Stavudine by Rising Pharmaceuticals, Inc. / Aurobindo Pharma Limited STAVUDINE capsule
Stavudine by
Drug Labeling and Warnings
Stavudine by is a Prescription medication manufactured, distributed, or labeled by Rising Pharmaceuticals, Inc., Aurobindo Pharma Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use STAVUDINE CAPSULES safely and effectively. See full prescribing information for STAVUDINE CAPSULES.
STAVUDINE capsules, for oral use
Initial U.S. Approval: 1994
WARNING: LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS; PANCREATITIS
See full prescribing information for complete boxed warning.
- Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases. Fatal lactic acidosis has been reported in pregnant individuals who received the combination of stavudine and didanosine. Coadministration of stavudine with didanosine is contraindicated. (4, 5.1)
- Fatal and nonfatal pancreatitis have occurred when stavudine was part of a combination regimen that included didanosine. Coadministration of stavudine with didanosine is contraindicated. (4, 5.4)
INDICATIONS AND USAGE
Stavudine is a nucleoside reverse transcriptase inhibitor for use in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV)-1 infection. (1)
DOSAGE AND ADMINISTRATION
- Recommended dosage for adults:
- Recommended dosage for pediatric patients:
- Renal impairment: Dose adjustment is recommended for CrCl ≤50 mL/min. (2.3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Hepatic toxicity: May be severe, fatal. Consider interruption or discontinuation. Avoid use in combination with hydroxyurea. Coadministration of stavudine with didanosine is contraindicated. Risk of hepatic decompensation exists when used in combination with interferon and ribavirin; closely monitor and consider discontinuation of stavudine. (4, 5.2, 7)
- Neurologic symptoms: Motor weakness, most often seen in the setting of lactic acidosis, may mimic Guillain-Barré syndrome; discontinue treatment. Monitor for peripheral neuropathy, which can be severe; treatment discontinuation should be considered. (5.3)
- Patients may develop localized loss of body fat, monitor for signs and symptoms of lipoatrophy. Alternative antiretrovirals should be considered. (5.5)
- Patients may develop immune reconstitution syndrome. (5.6)
ADVERSE REACTIONS
- In adults, the most common adverse reactions are headache, diarrhea, neuropathy, rash, nausea, and vomiting. (6.1)
- Adverse reactions in pediatric patients were consistent with those seen in adults. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Rising Pharmaceuticals, Inc. at 1-866-562-4597 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS; PANCREATITIS
1. INDICATIONS AND USAGE
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Adult Dosage
2.2 Recommended Pediatric Dosage
2.3 Dosage Adjustment
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Lactic Acidosis/Severe Hepatomegaly with Steatosis
5.2 Hepatic Toxicity
5.3 Neurologic Symptoms
5.4 Pancreatitis
5.5 Lipoatrophy
5.6 Immune Reconstitution Syndrome
6. ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7. DRUG INTERACTIONS
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: LACTIC ACIDOSIS and HEPATOMEGALY with STEATOSIS; PANCREATITIS
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination, including stavudine and other antiretrovirals. Fatal lactic acidosis has been reported in pregnant individuals who received the combination of stavudine and didanosine with other antiretroviral agents. Coadministration of stavudine and didanosine is contraindicated because of increased risk of serious and/or life-threatening events [see Warnings and Precautions (5.1)]. Suspend treatment if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur.
Fatal and nonfatal pancreatitis have occurred during therapy when stavudine was part of a combination regimen that included didanosine in both treatment-naive and treatment-experienced patients, regardless of degree of immunosuppression [see Warnings and Precautions (5.4)]. -
1. INDICATIONS AND USAGE
Stavudine capsules, in combination with other antiretroviral agents, is indicated for the treatment of human immunodeficiency virus (HIV)-1 infection [see Clinical Studies (14)].
-
2. DOSAGE AND ADMINISTRATION
The interval between doses of stavudine capsules should be 12 hours. Stavudine capsules may be taken with or without food.
2.1 Recommended Adult Dosage
The recommended adult dosage is based on body weight as follows:
- For patients weighing less than 60 kg: 30 mg every 12 hours.
- For patients weighing at least 60 kg: 40 mg every 12 hours.
2.2 Recommended Pediatric Dosage
- For newborns from birth to 13 days old: 0.5 mg/kg given every 12 hours.
- For pediatric patients at least 14 days old and weighing less than 30 kg: 1 mg/kg given every 12 hours.
- For pediatric patients weighing at least 30 kg: use the recommended adult dosage.
2.3 Dosage Adjustment
Renal Impairment
Adult Patients: Stavudine capsules may be administered to adult patients with impaired renal function with an adjustment in dosage as shown in Table 1.
Table 1: Recommended Dosage Adjustment for Adult Patients with Renal Impairment Creatinine Clearance
(mL/min)Recommended Stavudine Capsules Dose
by Patient Weightat least 60 kg less than 60 kg * Administered after the completion of hemodialysis on dialysis days and at the same time of day on non-dialysis days.
greater than 50
40 mg every 12 hours
30 mg every 12 hours
26 to 50
20 mg every 12 hours
15 mg every 12 hours
10 to 25
20 mg every 24 hours
15 mg every 24 hours
Hemodialysis
20 mg every 24 hours*
15 mg every 24 hours*
Pediatric Patients: Since urinary excretion is also a major route of elimination of stavudine in pediatric patients, the clearance of stavudine may be altered in children with renal impairment. There are insufficient data to recommend a specific dose adjustment of stavudine capsules in this patient population.
-
3. DOSAGE FORMS AND STRENGTHS
- Stavudine Capsules USP, 15 mg are dark red opaque/light yellow opaque size ‘4’ hard gelatin capsule filled with white to off white granular powder and imprinted with ‘E’ on dark red opaque cap and ‘76’ on light yellow opaque body with black ink.
- Stavudine Capsules USP, 20 mg are light brown opaque/light brown opaque size ‘3’ hard gelatin capsule filled with white to off white granular powder and imprinted with ‘E’ on light brown opaque cap and ‘77’ on light brown opaque body with black ink.
- Stavudine Capsules USP, 30 mg are dark orange opaque/light orange opaque size “2” hard gelatin capsule filled with white to off white granular powder and imprinted with “C” on dark orange opaque cap and “36” on light orange opaque body with black ink.
- Stavudine Capsules USP, 40 mg are dark orange opaque/dark orange opaque size “1” hard gelatin capsule filled with white to off white granular powder and imprinted with “C” on dark orange opaque cap and “37” on dark orange opaque body with black ink.
-
4. CONTRAINDICATIONS
Stavudine capsules are contraindicated in patients with clinically significant hypersensitivity to stavudine or to any of the components contained in the formulation.
Co-administration of stavudine capsules with didanosine is contraindicated due to the potential for serious and/or life-threatening events notably lactic acidosis, hepatotoxicity, peripheral neuropathy, and pancreatitis [see Warnings and Precautions (5.1, 5.2, 5.3, 5.4)].
-
5. WARNINGS AND PRECAUTIONS
5.1 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues alone or in combination, including stavudine and other antiretrovirals. Although relative rates of lactic acidosis have not been assessed in prospective well-controlled trials, longitudinal cohort and retrospective studies suggest that this infrequent event may be more often associated with antiretroviral combinations containing stavudine. Female gender, obesity, and prolonged nucleoside exposure may be risk factors. Fatal lactic acidosis has been reported in pregnant individuals who received the combination of stavudine and didanosine with other antiretroviral agents. Coadministration of stavudine and didanosine is contraindicated [see Contraindications (4) and Use in Specific Populations (8.1)].
Particular caution should be exercised when administering stavudine to any patient with known risk factors for liver disease; however, cases of lactic acidosis have also been reported in patients with no known risk factors. Generalized fatigue, digestive symptoms (nausea, vomiting, abdominal pain, and unexplained weight loss); respiratory symptoms (tachypnea and dyspnea); or neurologic symptoms, including motor weakness [see Warnings and Precautions (5.3)] might be indicative of the development of symptomatic hyperlactatemia or lactic acidosis syndrome.
Treatment with stavudine should be suspended in any patient who develops clinical or laboratory findings suggestive of symptomatic hyperlactatemia, lactic acidosis, or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations). Permanent discontinuation of stavudine should be considered for patients with confirmed lactic acidosis.
5.2 Hepatic Toxicity
The safety and efficacy of stavudine have not been established in HIV-infected patients with significant underlying liver disease. During combination antiretroviral therapy, patients with preexisting liver dysfunction, including chronic active hepatitis, have an increased frequency of liver function abnormalities, including severe and potentially fatal hepatic adverse events, and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
Hepatotoxicity and hepatic failure resulting in death were reported during postmarketing surveillance in HIV-infected patients treated with hydroxyurea and other antiretroviral agents. Fatal hepatic events were reported most often in patients treated with the combination of hydroxyurea, didanosine, and stavudine. Coadministration of stavudine and didanosine is contraindicated; and the combination of stavudine and hydroxyurea should be avoided [see Contraindications (4) and Drug Interactions (7)].
Use with Interferon and Ribavirin-Based Regimens
In vitro studies have shown ribavirin can reduce the phosphorylation of pyrimidine nucleoside analogues such as stavudine. Although no evidence of a pharmacokinetic or pharmacodynamic (e.g., loss of HIV-1/HCV virologic suppression) interaction was seen when ribavirin was coadministered with stavudine in HIV-1/HCV co-infected patients [see Drug Interactions (7)], hepatic decompensation (some fatal) has occurred in HIV-1/HCV co-infected patients receiving combination antiretroviral therapy for HIV-1 and interferon and ribavirin. Patients receiving interferon with or without ribavirin and stavudine should be closely monitored for treatment-associated toxicities, especially hepatic decompensation. Discontinuation of stavudine should be considered as medically appropriate. Dose reduction or discontinuation of interferon, ribavirin, or both should also be considered if worsening clinical toxicities are observed, including hepatic decompensation (e.g., Child-Pugh >6) (see the full prescribing information for interferon and ribavirin).
5.3 Neurologic Symptoms
Motor weakness has been reported rarely in patients receiving combination antiretroviral therapy including stavudine. Most of these cases occurred in the setting of lactic acidosis. The evolution of motor weakness may mimic the clinical presentation of Guillain-Barré syndrome (including respiratory failure). If motor weakness develops, stavudine should be discontinued. Symptoms may continue or worsen following discontinuation of therapy.
Peripheral sensory neuropathy, manifested by numbness, tingling, or pain in the hands or feet, has been reported in patients receiving stavudine therapy. Peripheral neuropathy, which can be severe, is dose-related and occurs more frequently in patients with advanced HIV-1 disease, a history of peripheral neuropathy, or in patients receiving other drugs that have been associated with neuropathy [see Drug Interactions (7)].
Patients should be monitored for the development of peripheral neuropathy. Stavudine-related peripheral neuropathy may resolve if therapy is withdrawn promptly. If peripheral neuropathy develops permanent discontinuation of stavudine should be considered. In some cases, symptoms may worsen temporarily following discontinuation of therapy.
5.4 Pancreatitis
Fatal and nonfatal pancreatitis have occurred during therapy when stavudine was part of a combination regimen that included didanosine in both treatment-naive and treatment-experienced patients, regardless of degree of immunosuppression. The combination of stavudine and any other agents that are toxic to the pancreas should be suspended in patients with suspected pancreatitis. Coadministration of stavudine and didanosine is contraindicated [see Contraindications (4)]. Reinstitution of stavudine after a confirmed diagnosis of pancreatitis should be undertaken with particular caution and close patient monitoring.
5.5 Lipoatrophy
In randomized controlled trials of treatment-naive patients, clinical lipoatrophy developed in a higher proportion of patients treated with stavudine compared to other nucleosides (tenofovir or abacavir). Dual energy x-ray absorptiometry (DEXA) scans demonstrated overall limb fat loss in stavudine-treated patients compared to limb fat gain or no gain in patients treated with other nucleosides (abacavir, tenofovir, or zidovudine). The incidence and severity of lipoatrophy are cumulative over time with stavudine-containing regimens. In clinical trials, switching from stavudine to other nucleosides (tenofovir or abacavir) resulted in increases in limb fat with modest to no improvements in clinical lipoatrophy.
Patients receiving stavudine should be monitored for symptoms or signs of lipoatrophy and questioned about body changes related to lipoatrophy. Given the potential risks of using stavudine including lipoatrophy, a benefit-risk assessment for each patient should be made and an alternative antiretroviral should be considered.5.6 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including stavudine. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jiroveci pneumonia (PCP), or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment. -
6. ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- lactic acidosis and severe hepatomegaly with steatosis [see Warnings and Precautions (5.1)]
- hepatic toxicity [see Warnings and Precautions (5.2)]
- neurologic symptoms and motor weakness [see Warnings and Precautions (5.3)]
- pancreatitis [see Warnings and Precautions (5.4)]
- lipoatrophy [see Warnings and Precautions (5.5)]
When stavudine is used in combination with other agents with similar toxicities, the incidence of adverse reactions may be higher than when stavudine is used alone.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Clinical Trials Experience in Adults
Selected adverse reactions that occurred in adult patients receiving stavudine in a controlled monotherapy study (Study AI455-019) are provided in Table 2.Table 2: Selected Adverse Reactions in Study AI455-019a (Monotherapy) Adverse Reaction Percent (%) Stavudineb
(40 mg twice daily)
(n=412)Zidovudine
(200 mg 3 times daily) (n=402)a The incidences reported included all severity grades and all reactions regardless of causality.
b Median duration of stavudine therapy = 79 weeks; median duration of zidovudine therapy = 53 weeks.
Headache
54
49
Diarrhea
50
44
Peripheral Neurologic Symptoms/Neuropathy
52
39
Rash
40
35
Nausea and Vomiting
39
44
Pancreatitis was observed in 3 of the 412 adult patients who received stavudine in study AI455-019.
Selected adverse reactions that occurred in antiretroviral-naive adult patients receiving stavudine from two controlled combination studies are provided in Table 3.
Table 3: Selected Adverse Reactionsa in START 1 and START 2b Studies (Combination Therapy) Adverse Reaction Percent (%) START 1 START 2b Stavudine + Lamivudine + Indinavir (n=100c) Zidovudine + Lamivudine + Indinavir
(n=102)Stavudine + Didanosine + Indinavir (n=102c) Zidovudine + Lamivudine + Indinavir (n=103) a The incidences reported included all severity grades and all reactions regardless of causality.
b START 2 compared two triple-combination regimens in 205 treatment-naive patients. Patients received either stavudine (40 mg twice daily) plus didanosine plus indinavir or zidovudine plus lamivudine plus indinavir.
c Duration of stavudine therapy = 48 weeks.
Nausea
43
63
53
67
Diarrhea
34
16
45
39
Headache
25
26
46
37
Rash
18
13
30
18
Vomiting
18
33
30
35
Peripheral Neurologic Symptoms/Neuropathy
8
7
21
10
Selected laboratory abnormalities reported in a controlled monotherapy study (Study AI455-019) are provided in Table 4.
Table 4: Selected Laboratory Abnormalities in Study AI455-019a,b Parameter Percent (%) Stavudine
(40 mg twice daily)
(n=412)Zidovudine
(200 mg 3 times daily) (n=402)a Data presented for patients for whom laboratory evaluations were performed.
b Median duration of stavudine therapy = 79 weeks; median duration of zidovudine therapy = 53 weeks.
ULN = upper limit of normal.
AST (SGOT) (>5 x ULN)
11
10
ALT (SGPT) (>5 x ULN)
13
11
Amylase (≥1.4 x ULN)
14
13
Selected laboratory abnormalities reported in two controlled combination studies are provided in Tables 5 and 6.
Table 5: Selected Laboratory Abnormalities in START 1 and START 2 Studies (Grades 3 to 4) Parameter Percent (%) START 1 START 2 Stavudine + Lamivudine + Indinavir (n=100) Zidovudine + Lamivudine + Indinavir (n=102) Stavudine + Didanosine + Indinavir (n=102) Zidovudine + Lamivudine + Indinavir (n=103) ULN = upper limit of normal.
Bilirubin (>2.6 x ULN)
7
6
16
8
AST (SGOT) (>5 x ULN)
5
2
7
7
ALT (SGPT) (>5 x ULN)
6
2
8
5
GGT (>5 x ULN)
2
2
5
2
Lipase (>2 x ULN)
6
3
5
5
Amylase (>2 x ULN)
4
<1
8
2
Table 6: Selected Laboratory Abnormalities in START 1 and START 2 Studies (All Grades) Parameter Percent (%) START 1 START 2 Stavudine + Lamivudine + Indinavir (n=100) Zidovudine + Lamivudine + Indinavir (n=102) Stavudine + Didanosine + Indinavir (n=102) Zidovudine + Lamivudine + Indinavir (n=103) Total Bilirubin
65
60
68
55
AST (SGOT)
42
20
53
20
ALT (SGPT)
40
20
50
18
GGT
15
8
28
12
Lipase
27
12
26
19
Amylase
21
19
31
17
Clinical Trials Experience in Pediatric Patients
Adverse reactions and serious laboratory abnormalities reported in pediatric patients from birth through adolescence during clinical trials were similar in type and frequency to those seen in adult patients [see Use in Specific Populations (8.4)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of stavudine. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions have been chosen for inclusion due to their seriousness, frequency of reporting, causal connection to stavudine, or a combination of these factors.
Body as a Whole: abdominal pain, allergic reaction, chills/fever.
Digestive Disorders: anorexia.
Exocrine Gland Disorders: pancreatitis, including fatal cases [see Warnings and Precautions (5.4)].
Hematologic Disorders: anemia, leukopenia, thrombocytopenia, neutropenia, and macrocytosis.
Liver: symptomatic hyperlactatemia/lactic acidosis and hepatic steatosis [see Warnings and Precautions (5.1)], hepatitis and liver failure.
Metabolic Disorders: lipoatrophy [see Warnings and Precautions (5.5)], diabetes mellitus and hyperglycemia.
Musculoskeletal: myalgia.
Nervous System: insomnia, severe motor weakness (most often reported in the setting of lactic acidosis) [see Warnings and Precautions (5.1, 5.3)].
-
7. DRUG INTERACTIONS
Stavudine is unlikely to interact with drugs metabolized by cytochrome P450 isoenzymes.
Hydroxyurea: When stavudine is used in combination with other agents with similar toxicities, the incidence of these toxicities may be higher than when stavudine is used alone. Thus, patients treated with stavudine in combination with hydroxyurea, may be at increased risk for pancreatitis and hepatotoxicity, which may be fatal, and severe peripheral neuropathy [see Warnings and Precautions (5.2)]. The combination of stavudine and hydroxyurea should be avoided.
Zidovudine: Zidovudine competitively inhibits the intracellular phosphorylation of stavudine. Therefore, use of zidovudine in combination with stavudine should be avoided.
Doxorubicin: In vitro data indicate that the phosphorylation of stavudine is inhibited at relevant concentrations by doxorubicin. The clinical significance of this interaction is unknown; therefore, concomitant use of stavudine with doxorubicin should be undertaken with caution.
Ribavirin: In vitro data indicate ribavirin reduces phosphorylation of lamivudine, stavudine, and zidovudine. The clinical significance of the interaction with stavudine is unknown; therefore, concomitant use of stavudine with ribavirin should be undertaken with caution. No pharmacokinetic (e.g., plasma concentrations or intracellular triphosphorylated active metabolite concentrations) or pharmacodynamic (e.g., loss of HIV-1/HCV virologic suppression) interaction was observed when ribavirin and lamivudine (n=18), stavudine (n=10), or zidovudine (n=6) were coadministered as part of a multi-drug regimen to HIV-1/HCV co-infected patients [see Warnings and Precautions (5.2)].
-
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to stavudine during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Fatal lactic acidosis has been reported in pregnant individuals who received the combination of stavudine and didanosine with other antiretroviral agents. It is unclear if pregnancy augments the risk of lactic acidosis/hepatic steatosis syndrome reported in non-pregnant individuals receiving nucleoside analogues [see Warnings and Precautions (5.1)]. The combination of stavudine and didanosine is contraindicated [see Contraindications (4)].
Prospective pregnancy data from APR are not sufficient to adequately assess the risk of major birth defects, miscarriage or adverse developmental outcomes. Available data from the APR show no increase in overall risk of major birth defects compared with 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP). The rate of miscarriage is not reported in the APR. In the U.S. general population, the estimated background risks of miscarriage in clinically recognized pregnancies is 15 to 20%.
In animal reproduction studies, no adverse developmental effects were observed with oral administration of stavudine at clinically relevant exposures. No developmental toxicities were observed in rats and rabbits at systemic exposures 112 (AUC) and 183 (Cmax) times, respectively, the exposures in humans at the recommended human dose (RHD) of stavudine (see Data).
Clinical Considerations
Maternal Adverse Reactions
Cases of lactic acidosis syndrome, sometimes fatal have occurred in pregnant individuals using stavudine in combination with didanosine. Stavudine is associated with an increased risk of lactic acidosis syndrome/hepatic steatosis syndrome [see Warnings and Precautions (5.1)].
Data
Human Data
Based on prospective reports to the APR of live births following exposure to stavudine-containing regimens during pregnancy (including 811 exposed in the first trimester and 196 exposed in the second/third trimester), the prevalence of birth defects in live births for stavudine was 2.6% (95% CI: 1.6 % to 3.9%) with first trimester exposure and 3.1% (95% CI: 1.1% to 6.5%) with second/third trimester exposure compared to the background birth defect rate of 2.7% in the U.S. reference population of the MACDP.
Prospective reports from the APR of overall major birth defects in pregnancies exposed to stavudine is compared with a U.S. background major birth defect rate. Methodological limitations of the APR include the use of MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations, as well as confounding due to the underlying disease.
Animal Data
Stavudine was administered orally to pregnant rats (0, 50, 250, and 1000 mg/kg/day from gestation day 6 to 17) and rabbits (0, 60, 150, 300 and 600 mg/kg/day from gestation day 6 to 18). In rats, fetal skeletal variations, including increased unossified or incomplete ossification of sternebra, were observed at the highest dose (1000 mg/kg/day) (approximately 488 times human AUC exposure at the RHD). In rabbits, there were no developmental effects up to the highest dose of 600 mg/kg (approximately 183 times human Cmax exposure at the RHD).
In the pre/post-natal development study, stavudine was administered orally to rats at 0, 50, 250, and 1000 mg/kg/day from gestation day 17 to postnatal day 21. Post-implantation loss and an increase in early neonatal mortality was observed at 1000 mg/kg/day (approximately 488 times human AUC exposure at the RHD). No developmental effects were observed at 250 mg/kg/day (approximately 112 times human AUC exposure at the RHD).
Stavudine was transferred to the fetus through the placenta in rats with concentrations in fetal tissues approximately half the concentration detected in maternal plasma.8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV.
Based on limited data, stavudine has been detected in human milk. No data are available regarding the effects of stavudine on the breastfed infant, or the effects on milk production.
Because of the potential for (1) HIV transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants) and (3) adverse reactions in breastfed infants similar to those seen in adults, instruct mothers not to breastfeed if they are receiving stavudine.8.4 Pediatric Use
Use of stavudine in pediatric patients from birth through adolescence is supported by evidence from adequate and well-controlled studies of stavudine in adults with additional pharmacokinetic and safety data in pediatric patients [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
Adverse reactions and laboratory abnormalities reported to occur in pediatric patients in clinical studies were generally consistent with the safety profile of stavudine in adults. These studies include ACTG 240, where 105 pediatric patients ages 3 months to 6 years received stavudine 2 mg/kg/day for a median of 6.4 months; a controlled clinical trial where 185 newborns received stavudine 2 mg/kg/day either alone or in combination with didanosine from birth through 6 weeks of age; and a clinical trial where 8 newborns received stavudine 2 mg/kg/day in combination with didanosine and nelfinavir from birth through 4 weeks of age.
Stavudine pharmacokinetics have been evaluated in 25 HIV-1-infected pediatric patients ranging in age from 5 weeks to 15 years and in weight from 2 to 43 kg after IV or oral administration of single doses and twice-daily regimens and in 30 HIV-1-exposed or -infected newborns ranging in age from birth to 4 weeks after oral administration of twice-daily regimens [see Clinical Pharmacology (12.3, Table 9)].8.5 Geriatric Use
Clinical studies of stavudine did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently than younger patients. Greater sensitivity of some older individuals to the effects of stavudine cannot be ruled out.
In a monotherapy Expanded Access Program for patients with advanced HIV-1 infection, peripheral neuropathy or peripheral neuropathic symptoms were observed in 15 of 40 (38%) elderly patients receiving 40 mg twice daily and 8 of 51 (16%) elderly patients receiving 20 mg twice daily. Of the approximately 12,000 patients enrolled in the Expanded Access Program, peripheral neuropathy or peripheral neuropathic symptoms developed in 30% of patients receiving 40 mg twice daily and 25% of patients receiving 20 mg twice daily. Elderly patients should be closely monitored for signs and symptoms of peripheral neuropathy.
Stavudine is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, it may be useful to monitor renal function. Dose adjustment is recommended for patients with renal impairment [see Dosage and Administration (2.3)].8.6 Renal Impairment
Data from two studies in adults indicated that the apparent oral clearance of stavudine decreased and the terminal elimination half-life increased as creatinine clearance decreased. Based on these observations, it is recommended that the stavudine dosage be modified in patients with reduced creatinine clearance and in patients receiving maintenance hemodialysis [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Experience with adults treated with 12 to 24 times the recommended daily dosage revealed no acute toxicity. Complications of chronic overdosage include peripheral neuropathy and hepatic toxicity. Stavudine can be removed by hemodialysis; the mean ± SD hemodialysis clearance of stavudine is 120 ± 18 mL/min. Whether stavudine is eliminated by peritoneal dialysis has not been studied.
-
11 DESCRIPTION
Stavudine capsules, USP contain stavudine (d4T), which is a synthetic thymidine nucleoside analogue, active against the human immunodeficiency virus type 1 (HIV-1). The chemical name for stavudine is 2′,3′-didehydro-3′-deoxythymidine. Stavudine has the following structural formula:
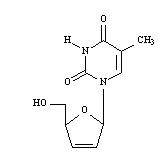
Stavudine USP is a white to off-white crystalline solid with the molecular formula C10H12N2O4 and a molecular weight of 224.2. The solubility of stavudine at 23°C is approximately 83 mg/mL in water and 30 mg/mL in propylene glycol. The n-octanol/water partition coefficient of stavudine at 23°C is 0.144.
Stavudine is available as capsules for oral administration containing either 15 mg, 20 mg, 30 mg, or 40 mg of stavudine. Each capsule also contains inactive ingredients microcrystalline cellulose, sodium starch glycolate, anhydrous lactose, and magnesium stearate. The hard gelatin shell consists of gelatin, sodium lauryl sulfate, titanium dioxide, yellow iron oxide, and red iron oxide. The capsules are printed with black ink containing black iron oxide. -
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
The pharmacokinetics of stavudine have been evaluated in HIV-1-infected adult and pediatric patients (Tables 7, 8, and 9). Peak plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC) increased in proportion to dose after both single and multiple doses ranging from 0.03 to 4 mg/kg. There was no significant accumulation of stavudine with repeated administration every 6, 8, or 12 hours.
Absorption
Following oral administration, stavudine is rapidly absorbed, with peak plasma concentrations occurring within 1 hour after dosing. The systemic exposure to stavudine is the same following administration as capsules or solution. Steady-state pharmacokinetic parameters of stavudine in HIV-1-infected adults are shown in Table 7.
Table 7: Steady-State Pharmacokinetic Parameters of Stavudine in HIV-1-Infected Adults Parameter Stavudine 40 mg BID
Mean ± SD (n=8)AUC0-24 = Area under the curve over 24 hours.
Cmax = Maximum plasma concentration.
Cmin = Trough or minimum plasma concentration.
AUC0-24 (ngh/mL)
2568 ± 454
Cmax (ng/mL)
536 ± 146
Cmin (ng/mL)
8 ± 9
Distribution
Binding of stavudine to serum proteins was negligible over the concentration range of 0.01 to 11.4 mcg/mL. Stavudine distributes equally between red blood cells and plasma. Volume of distribution is shown in Table 8.
Metabolism
Metabolism plays a limited role in the clearance of stavudine. Unchanged stavudine was the major drug-related component circulating in plasma after an 80 mg dose of 14C-stavudine, while metabolites constituted minor components of the circulating radioactivity. Minor metabolites include oxidized stavudine, glucuronide conjugates of stavudine and its oxidized metabolite, and an N-acetylcysteine conjugate of the ribose after glycosidic cleavage, suggesting that thymine is also a metabolite of stavudine.
Elimination
Following an 80 mg dose of 14C-stavudine to healthy subjects, approximately 95% and 3% of the total radioactivity was recovered in urine and feces, respectively. Radioactivity due to parent drug in urine and feces was 73.7% and 62%, respectively. The mean terminal elimination half-life is approximately 2.3 hours following single oral doses. Mean renal clearance of the parent compound is approximately 272 mL/min, accounting for approximately 67% of the apparent oral clearance.
In HIV-1-infected patients, renal elimination of unchanged drug accounts for about 40% of the overall clearance regardless of the route of administration (Table 8). The mean renal clearance was about twice the average endogenous creatinine clearance, indicating active tubular secretion in addition to glomerular filtration.
Table 8: Pharmacokinetic Parameters of Stavudine in HIV-1-Infected Adults: Bioavailability, Distribution, and Clearance Parameter Mean ± SD n a Following 1-hour IV infusion.
b Following single oral dose.
c Assuming a body weight of 70 kg.
d Over 12 to 24 hours.
Oral bioavailability (%)
86.4 ± 18.2
25
Volume of distribution (L)a
46 ± 21
44
Total body clearance (mL/min)a
594 ± 164
44
Apparent oral clearance (mL/min)b
560 ± 182c
113
Renal clearance (mL/min)a
237 ± 98
39
Elimination half-life, IV dose (h)a
1.15 ± 0.35
44
Elimination half-life, oral dose (h)b
1.6 ± 0.23
8
Urinary recovery of stavudine (% of dose)a,d
42 ± 14
39
Special Populations
Pediatric
Pharmacokinetic parameters of stavudine in pediatric patients are presented in Table 9.
Table 9: Pharmacokinetic Parameters (Mean ± SD) of Stavudine in HIV-1-Exposed or -Infected Pediatric Patients Parameter Ages 5 weeks to 15 years n Ages 14 to 28 days n Day of Birth n a Following 1-hour IV infusion.
b At median time of 2.5 hours (range 2 to 3 hours) following multiple oral doses.
c Following single oral dose.
d Over 8 hours.
ND = Not determined.
Oral bioavailability (%)
76.9 ± 31.7
20
ND
ND
Volume of distribution (L/kg)a
0.73 ± 0.32
21
ND
ND
Ratio of CSF: plasma concentrations (as %)b
59 ± 35
8
ND
ND
Total body clearance (mL/min/kg)a
9.75 ± 3.76
21
ND
ND
Apparent oral clearance (mL/min/kg)c
13.75 ± 4.29
20
11.52 ± 5.93
30
5.08 ± 2.8
17
Elimination half-life, IV dose (h)a
1.11 ± 0.28
21
ND
ND
Elimination half-life, oral dose (h)c
0.96 ± 0.26
20
1.59 ± 0.29
30
5.27 ± 2.01
17
Urinary recovery of stavudine
(% of dose)c,d
34 ± 16
19
ND
ND
Renal Impairment
Data from two studies in adults indicated that the apparent oral clearance of stavudine decreased and the terminal elimination half-life increased as creatinine clearance decreased (see Table 10). Cmax and Tmax were not significantly altered by renal impairment. The mean ± SD hemodialysis clearance value of stavudine was 120 ± 18 mL/min (n=12); the mean ± SD percentage of the stavudine dose recovered in the dialysate, timed to occur between 2 to 6 hours post-dose, was 31 ± 5%. Based on these observations, it is recommended that stavudine dosage be modified in patients with reduced creatinine clearance and in patients receiving maintenance hemodialysis [seeDosage and Administration (2.3)].
Table 10: Mean ± SD Pharmacokinetic Parameter Values of Stavudinea in Adults with Varying Degrees of Renal Function a Single 40 mg oral dose.
b Determined while patients were off dialysis.
T½ = Terminal elimination half-life.
NA = Not applicable.
Creatinine Clearance
Hemodialysis
Patientsb (n=11)
>50 mL/min
(n=10)
26 to
50 mL/min
(n=5)
9 to 25 mL/min
(n=5)
Creatinine clearance (mL/min)
104 ± 28
41 ± 5
17 ± 3
NA
Apparent oral clearance (mL/min)
335 ± 57
191 ± 39
116 ± 25
105 ± 17
Renal clearance (mL/min)
167 ± 65
73 ± 18
17 ± 3
NA
T½ (h)
1.7 ± 0.4
3.5 ± 2.5
4.6 ± 0.9
5.4 ± 1.4
Hepatic Impairment
Stavudine pharmacokinetics were not altered in five non-HIV-infected patients with hepatic impairment secondary to cirrhosis (Child-Pugh classification B or C) following the administration of a single 40 mg dose.
Geriatric
Stavudine pharmacokinetics have not been studied in patients >65 years of age [See Use in Specific Populations (8.5)].
Gender
A population pharmacokinetic analysis of data collected during a controlled clinical study in HIV-1-infected patients showed no clinically important differences between males (n=291) and females (n=27).
Race
A population pharmacokinetic analysis of data collected during a controlled clinical study in HIV-1-infected patients showed no clinically important differences between races (n=233 Caucasian, 39 African-American, 41 Hispanic, 1 Asian, and 4 other).
Drug Interaction Studies
Stavudine does not inhibit the major cytochrome P450 isoforms CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4; therefore, it is unlikely that clinically significant drug interactions will occur with drugs metabolized through these pathways. Because stavudine is not protein-bound, it is not expected to affect the pharmacokinetics of protein-bound drugs.
Tables 11 and 12 summarize the effects on AUC and Cmax, with a 95% confidence interval (CI) when available, following coadministration of stavudine with didanosine, lamivudine, and nelfinavir. No clinically significant pharmacokinetic interactions were observed.
Table 11: Results of Drug Interaction Studies with Stavudine: Effects of Coadministered Drug on Stavudine Plasma AUC and Cmax Values Drug Stavudine Dosage na AUC of Stavudine (95% CI) Cmax of Stavudine
(95% CI)↑ Indicates increase.
↔ Indicates no change, or mean increase or decrease of <10%.
a HIV-1-infected patients.
Didanosine, 100 mg q12h for 4 days
40 mg q12h for 4 days
10
↔
↑ 17%
Lamivudine, 150 mg single dose
40 mg single dose
18
↔
(92.7 to 100.6%)
↑ 12%
(100.3 to 126.1%)
Nelfinavir, 750 mg q8h for 56 days
30 to 40 mg q12h
for 56 days
8
↔
↔
Table 12: Results of Drug Interaction Studies with Stavudine: Effects of Stavudine on Coadministered Drug Plasma AUC and Cmax Values Drug Stavudine Dosage na AUC of Coadministered Drug
(95% CI)Cmax of Coadministered Drug
(95% CI)↔ Indicates no change, or mean increase or decrease of <10%.
a HIV-1-infected patients.
Didanosine, 100 mg q12h for 4 days
40 mg q12h for 4 days
10
↔
↔
Lamivudine, 150 mg single dose
40 mg single dose
18
↔
(90.5 to 107.6%)
↔
(87.1 to 110.6%)
Nelfinavir, 750 mg q8h for 56 days
30 to 40 mg q12h
for 56 days
8
↔
↔
12.4 Microbiology
Mechanism of Action
Stavudine, a nucleoside analogue of thymidine, is phosphorylated by cellular kinases to the active metabolite stavudine triphosphate. Stavudine triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) by competing with the natural substrate thymidine triphosphate (Ki=0.0083 to 0.032 μM) and by causing DNA chain termination following its incorporation into viral DNA. Stavudine triphosphate inhibits cellular DNA polymerases β and γ and markedly reduces the synthesis of mitochondrial DNA.
Antiviral Activity in Cell Culture
The cell culture antiviral activity of stavudine was measured in peripheral blood mononuclear cells, monocytic cells, and lymphoblastoid cell lines. The concentration of drug necessary to inhibit HIV-1 replication by 50% (EC50) ranged from 0.009 to 4 μM against laboratory and clinical isolates of HIV-1. In cell culture, stavudine exhibited antagonistic activity in combination with zidovudine. The anti-HIV-1 activity of stavudine in combination with either abacavir, didanosine, tenofovir, or zalcitabine was not antagonistic. Ribavirin, at the 9 to 45 μM concentrations tested, reduced the anti-HIV-1 activity of stavudine by 2.5- to 5-fold. The relationship between cell culture susceptibility of HIV-1 to stavudine and the inhibition of HIV-1 replication in humans has not been established.
Resistance
HIV-1 isolates with reduced susceptibility to stavudine have been selected in cell culture (strain-specific) and were also obtained from patients treated with stavudine. Phenotypic analysis of HIV-1 isolates from 61 patients receiving prolonged (6 to 29 months) stavudine monotherapy showed that post-therapy isolates from four patients exhibited EC50 values more than 4-fold (range 7- to 16-fold) higher than the average pretreatment susceptibility of baseline isolates. Of these, HIV-1 isolates from one patient contained the zidovudine-resistance-associated substitutions T215Y and K219E, and isolates from another patient contained the multiple-nucleoside-resistance-associated substitution Q151M. Mutations in the RT gene of HIV-1 isolates from the other two patients were not detected. The genetic basis for stavudine susceptibility changes has not been identified.
Cross-resistance
Cross-resistance among HIV-1 reverse transcriptase inhibitors has been observed. Several studies have demonstrated that prolonged stavudine treatment can select and/or maintain thymidine analogue mutation (TAMs) substitutions in the HIV-1 RT (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) associated with zidovudine resistance. HIV-1 isolates with one or more TAMs substitutions exhibited reduced susceptibility to stavudine in cell culture. These TAMs substitutions are seen at a similar frequency with stavudine and zidovudine in virological treatment. The clinical relevance of these findings suggests that stavudine should be avoided in the presence of thymidine analogue mutation substitutions.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In 2-year carcinogenicity studies in mice and rats, stavudine was noncarcinogenic at doses which produced exposures (AUC) 39 and 168 times, respectively, human exposure at the recommended clinical dose. Benign and malignant liver tumors in mice and rats and malignant urinary bladder tumors in male rats occurred at levels of exposure 250 (mice) and 732 (rats) times human exposure at the recommended clinical dose.
Stavudine was not mutagenic in the Ames, E. coli reverse mutation, or the CHO/HGPRT mammalian cell forward gene mutation assays, with and without metabolic activation. Stavudine produced positive results in the in vitro human lymphocyte clastogenesis and mouse fibroblast assays, and in the in vivo mouse micronucleus test. In the in vitro assays, stavudine elevated the frequency of chromosome aberrations in human lymphocytes (concentrations of 25 to 250 mcg/mL, without metabolic activation) and increased the frequency of transformed foci in mouse fibroblast cells (concentrations of 25 to 2500 mcg/mL, with and without metabolic activation). In the in vivo micronucleus assay, stavudine was clastogenic in bone marrow cells following oral stavudine administration to mice at dosages of 600 to 2000 mg/kg/day for 3 days.
No evidence of impaired fertility was seen in rats with exposures (based on AUC) up to 137 times human exposure at the RHD. -
14 CLINICAL STUDIES
Combination Therapy
The combination use of stavudine is based on the results of clinical studies in HIV-1-infected patients in double- and triple-combination regimens with other antiretroviral agents.
One of these studies (START 1) was a multicenter, randomized, open-label study comparing stavudine (40 mg twice daily) plus lamivudine plus indinavir to zidovudine plus lamivudine plus indinavir in 202 treatment-naive patients. Both regimens resulted in a similar magnitude of inhibition of HIV-1 RNA levels and increases in CD4+cell counts through 48 weeks.
Monotherapy
The efficacy of stavudine was demonstrated in a randomized, double-blind study (AI455-019, conducted 1992 to 1994) comparing stavudine with zidovudine in 822 patients with a spectrum of HIV-1-related symptoms. The outcome in terms of progression of HIV-1 disease and death was similar for both drugs. -
16 HOW SUPPLIED/STORAGE AND HANDLING
Capsules
Stavudine Capsules USP, 15 mg are dark red opaque/light yellow opaque size ‘4’ hard gelatin capsule filled with white to off white granular powder and imprinted with ‘E’ on dark red opaque cap and ‘76’ on light yellow opaque body with black ink.
Bottle of 60 Capsules NDC: 64980-354-06
Stavudine Capsules USP, 20 mg are light brown opaque/light brown opaque size ‘3’ hard gelatin capsule filled with white to off white granular powder and imprinted with ‘E’ on light brown opaque cap and ‘77’ on light brown opaque body with black ink.
Bottle of 60 Capsules NDC: 64980-355-06
Stavudine Capsules USP, 30 mg are dark orange opaque/light orange opaque size “2” hard gelatin capsule filled with white to off white granular powder and imprinted with “C” on dark orange opaque cap and “36” on light orange opaque body with black ink.
Bottle of 60 Capsules NDC: 64980-356-06
Stavudine Capsules USP, 40 mg are dark orange opaque/dark orange opaque size “1” hard gelatin capsule filled with white to off white granular powder and imprinted with “C” on dark orange opaque cap and “37” on dark orange opaque body with black ink.
Bottle of 60 Capsules NDC: 64980-357-06
Storage
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Lactic Acidosis
Inform patients of the importance of early recognition of symptoms of symptomatic hyperlactatemia or lactic acidosis syndrome, which include unexplained weight loss, abdominal discomfort, nausea, vomiting, fatigue, dyspnea, and motor weakness. Patients in whom these symptoms develop should seek medical attention immediately. Discontinuation of stavudine therapy may be required. Advise pregnant individuals of the potential risks of lactic acidosis syndrome/hepatic steatosis syndrome [see Contraindications (4), Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
Hepatic Toxicity
Inform patients that hepatotoxicity, which may be fatal, may occur in patients treated with stavudine in combination with didanosine and hydroxyurea. Stavudine is contraindicated in combination with didanosine [see Contraindications (4)]. Avoid coadministration of stavudine with hydroxyurea [see Warnings and Precautions (5.2) and Drug Interactions (7)].
Peripheral Neuropathy
Inform patients that an important toxicity of stavudine is peripheral neuropathy. Make patients aware that peripheral neuropathy is manifested by numbness, tingling, or pain in hands or feet, and that these symptoms should be reported to their physicians. Counsel patients that peripheral neuropathy occurs with greatest frequency in patients who have advanced HIV-1 disease or a history of peripheral neuropathy, and discontinuation of stavudine may be required if toxicity develops.
Instruct caregivers of young children receiving stavudine therapy regarding detection and reporting of peripheral neuropathy [see Warnings and Precautions (5.3)].
PancreatitisInform patients that an increased risk of pancreatitis, which may be fatal, may occur in patients treated with the combination of stavudine and didanosine. Stavudine is contraindicated in combination with didanosine [see Contraindications (4)]. Closely monitor patients for symptoms of pancreatitis such as severe abdominal pain, nausea and vomiting, and fever.
Instruct patients to avoid alcohol while taking stavudine. Alcohol may increase the patient’s risk of pancreatitis or liver damage [see Warnings and Precautions (5.4)].
Lipoatrophy
Inform patients that loss of body fat (e.g., loss of fat from arms, legs, or face) may occur in individuals receiving stavudine. Monitor patients receiving stavudine for clinical signs and symptoms of lipoatrophy. Patients should be questioned routinely about body changes related to lipoatrophy [see Warnings and Precautions (5.5)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant individuals exposed to stavudine [see Use in Specific Populations (8.1)].
Lactation
Advise mothers with HIV-1 not to breastfeed because HIV-1 can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].
Dosing Information
Instruct patients not to miss a dose but if they do, patients should take stavudine as soon as possible. Inform patients that it is important to take stavudine on a regular dosing schedule and to avoid missing doses as it can result in development of resistance.
Patients should be instructed if they take too much stavudine, they should contact a poison control center or emergency room right away.
Dispense with Medication Guide available at: http://www.risingpharma.com/med-guides.html
Distributed by:
Rising Pharmaceuticals, Inc.
Saddle Brook, NJ 07663
Made in India
Code No.: TS/DRUGS/19/1993
Revised: 02/2019 -
Medication Guide
Stavudine Capsules, USP
(stav' ue deen)
What is the most important information I should know about stavudine capsules?Stavudine capsules can cause serious side effects, including:
Build-up of an acid in your blood (lactic acidosis). Lactic acidosis can happen in some people who take stavudine capsules or similar medicines (nucleoside analogues). Lactic acidosis is a serious medical emergency that can lead to death. Do not take stavudine capsules with didanosine.
Call your healthcare provider right away if you get any of the following symptoms which could be signs of lactic acidosis:
- feel very weak or tired
- have unusual (not normal) muscle pain
- have trouble breathing
- have stomach pain with nausea and vomiting
- feel cold, especially in your arms and legs
- feel dizzy or lightheaded
- have a fast or irregular heartbeat
- weight loss
- Severe liver problems. Severe liver problems, including liver failure can happen in people who take stavudine capsules. In some cases, these liver problems can lead to death. Your liver may become large (hepatomegaly) and you may develop fat in your liver (steatosis). Taking stavudine capsules with medicines that contain didanosine or hydroxyurea may increase your risk for liver problems.
Call your healthcare provider right away if you have any of the following symptoms of liver problems:
- your skin or the white part of your eyes turns yellow (jaundice)
- nausea
- light colored stools (bowel movements)
- loss of appetite
- dark or “tea-colored” urine
- pain, aching, or tenderness on the right side of your stomach area
You may be more likely to get lactic acidosis or severe liver problems if you are female, are very overweight (obese), or have been taking nucleoside analogue medicines for a long time.
- Neurologic problems including weakness of your legs, feet, arms, or hands (motor weakness) and numbness, tingling or pain in your hands or feet (peripheral neuropathy). Peripheral neuropathy can be common and severe and happens more often in people who have advanced HIV-1 disease, have a history of peripheral neuropathy, or in people who take other medicines that can cause peripheral neuropathy. In some cases, symptoms of neurologic problems may continue, worsen or temporarily worsen after you stop treatment with stavudine capsules.
Neurologic problems can be difficult to notice in children who take stavudine capsules. Ask your child’s healthcare provider for the signs and symptoms of neurologic problems in children.
- Inflammation of your pancreas (pancreatitis) can happen in people who take stavudine capsules in combination with didanosine and can lead to death. Do not take stavudine capsules with didanosine.
Call your healthcare provider right away if you have any of the following symptoms of pancreatitis:
- severe stomach (abdomen) pain
- swelling of your stomach
- nausea and vomiting
- fever
For more information about side effects, see “What are the possible side effects of stavudine capsules?”
What are stavudine capsules?
Stavudine capsules are a prescription medicine that is used with other antiretroviral medicines to treat Human Immunodeficiency Virus (HIV)-1 infection.
HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS).
Do not take stavudine capsules if you:
- are allergic to stavudine or any of the ingredients in stavudine capsules. See the end of this Medication Guide for a complete list of the ingredients in stavudine capsules.
- take a medicine that contains didanosine.
Before taking stavudine capsules, tell your healthcare provider about all of your medical conditions, including if you:
- have or had liver problems, including hepatitis C virus infection
- have or had problems with your pancreas
- have or had kidney problems
- are receiving dialysis
- have or had numbness, tingling, or pain in the hands or feet (peripheral neuropathy)
- drink alcoholic beverages
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if stavudine capsules will harm your unborn baby.
Pregnancy Registry: There is a pregnancy registry for women who take antiretroviral medicines, including stavudine capsules during pregnancy. The purpose of the registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
-
are breastfeeding or plan to breastfeed. Do not breastfeed if you take stavudine capsules.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- Stavudine can pass into your breast milk and it could harm your baby.
Talk with your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines that you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements. Especially tell your healthcare provider if you take a medicine called hydroxyurea.
Some medicines interact with stavudine capsules. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
- You can ask your healthcare provider or pharmacist for a list of medicines that interact with stavudine capsules.
- Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take stavudine capsules with other medicines.
How should I take stavudine capsules?
- Take stavudine capsules exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much stavudine capsules to take and when to take it.
- Stavudine capsules may be taken with or without food.
- Stavudine capsules should be taken every 12 hours.
- Your child’s healthcare provider should give you instructions on how to give stavudine capsules to your child.
- Your healthcare provider may change your dose. Do not change your dose of stavudine capsules without talking to your healthcare provider.
- Do not miss a dose of stavudine capsules. If you miss a dose of stavudine capsules, take it as soon as possible.
- It is important to take stavudine capsules on a regular schedule. The virus in your blood may increase and the virus may become harder to treat if you miss doses.
- If you take too much stavudine capsules, contact a poison control center or go to the nearest hospital emergency room right away.
What should I avoid while taking stavudine capsules?
- Avoid drinking alcohol while taking stavudine capsules. Alcohol may increase your risk of side effects during treatment with stavudine capsules.
What are the possible side effects of stavudine capsules?
Stavudine capsules can cause serious side effects including:
- See “What is the most important information I should know about stavudine capsules?”
- Loss of body fat (lipoatrophy) from the arms, legs, or face. Loss of body fat (lipoatrophy) happens more often in people who take stavudine capsules than in people who take other similar HIV-1 medicines. Your healthcare provider will monitor you for changes in your body fat. It is important to tell your healthcare provider if you notice any changes.
- Changes in your immune system (immune reconstitution syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine.
The most common side effects of stavudine capsules include:
- headache
- diarrhea
- rash
- nausea
- vomiting
These are not all the possible side effects of stavudine capsules.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store stavudine capsules?
Capsules:
- Store stavudine capsules in a tightly closed container at room temperature at 15° to 30°C (59° to 86°F).
Keep stavudine capsules and all medicines out of the reach of children.
General information about the safe and effective use of stavudine capsules.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use stavudine capsules for a condition for which it was not prescribed. Do not give stavudine capsules to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about stavudine capsules that is written for health professionals.
What are the ingredients in stavudine capsules?
Active ingredient: stavudine
Inactive ingredients:microcrystalline cellulose, sodium starch glycolate, anhydrous lactose, and magnesium stearate. The hard gelatin shell consists of gelatin, sodium lauryl sulfate, titanium dioxide, yellow iron oxide, and red iron oxide. The capsules are printed with black ink containing black iron oxide.
Dispense with Medication Guide available at: http://www.risingpharma.com/med-guides.html
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Distributed by:
Rising Pharmaceuticals, Inc.
Saddle Brook, NJ 07663
Made in India
Code No.: TS/DRUGS/19/1993
Revised: 02/2019 -
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 15 mg (60 Capsule Bottle)
Rising® NDC: 64980-354-06
Stavudine
Capsules, USP
15 mg
PHARMACIST: Dispense the
accompanying Medication Guide to
each patient.
60 Capsules Rx only

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 20 mg (60 Capsule Bottle)
Rising® NDC: 64980-355-06
Stavudine
Capsules, USP
20 mg
PHARMACIST: Dispense the
accompanying Medication Guide to
each patient.
60 Capsules Rx only

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 30 mg (60 Capsule Bottle)
Rising® NDC: 64980-356-06
Stavudine
Capsules, USP
30 mg
PHARMACIST: Dispense the
accompanying Medication Guide to
each patient.
60 Capsules Rx only

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 40 mg (60 Capsule Bottle)
Rising® NDC: 64980-357-06
Stavudine
Capsules, USP
40 mg
PHARMACIST: Dispense the
accompanying Medication Guide to
each patient.
60 Capsules Rx only

-
INGREDIENTS AND APPEARANCE
STAVUDINE
stavudine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64980-354 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength STAVUDINE (UNII: BO9LE4QFZF) (STAVUDINE - UNII:BO9LE4QFZF) STAVUDINE 15 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color RED (Dark Red Opaque) , YELLOW (Light Yellow Opaque) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code E;76 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64980-354-06 60 in 1 BOTTLE; Type 0: Not a Combination Product 12/29/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077672 12/29/2008 STAVUDINE
stavudine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64980-355 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength STAVUDINE (UNII: BO9LE4QFZF) (STAVUDINE - UNII:BO9LE4QFZF) STAVUDINE 20 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color BROWN (Light Brown Opaque) Score no score Shape CAPSULE Size 16mm Flavor Imprint Code E;77 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64980-355-06 60 in 1 BOTTLE; Type 0: Not a Combination Product 12/29/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077672 12/29/2008 STAVUDINE
stavudine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64980-356 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength STAVUDINE (UNII: BO9LE4QFZF) (STAVUDINE - UNII:BO9LE4QFZF) STAVUDINE 30 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color ORANGE (Dark Orange Opaque) , ORANGE (Light Orange Opaque) Score no score Shape CAPSULE Size 18mm Flavor Imprint Code C;36 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64980-356-06 60 in 1 BOTTLE; Type 0: Not a Combination Product 12/29/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077672 12/29/2008 STAVUDINE
stavudine capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64980-357 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength STAVUDINE (UNII: BO9LE4QFZF) (STAVUDINE - UNII:BO9LE4QFZF) STAVUDINE 40 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color ORANGE (Dark Orange Opaque) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code C;37 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64980-357-06 60 in 1 BOTTLE; Type 0: Not a Combination Product 12/29/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077672 12/29/2008 Labeler - Rising Pharmaceuticals, Inc. (041241766) Registrant - Aurobindo Pharma Limited (650082092) Establishment Name Address ID/FEI Business Operations Aurobindo Pharma Limited 918917642 ANALYSIS(64980-354, 64980-355, 64980-356, 64980-357) , MANUFACTURE(64980-354, 64980-355, 64980-356, 64980-357)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.