
- For females of reproductive potential, verify that the patient is not pregnant prior to initiating treatment. (2.1)
- Recommended dosage (2.2)
Patient Body Weight
Morning Dose
Afternoon Dose
Evening Dose
15 to <25 kg
1 g
No Dose
1 g
25 to <35 kg
1 g
1 g
1 g
35 kg or more
2 g
1 g
1 g
- See the full prescribing information for administration instructions. (2.3)
DOSAGE FORMS AND STRENGTHS
For oral suspension: 1 gram levacetylleucine in a unit-dose packet. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential to use effective contraception during treatment and for 7 days after the last dose if AQNEURSA is discontinued. (5.1)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥5% and greater than placebo) are abdominal pain, dysphagia, upper respiratory tract infections, and vomiting. (6)
To report SUSPECTED ADVERSE REACTIONS, contact IntraBio Inc. at 1-833-306-9677 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to AQNEURSA Treatment Initiation
2.2 Recommended Dosage
2.3 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on AQNEURSA
7.2 Effect of AQNEURSA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Recommendation Prior to AQNEURSA Treatment Initiation
For females of reproductive potential, verify that the patient is not pregnant [see Use in Specific Populations (8.1, 8.3)].
2.2 Recommended Dosage
The recommended dosage of AQNEURSA is based on the patient’s actual body weight (kg) to be administered orally up to three times daily. See Table 1.
AQNEURSA can be taken with or without food [see Clinical Studies (14)].
For 2 gram levacetylleucine doses, prepare two AQNEURSA packets individually [see Dosage and Administration (2.3)].
Table 1 Recommended Dosage of Levacetylleucine Based on Body Weight (kg) Patient’s Body Weight
Morning Dose
Afternoon Dose
Evening Dose
15 kg to less than 25 kg
1 gram
No Dose
1 gram
25 kg to less than 35 kg
1 gram
1 gram
1 gram
35 kg or more
2 gram
1 gram
1 gram
One AQNEURSA packet contains 1 gram levacetylleucine.
Missed Dose
If a dose of AQNEURSA is missed, skip the missed dose and take the next dose at the scheduled time. Do not take 2 doses at the same time to make up for a missed dose.
2.3 Preparation and Administration Instructions
Oral Administration
For oral administration, administer AQNEURSA as follows:
- 1. Obtain the required number of AQNEURSA packets for the prescribed dose (one or two packets).
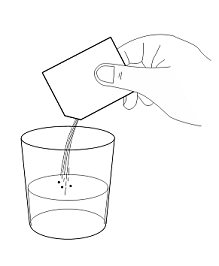
- 2. Open and empty the entire contents of one AQNEURSA packet into a container with 40 mL of water, orange juice, or almond milk. Do not use hot liquid.
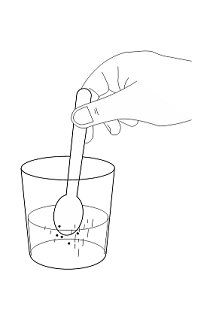
- 3. Stir to form a suspension.
- 4. Swallow the suspension immediately (within 30 minutes).
- 5. For doses requiring two AQNEURSA packets, repeat steps 2 to 4.
- 6. Discard unused AQNEURSA suspension if not administered within 30 minutes.
Use of Gastrostomy Tube (G-Tube) for Feeding Tube Administration
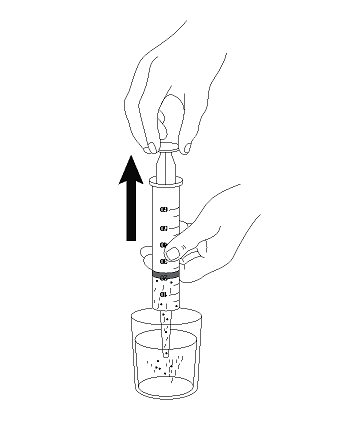
For patients who have a G-tube (French size 18 or larger) in place, administer AQNEURSA as follows:
- 1. Prepare AQNEURSA suspension immediately before administration via gastrostomy tube.
- 2. Obtain the required number of AQNEURSA packets for the prescribed dose (one or two packets).
- 3. Open and empty the entire contents of one AQNEURSA packet into a container with 40 mL of water ONLY. Do not use hot liquid.
- 4. Stir to form a suspension.
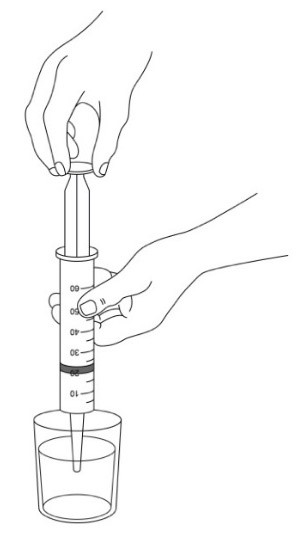
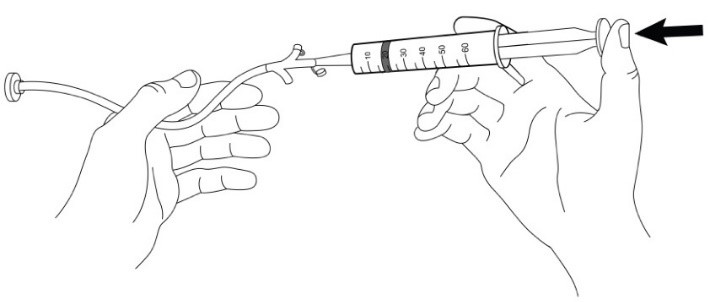
- 5. Draw up the suspension into a catheter tip syringe.
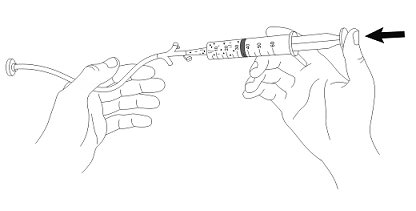
- 6. Administer the suspension immediately through the G-tube.
- 7. Flush any residual suspension in the catheter tip syringe with an additional 20 mL of water.
- 8. Flush the G-tube again, as needed, until no residual suspension is left in the syringe or feeding tube.
- 9. For doses requiring two AQNEURSA packets, repeat steps 3 to 8.
- 10. Discard unused AQNEURSA suspension if not administered immediately.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, AQNEURSA may cause embryo-fetal harm when administered during pregnancy. Administration of levacetylleucine to pregnant rats and rabbits during the period of organogenesis caused an increase in embryo-fetal death (post implantation loss/resorption) and skeletal malformations at a dose that was approximately 1.4-fold and 6-fold, respectively, the maximum recommended human dose (MRHD) of 4 g/day of levacetylleucine (based on body surface area).
The decision to continue or discontinue AQNEURSA treatment during pregnancy should consider the female’s need for AQNEURSA, the potential drug-related risks to the fetus, and the potential adverse outcomes from untreated maternal disease.
For females of reproductive potential, verify that the patient is not pregnant prior to initiating treatment with AQNEURSA. Advise females of reproductive potential to use effective contraception during treatment with AQNEURSA and for 7 days after the last dose if AQNEURSA is discontinued [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of AQNEURSA was evaluated in Trial 1, which included a total of 60 patients with Niemann-Pick disease Type C (NPC), in a placebo-controlled, randomized, crossover trial [see Clinical Studies (14)]. The mean (SD) treatment duration of AQNEURSA was 86.2 (4.7) days (69 min, 97 max); the mean (SD) treatment duration on placebo was 87.3 (4.8) days (78 min, 113 max).
Table 2 summarizes adverse reactions that occurred in patients who were treated with AQNEURSA in Treatment Period I of Trial 1.
Table 2 Adverse Reactions that Occurred in Adult and Pediatric Patients with NPC at an Incidence of ≥5% in Treatment Period I of Trial 1 Adverse Reaction
AQNEURSA
N=30
n (%)Placebo
N=30
n (%)Upper respiratory tract infection
5 (17)
1 (3)
Abdominal pain
2 (7)
0 (0)
Dysphagia
2 (7)
0 (0)
Vomiting
2 (7)
0 (0)
Rosacea
One patient experienced an exacerbation of rosacea during Trial 1 that responded to treatment. AQNEURSA was not discontinued.
Laboratory Findings
Thrombocytopenia with platelets < 100 10^3 cells/µL was observed in four patients during Treatment Period 1, all of whom were receiving miglustat for 42 days or longer at the time of enrollment. In two of these patients, the thrombocytopenia was present at baseline. In the other two patients, the thrombocytopenia developed during the trial.
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on AQNEURSA
N-acetyl-DL-leucine and N-acetyl-D-leucine
Avoid concomitant use of AQNEURSA with N-acetyl-DL-leucine and N-acetyl-D-leucine. The D-enantiomer, N-acetyl-D-leucine, competes with levacetylleucine for monocarboxylate transporter uptake, which may reduce the levacetylleucine efficacy.
7.2 Effect of AQNEURSA on Other Drugs
P-glycoprotein (P-gp) Transporter Substrates
Monitor more frequently for P-gp substrate related adverse reactions when used concomitantly with AQNEURSA.
Levacetylleucine inhibits P-gp [see Clinical Pharmacology (12.3)]. However, the clinical significance of this finding has not been fully characterized.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, AQNEURSA may cause embryo-fetal harm when administered during pregnancy. In animal reproduction studies, an increase in embryo-fetal death (post implantation loss/resorption), decrease in fetal body weight, and increase in external and skeletal malformations were observed in rats and rabbits when levacetylleucine was administered in pregnant rats and rabbits during the period of organogenesis. These effects were observed in rats and rabbits at the doses that were approximately 1.4-fold and 6-fold, respectively, the maximum recommended human dose (MRHD) in patients taking 4 grams of AQNEURSA per day (see Data).
There are no available data on AQNEURSA use in pregnant females to evaluate a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. Advise a pregnant female of the potential risk to the fetus. The decision to continue or discontinue AQNEURSA treatment during pregnancy should consider the female’s need for AQNEURSA, the potential drug-related risks to the fetus, and the potential adverse outcomes from untreated maternal disease.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
In a dose-range finding rat embryo-fetal development study, doses of up to 1000 mg/kg/day oral levacetylleucine were administered daily to pregnant females during organogenesis (Gestation Day [GD] 6 through GD 17). Increases in the mean number of resorptions, mean post-implantation losses and skeletal malformations (thoracic arch of the vertebra-fused thoracic arch, misaligned thoracic arch, hemicentric thoracic centrum, and misaligned thoracic centrum; vertebral column-scoliosis, and ribs-absent costal cartilage and short rib) were observed at a dose of 600 mg/kg/day, which is 1.4-fold the 4 g/day MRHD of levacetylleucine based on body surface area.
In a dose-range finding rabbit embryo-fetal development study, doses of up to 2500 mg/kg/day oral levacetylleucine were administered daily to pregnant females during organogenesis (GD 7 through GD 19). Increased embryo-fetal death (post-implantation loss/resorption), decreased fetal body weight, and increase in external (open or partially open eyes, hyperflexion of limbs) and skeletal malformations (misshapen maxillae and premaxillae, and small interparietal bones) were observed at the dose of 1250 mg/kg/day, which is 6-fold the MRHD of levacetylleucine based on body surface area.
8.2 Lactation
Risk Summary
There are no data on the presence of levacetylleucine or its metabolites in either human or animal milk, the effects on the breastfed infant or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for AQNEURSA and any potential adverse effects on the breastfed infant from levacetylleucine or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
AQNEURSA may cause embryo-fetal harm when administered to a pregnant female [see Use in Specific Populations (8.1)].
Pregnancy Testing
For a female of reproductive potential, verify that the patient is not pregnant prior to initiating treatment with AQNEURSA.
Contraception
Females
Advise a female of reproductive potential to use effective contraception during treatment and for 7 days after the last dose if AQNEURSA is discontinued.
8.4 Pediatric Use
The safety and effectiveness of AQNEURSA for the treatment of NPC have been established in 23 pediatric patients weighing ≥15 kg in Trial 1. Use of AQNEURSA for this indication is supported by evidence from one adequate and well-controlled study in adults and pediatric patients weighing ≥15 kg with additional pharmacokinetic data from 40 adults and 17 pediatric patients who participated in two open-label studies [see Clinical Studies (14) and Clinical Pharmacology (12.3)]. The safety and effectiveness of AQNEURSA have not been established in pediatric patients weighing <15 kg.
-
11 DESCRIPTION
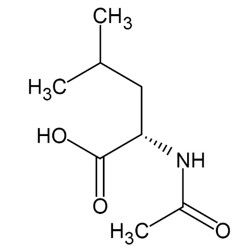
AQNEURSA (levacetylleucine) for oral suspension contains the drug substance levacetylleucine, a modified amino acid. Levacetylleucine is slightly soluble in aqueous solutions. The chemical name is 2-acetamido-4-methylpentanoic acid. The empirical formula is C8H15NO3 and the molecular weight is 173.21. The chemical structure is:
Each packet of AQNEURSA granules contains 1 gram levacetylleucine and the inactive ingredients hypromellose, isomalt and strawberry flavor.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The distinct molecular target for levacetylleucine in the treatment of NPC is unknown.
12.2 Pharmacodynamics
Clinical pharmacodynamic studies have not been conducted for levacetylleucine.
A relationship was not observed between increasing levacetylleucine exposure and clinical efficacy. The time course of pharmacodynamic response is unknown.
12.3 Pharmacokinetics
Levacetylleucine pharmacokinetic parameters at steady state are presented as mean (SD) unless otherwise specified. Levacetylleucine maximum concentration (Cmax) and area under the curve from time 0 to 24 hours (AUC0-24hrs) were 8.3 (3.3) µg/mL and 33.2 (12.5) h*μg/mL.
No levacetylleucine accumulation occurs after repeated administration.
Absorption
Levacetylleucine time to Cmax (Tmax) is 1 hour (ranging from 0.5 to 2.5 hours).
Distribution
Levacetylleucine apparent (oral) volume of distribution (VSS/F) is 253 (125) L.
Elimination
Levacetylleucine estimated half-life is around 1 hour and the apparent (oral) clearance is 139 (59) L/h.
Metabolism
Levacetylleucine is metabolized into acetate and L-leucine by ubiquitously expressed enzymes, which are used endogenously in catabolic and metabolic pathways. Cytochrome P450 enzymes are not involved in the metabolism of levacetylleucine.
Specific Populations
No clinically significant differences in pharmacokinetics of levacetylleucine were observed based on age (range 5 to 67 years), gender (female 45.5%, male 54.5%), or race/ethnicity (White 91%, Asian 4%, Other or not specified 5%). The effect of renal impairment, hepatic impairment, or pregnancy on levacetylleucine pharmacokinetics is unknown.
Body Weight
The volume of distribution and clearance of levacetylleucine increases with increasing body weight (20.5 kg to 98.4 kg), but these effects on pharmacokinetics are minimized by the recommended weight-based dosing.
Drug Interaction Studies
In Vitro Studies
Cytochrome P450 (CYP450) enzymes: Levacetylleucine does not inhibit CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4 and does not induce CYP 1A2, 2B6, and or 3A4.
Transporter Systems
Levacetylleucine is a substrate of organic anion transporter (OAT)1 and OAT3, but not organic cation transporter (OCT)2, breast cancer resistance protein (BCRP), or P-glycoprotein (P-gp). Levacetylleucine inhibits P-gp, BCRP, bile salt export pump (BSEP), OAT1 and OAT3, but does not inhibit organic anion transporting polypeptide (OATP)1B1 and OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Animal studies to evaluate the carcinogenic potential of levacetylleucine have not been conducted.
Mutagenesis
Levacetylleucine was not mutagenic or clastogenic in a battery of in vitro and in vivo assays including bacterial reverse mutation (Ames), chromosomal aberration (in human lymphocytes) and mouse micronucleus assays.
Impairment of Fertility
Animal studies to evaluate effects of levacetylleucine on fertility have not been conducted.
-
14 CLINICAL STUDIES
The safety and efficacy of AQNEURSA for the treatment of NPC were evaluated in a randomized, double-blind, placebo-controlled, two-period crossover study (NCT05163288) that evaluated the efficacy of AQNEURSA in 60 patients. To be eligible for the study, patients had to be aged 4 years or older with a confirmed diagnosis of NPC. Patients were required to have at least mild disease-related neurological symptoms.
Patients were assessed over a 2-week baseline period. Patients were then randomized in a 1:1 ratio to one of the two treatment sequences:
- Treatment Sequence 1 (N=30): AQNEURSA in Treatment Period I, followed by immediate crossover to placebo in Treatment Period II
- Treatment Sequence 2 (N=30): placebo in Treatment Period I, followed by immediate crossover to AQNEURSA in Treatment Period II.
AQNEURSA and placebo were administered orally with or without food for 12 weeks in each period.
Patients aged ≥13 years received 4 gram per day (as 2 gram morning dose, 1 gram afternoon dose, and 1 gram evening dose). The AQNEURSA dosage in pediatric patients under 13 years was based on patient’s body weight [see Dosage and Administration (2.2)].The approved dosage regimen is based on body weight and not age. Fifty-nine patients (98%) completed the study and received both placebo and AQNEURSA. One patient withdrew based on healthcare provider decision during AQNEURSA treatment.
Of the 60 randomized patients (37 adults and 23 pediatric patients), 27 were female and 33 were male. The median age at treatment initiation was 25 years (range: 5 to 67 years). 90% of the patients were White, 3% Asian, and 7% Other. The majority of the patients (n=51, 85%) received miglustat treatment prior to randomization and during the trial.
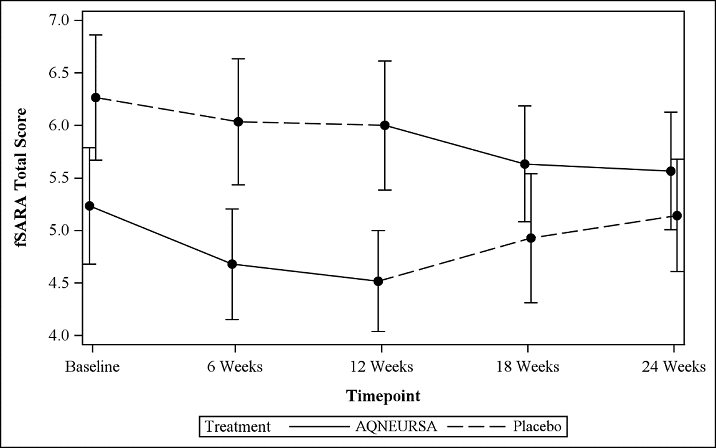
The primary efficacy outcome was assessed using a modified version of the Scale for Assessment and Rating of Ataxia (SARA), referred to as the functional SARA (fSARA). The SARA is a clinical assessment tool that assesses gait, stability, speech, and upper and lower limb coordination across 8 individual domains. The fSARA consists only of gait, sitting, stance, and speech disturbance domains of the original SARA with modifications to the scoring responses. Each domain was rescored from 0 to 4, where 0 is the best neurological status and 4 the worst, with a total score ranging from 0 to 16.
The fSARA score was assessed at baseline, 6 weeks, 12 weeks (the end of Period I), 18 weeks, and 24 weeks (the end of Period II). The estimated mean fSARA total score was 5.1 when patients were treated with AQNEURSA and 5.6 when patients were treated with placebo. The estimated treatment difference for the fSARA total score was -0.4 (95% CI: -0.7, -0.2) (Table 3).
Table 3 Summary of fSARA Efficacy Results CI = confidence interval; SD = standard deviation; SE = standard error. - * Two patients did not have an assessment at the end of Period II (week 24).
- † Two-sided p-value <0.001
Variable
fSARA Total Score
Treatment Sequence 1:
AQNEURSA - Placebo
Treatment Sequence 2:
Placebo - AQNEURSA
Baseline
N=30
N=30
- Mean (SD)
5.2 (3.0)
6.3 (3.3)
Period I
N=29
N=30
- Mean (SD)
4.5 (2.6)
6.0 (3.4)
Period II
N=28*
N=30
- Mean (SD)
5.1 (2.8)
5.6 (3.1)
Estimated Mean fSARA Score (SE) by Treatment
AQNEURSA
5.1 (0.1)
Placebo
5.6 (0.1)
Treatment Difference (95% CI)
-0.4 (-0.7, -0.2)†
Patients who received AQNEURSA in Period I followed by placebo in Period II (Treatment Sequence 1) showed a greater improvement in the fSARA score in Period I with a mean change from baseline of -0.5 (SD 1.2), compared to Period II with a mean change from baseline of 0 (1.5). Similarly, patients who received placebo in Period I followed by AQNEURSA in Period II (Treatment Sequence 2) experienced greater improvement in the fSARA score while receiving AQNEURSA in Period II with a mean change of -0.7 (0.9), compared to a mean change of -0.3 (0.9) in Period I.
Figure 1 Mean (+/- standard error) plot of the fSARA total score by time and treatment sequence
Results on the fSARA were supported by consistent results demonstrated on the original SARA.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
AQNEURSA (levacetylleucine) for oral suspension is supplied as white to off-white granules in a unit-dose multi-layer aluminum/polyethylene packet. Each packet contains 1.7 gram white to off-white granules, equivalent to 1 gram levacetylleucine.
NDC: 83853-101-01: Carton containing 28 unit-dose packets
Storage and Handling
Store AQNEURSA at room temperature between 20°C to 25°C (68°F to 77°F); excursion permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Instructions for Use).
Embryo-Fetal Toxicity
AQNEURSA may cause embryo-fetal harm. Advise a pregnant female of the potential risk to the fetus. Advise a female of reproductive potential and caregiver to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)]. Advise a female of reproductive potential to use effective contraception during treatment and for 7 days after the last dose if AQNEURSA is discontinued [see Use in Specific Populations (8.1, 8.3)].
Distributed by: IntraBio Inc., Austin TX 78701
AQNEURSA is a trademark of IntraBio.
-
INSTRUCTIONS FOR USE
AQNEURSA [ak nur' sah]
(levacetylleucine)
for oral suspension
This Instructions for Use contains information on how to prepare and take or give AQNEURSA oral suspension. Read this Instructions for Use before you prepare and take or give the first dose of AQNEURSA oral suspension and each time you get a refill. There may be new information. This Instructions for Use does not take the place of talking to you or your child’s healthcare provider about your or your child’s medical condition or treatment.
Important Information You Need to Know Before Taking or Giving AQNEURSA Oral Suspension
- Take or give AQNEURSA oral suspension exactly as instructed by your healthcare provider.
- Take or give AQNEURSA oral suspension with or without food.
- AQNEURSA oral suspension can be taken or given by mouth or through a gastrostomy tube (G-tube) feeding tube (French size 18 or larger).
- Do not use hot liquid with AQNEURSA oral suspension.
- For instructions on disposing of expired AQNEURSA packets or unused AQNEURSA oral suspension, see the end of this Instructions for Use.
Supplies Needed to Prepare and Take or Give AQNEURSA Oral Suspension
Gather the following supplies on a clean flat surface:
- the number of AQNEURSA packets needed for your prescribed dose
- a clean spoon
- a container
- a measuring cup from your pharmacist
- 40 mL of water, orange juice, or almond milk
- catheter tip syringe (if giving AQNEURSA oral suspension through G-tube)
Storing AQNEURSA
- Store AQNEURSA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep AQNEURSA oral suspension and all medicines out of reach of children.
Disposing of Expired AQNEURSA Packets or Unused AQNEURSA Oral Suspension
Throw away (dispose of) expired AQNEURSA packets or unused AQNEURSA oral suspension following the steps below:
- Mix medicine with a substance such as dirt, cat litter, or used coffee grounds.
- Place the mixture in a container such as a sealed plastic bag.
- Throw away (dispose of) the container in your household trash.
Distributed by: IntraBio Inc., Austin, TX 78701
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Issued: 9/2024
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
AQNEURSA
levacetylleucine granule, for suspensionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 83853-101 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEVACETYLLEUCINE (UNII: E915HL7K2O) (LEVACETYLLEUCINE - UNII:E915HL7K2O) LEVACETYLLEUCINE 1000 mg Product Characteristics Color Score Shape Size Flavor STRAWBERRY Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 83853-101-01 28 in 1 CARTON 09/24/2024 1 1 in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219132 09/24/2024 Labeler - IntraBio Inc (080729384)



Trademark Results [AQNEURSA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 AQNEURSA 98104776 not registered Live/Pending |
IntraBio Ltd 2023-07-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.