FORZINITY- elamipretide hydrochloride injection
FORZINITY by
Drug Labeling and Warnings
FORZINITY by is a Prescription medication manufactured, distributed, or labeled by Stealth Biotherapeutics Inc., PCI San Diego, Inc., Sharp Clinical Services, Inc., AMPAC Fine Chemicals LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FORZINITY™ safely and effectively. See full prescribing information for FORZINITY.
FORZINITY (elamipretide) injection, for subcutaneous use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
FORZINITY™ is a mitochondrial cardiolipin binder indicated to improve muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kg. ( 1)
This indication is approved under accelerated approval based on an improvement in knee extensor muscle strength, an intermediate clinical endpoint. ( 14) Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 280 mg/3.5 mL (80 mg/mL) solution for injection in single-patient-use vials. ( 3)
WARNINGS AND PRECAUTIONS
- Benzyl alcohol toxicity: Do not use in neonates. ( 5.1)
ADVERSE REACTIONS
Most common adverse reactions are injection site reactions. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Stealth BioTherapeutics Inc. at 1-844-444-6486 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications For Renal Impairment
2.3 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Benzyl Alcohol Toxicity in Neonates
5.2 Hypersensitivity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
FORZINITY is indicated to improve muscle strength in adult and pediatric patients with Barth syndrome weighing at least 30 kg.
This indication is approved under accelerated approval based on an improvement in knee extensor muscle strength, an intermediate clinical endpoint [see Clinical Studies (14)] . Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of FORZINITY in patients weighing at least 30 kg is 40 mg subcutaneously once daily.
Patients should receive the dose at the same time each day.
Missed Dose
If a dose is missed, skip the dose and take the next dose of FORZINITY at the scheduled time. Do not take a double dose of FORZINITY.
2.2 Dosage Modifications For Renal Impairment
Refer to Table 1 for dosage modifications in adults with renal impairment.
Table 1: Recommended Dosage in Adults with Renal Impairment Estimated Glomerular Filtration Rate (eGFR) (mL/minute) Recommended Dosage Greater than or equal to 30 mL/minute 40 mg subcutaneously once daily Less than 30 mL/minute and NOT on dialysis 20 mg subcutaneously once daily There is insufficient information to recommend a dosage regimen in adults with eGFR less than 30 mL/minute and on dialysis.
There is insufficient information to recommend a dosage regimen in pediatric patients weighing 30 kg or greater with renal impairment.
2.3 Important Administration Instructions
FORZINITY is a clear, colorless to yellow aqueous solution that contains the preservative, benzyl alcohol. Visually inspect each vial of FORZINITY for particulate matter and cloudiness prior to administration, whenever solution and container permit. Do not use if the solution is cloudy or particulate matter is present.
Adult patients or caregivers may administer FORZINITY after proper training in preparing FORZINITY vials for administration, if a healthcare provider determines that it is appropriate, and with medical follow-up as necessary.
Use aseptic technique to prepare and administer FORZINITY. Administer FORZINITY by subcutaneous injection in the abdomen (at least 2 inches from the navel) or outer thigh and rotate the injection site daily. Do not inject where the skin is tender, bruised, red, or hard. Avoid injecting into scars or stretch marks. Refer to the Instructions for Use for preparation and administration.
FORZINITY is for single-patient-use only.
Do not mix other products in the same syringe.
Discard vials 8 days after first opening.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Serious hypersensitivity to elamipretide or any of the excipients in FORZINITY [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Benzyl Alcohol Toxicity in Neonates
FORZINITY is not approved for use in neonates. Serious adverse reactions, including fatal reactions, of new onset or worsening metabolic acidosis that progressed to neurotoxicity, and in some cases gasping syndrome, have been reported in low-birth weight neonates (less than 2,500 grams) and preterm neonates (gestational age less than 34 weeks) who received benzyl alcohol (BA)-containing drugs intravenously. FORZINITY is not approved for intravenous use [see Dosage and Administration (2.1)] . Gasping syndrome is a life-threatening condition in neonates caused by BA toxicity and is primarily characterized by multiorgan dysfunction secondary to metabolic acidosis, which leads to gasping respirations and death. The minimum amount of BA at which these serious adverse reactions, including fatal reactions, may occur is not known (FORZINITY contains 20 mg of BA per mL). [See Use in Specific Populations (8.4).]
5.2 Hypersensitivity
Hypersensitivity reactions, including serious allergic reactions requiring emergency medical intervention, have been reported in patients receiving FORZINITY. These reactions have included skin manifestations such as rash, papular lesions, and eczematous dermatitis, as well as respiratory symptoms including cough [see Adverse Reactions (6.1)] . Reactions may occur within minutes to months after treatment initiation.
Monitor patients for signs and symptoms of hypersensitivity reactions during treatment. If a serious hypersensitivity reaction occurs, do not administer further doses of FORZINITY and institute appropriate emergency treatment, including epinephrine, antihistamines, and corticosteroids as clinically indicated. Patients who have experienced a serious hypersensitivity reaction should not be rechallenged with FORZINITY [see Contraindications (4)] .
For mild to moderate skin reactions, consider treatment with topical corticosteroids and oral antihistamines. Consider discontinuation of FORZINITY if persistent or severe skin reactions develop.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the FORZINITY clinical development program, 12 male patients aged 12 to 35 years with genetically-confirmed Barth syndrome received treatment with daily subcutaneous injections of 40 mg FORZINITY. Eleven of these 12 patients were Caucasian.
Patients first participated in a double-blind, placebo-controlled crossover trial where they were randomized to one of two sequences:
- 12 weeks of FORZINITY in Period 1 then a 4-week washout followed by 12 weeks of placebo in Period 2 or
- 12 weeks of placebo in Period 1 then a 4-week washout followed by 12 weeks of FORZINITY in Period 2
Ten patients completed the randomized trial and entered the open-label extension period where they received FORZINITY once daily. Eight of these patients received FORZINITY for 168 weeks, three of whom received FORZINITY for a total of 192 weeks.
Adverse reactions occurring more commonly on FORZINITY than on placebo include injection site reactions such as injection site erythema, pain, induration, pruritus, bruising, and urticaria (Table 2).
Table 2: Summary of Adverse Drug Reactions in the Placebo-Controlled Crossover Study, Barth Safety Population Combined (Periods 1 and 2) Elamipretide Placebo N=12 N=12 n (%) n (%) Any local administration reaction 12 (100) 8 (67) Injection site erythema 12 (100) 3 (25) Injection site induration 8 (67) 2 (17) Injection site pruritus 8 (67) 2 (17) Injection site pain 9 (75) 5 (42) Injection site bruising 3 (25) 0 Injection site urticaria 3 (25) 0 Injection site hemorrhage 0 1 (8) Eosinophilia
Increases in absolute eosinophil counts were noted frequently in studies where duration of administration of FORZINITY was 30 days or greater. Eosinophil counts generally peaked around 90 days after initial exposure (mean increase from baseline ~0.5 to 0.6 × 10 3/uL) and returned to baseline levels after 6 to 12 months of continuous exposure or after discontinuation of FORZINITY. The elevation in eosinophils was not associated with clinical manifestations or changes in other laboratory parameters.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Barth Syndrome is a rare, X-linked, recessive, genetic disorder and is not likely to affect females. Therefore, there are no data with FORZINITY use in pregnant women to evaluate for a drug-related risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, no adverse developmental outcomes occurred at any dose tested (see Data). FORZINITY contains benzyl alcohol as a preservative. Because benzyl alcohol is rapidly metabolized by a pregnant woman, benzyl alcohol exposure in the fetus is unlikely.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
In pregnant rats, once-daily intravenous infusion of elamipretide during the period of organogenesis [gestation day (GD) 7 to GD 17] did not result in embryo-fetal developmental toxicity at doses tested up to 10 mg/kg/day, approximately 4 times the clinical exposure at the maximum recommended human dose (MRHD) of 40 mg/day, based on area under the concentration-time curve (AUC).
In pregnant rabbits, intravenous infusion with elamipretide once daily during the period of organogenesis (GD 7 to GD 19) did not result in embryo-fetal developmental toxicity at doses tested up to 50 mg/kg/day, approximately 10 times the clinical exposure at the MRHD, based on AUC.
In a pre- and postnatal study in rats, elamipretide was subcutaneously administered at doses of 0, 5, 10 or 15 mg/kg/day throughout pregnancy and lactation (GD 6 to Lactation Day 20). No adverse developmental effects were observed at doses up to 15 mg/kg/day, approximately 6-times the clinical exposure at the MRHD, based on AUC.
8.2 Lactation
Risk Summary
Barth Syndrome is a rare, X-linked, recessive, genetic disorder and is not likely to affect females. Therefore, there is no data to evaluate the presence of FORZINITY in human milk, the effect on the breastfed infant, or the effects on milk production.
FORZINITY contains benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a lactating female, benzyl alcohol exposure in the breastfed infant is unlikely. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for FORZINITY and any potential adverse effects on the breastfed infant from FORZINITY or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of FORZINITY to improve muscle strength have been established in pediatric patients with Barth syndrome weighing at least 30 kg. Use of FORZINITY for this indication is supported by improvement in knee extensor muscle strength, an intermediate clinical endpoint, observed in an open-label extension study of FORZINITY that included seven pediatric patients aged 12 years and older. [see Dosage and Administration (2.1), and Clinical Studies (14)] .
The safety and effectiveness of FORZINITY have not been established in pediatric patients weighing less than 30 kg.
FORZINITY is not approved for use in neonates. Serious adverse reactions, including fatal reactions, of new onset or worsening metabolic acidosis that progressed to neurotoxicity, and in some cases gasping syndrome, have been reported in low-birth weight neonates and preterm neonates who received BA containing drugs intravenously (FORZINITY is not approved for intravenous use) [see Dosage and Administration (2.1)] . Gasping syndrome is a life-threatening condition in neonates caused by BA toxicity that is characterized by new onset or worsening metabolic acidosis with gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and gasping respirations followed by death. In reported cases, BA in amounts of 99 to 234 mg/kg/day produced blood BA levels of 6.6 to 14.9 mg/dL, but the minimum amount of BA at which gasping syndrome may occur in neonates is not known (FORZINITY contains 20 mg of BA per mL) [see Warnings and Precautions (5.1)] .
8.5 Geriatric Use
Clinical studies of FORZINITY did not enroll subjects with Barth syndrome aged 65 years and older.
8.6 Renal Impairment
No dosage adjustment is required for patients with mild (eGFR 60 to 89 mL/min) or moderate (eGFR 30 to 59 mL/min) renal impairment. The FORZINITY dose should be reduced by one half administered subcutaneously once daily in adults with severe renal impairment (eGFR less than 30 mL/min) who are not on dialysis [see Dosage and Administration (2.2)] . FORZINITY has not been studied in patients with renal failure on dialysis [see Clinical Pharmacology (12.3)] .
-
10 OVERDOSAGE
There were no reports of overdose during clinical trials with FORZINITY. Symptoms and signs of overdose are likely to be histamine-related (e.g., decreased blood pressure, presyncope) and should be managed according to standard of care. Interrupt treatment with FORZINITY if there is suspicion of overdose.
-
11 DESCRIPTION
FORZINITY contains elamipretide, a mitochondrial cardiolipin binder. Elamipretide is isolated as a hydrochloride salt that is freely soluble in water. The chemical name for elamipretide hydrochloride is L-Phenylalaninamide, D-arginyl-2,6-dimethyl-L-tyrosyl-L-lysyl-, hydrochloride (1:3). Its molecular formula is C 32H 49N 9O 5∙3HCl and its molecular weight is 749.2.
The structure of elamipretide hydrochloride is:
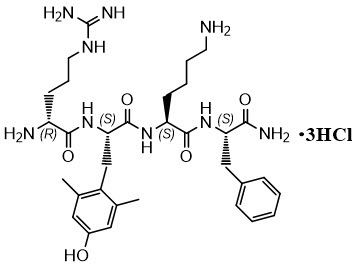
All amino acid residues in FORZINITY have the L configuration except for arginine which has the D configuration. The peptide sequence is denoted as D-Arg-2,6-dimethyl-Tyr-Lys-Phe-NH 2.
FORZINITY is a ready-to-use sterile, clear, colorless to yellow aqueous solution supplied as single-patient-use vials containing 3.5 mL solution for subcutaneous injection. Each 0.5 mL dose of FORZINITY contains 40 mg of elamipretide (equivalent to 46.8 mg elamipretide hydrochloride), 10 mg benzyl alcohol as a preservative, and 2.07 mg monobasic sodium phosphate (as monohydrate). The product may contain hydrochloric acid or sodium hydroxide to adjust pH. The pH of FORZINITY solution is 4.7 to 6.1.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
FORZINITY is a mitochondrial cardiolipin binder that localizes to the inner mitochondrial membrane and improves mitochondrial morphology and function.
12.2 Pharmacodynamics
Effects on QTc Interval
Cardiac electrophysiology
Clinically significant QTc interval prolongation was not observed at 3 times the peak concentration of the maximum recommended FORZINITY dose.
12.3 Pharmacokinetics
Elamipretide exposure increases proportionally over a dose range of 2 to 80 mg following daily subcutaneous injections with minimal accumulation.
Absorption
Maximum elamipretide concentrations were reached between 0.5 to 1 hour after subcutaneous administration. The absolute bioavailability following subcutaneous administration is approximately 92%. FORZINITY exposure is comparable after subcutaneous injection to the thigh or to the abdomen.
Distribution
Elamipretide is distributed throughout total body water with an approximate volume of distribution of 0.5 L/kg. There is low binding to plasma proteins (approximately 39%).
Elimination
Metabolism
Elamipretide is metabolized via sequential C-terminal degradation to the M1 tripeptide and M2 dipeptide metabolites, which do not have pharmacological activity.
Excretion
Elamipretide and its metabolites M1 and M2 are excreted in the urine. At 48 hours post-dose, approximately 100% of the FORZINITY dose was recovered in the urine as either elamipretide, M1, or M2 in patients with normal renal function.
Specific Populations
Patients with Renal Impairment
Elamipretide exposure (AUC) increased by 39% in subjects with creatinine clearance 60 to 89 mL/min, 75% in subjects with creatinine clearance 30 to 59 mL/min, and 125% in subjects with creatinine clearance less than 30 mL/min not on dialysis. Renal impairment was categorized based on 24-hour measured urinary creatinine clearance. There was minimal accumulation of elamipretide with daily dosing, regardless of the severity of renal impairment. There was a significant increase in exposure of the M1 and M2 metabolites, up to 280% and 640%, respectively, in subjects with severe renal impairment not on dialysis (creatinine clearance less than 30 mL/min).
While the effect of renal impairment on elamipretide pharmacokinetics was characterized using 24-hour measured urinary creatinine clearance, analyses conducted with estimated glomerular filtration (CKD-EPI equation) support the recommendations for use in this specific population [see Dosage and Administration (2.2)and Use in Specific Populations (8.6)] .
Patients with Hepatic Impairment
No hepatic metabolism was observed for elamipretide in vitro.Hepatic impairment is not expected to alter the pharmacokinetics (PK) of elamipretide.
Drug Interaction Studies
In Vitro Studies
CYP enzymes: Elamipretide does not inhibit CYP1A, CYP2D6, CYP2E1, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A. Elamipretide does not induce metabolism by CYP1A2, CYP2B6, or CYP3A4.
Drug transporters: Elamipretide does not inhibit the activity of OCT2, BCRP, OAT1, OAT3, OATP1B1, OATP1B3, P-gp, or MATE2-K. Elamipretide is an inhibitor of MATE1 (IC 503.53 μM).
In Vivo Clinical Studies
Aspirin: administration of elamipretide (0.1 mg/kg/hour for 4 hours via intravenous infusion) starting 4 hours after a single dose of 650 mg aspirin did not affect the PK of aspirin and its metabolite, salicylic acid. There was no impact on the antiplatelet activity of aspirin when co-administered with elamipretide.
Clopidogrel: administration of elamipretide (0.1 mg/kg/hour for 4 hours via intravenous infusion) starting 4 hours after a single dose of 300 mg clopidogrel did not significantly affect the PK of clopidogrel. There was no impact on the anti-thrombotic activity of clopidogrel when co-administered with elamipretide.
Unfractionated heparin: administration of elamipretide (0.25 mg/kg/hour for 4 hours via intravenous infusion) had no significant impact on activated partial thromboplastin time (aPTT) and anti-factor Xa activity of unfractionated heparin, when co-administered 7 hours after starting the unfractionated heparin infusion.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with elamipretide.
Elamipretide was negative in an in vitrobacterial reverse mutation assay, a chromosomal aberration assay in Chinese hamster ovary cells, and an in vivorat bone marrow micronucleus assay.
Subcutaneous doses of elamipretide in rats of up to 20 mg/kg/day, approximately 5-times the clinical exposure at the MRHD of 40 mg, did not adversely affect fertility or reproductive performance.
-
14 CLINICAL STUDIES
FORZINITY was evaluated in a randomized, double-blind, placebo-controlled, crossover trial and its 192-week, open-label, single-arm extension period.
The randomized trial evaluated the efficacy and safety of once daily FORZINITY 40 mg injected subcutaneously for 12 weeks in 12 subjects ≥12-years-old and >30 kg with genetically confirmed Barth syndrome. The primary endpoints for the randomized trial were distance walked during 6-minute walk test and Total Fatigue Score on the Barth syndrome Symptom Assessment. FORZINITY was not superior to placebo on these primary endpoints. Ten subjects completed the randomized trial and entered the extension period designed to evaluate long-term safety and tolerability of FORZINITY. Eight of these 10 subjects participated through Week 168 of the extension period.
Knee extensor muscle strength measured by handheld dynamometry was evaluated as one of the secondary endpoints in the randomized trial and in the extension period. Increases in knee extensor muscle strength were not observed during the randomized trial but were observed during the extension period. At the pre-dose baseline visit at the start of the randomized trial, median (min, max) muscle strength was 124 (92, 176) newtons. Table 3 shows descriptive changes from pre-dose baseline for knee extensor muscle strength during the randomized controlled trial and extension period.
Table 3. Descriptive Statistics, Changes from Baseline *in Muscle Strength (newtons) Visit N Median Change Min, Max Change - * All descriptive statistics use the muscle strength measurement obtained immediately prior to the first dose of study medication at the beginning of Period 1 of the randomized controlled trial as baseline
Randomized Controlled Trial Week 12 Placebo 12 -5 -31, 48 Week 12 Elamipretide 12 4 -41, 86 Open-Label Extension Period Week 12 10 34 7, 95 Week 24 9 68 3, 90 Week 36 8 57 9, 92 Week 48 8 41 -2, 144 Week 72 8 35 -4, 100 Week 168 8 63 38, 78 -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
FORZINITY (elamipretide) injection is a sterile, clear, colorless to yellow aqueous solution supplied as:
- Carton containing four 280 mg/3.5 mL (80 mg/mL) single-patient-use vials (NDC: 72507-800-04)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and caregiver to read the FDA-approved patient labeling (Instructions for Use).
Instruct patients and caregivers to discard vials 8 days after first opening.
Instruct patients and caregivers on how to prepare and administer the correct dose of FORZINITY using aseptic technique. Instruct patients and caregivers to administer FORZINITY by subcutaneous injection in the abdomen (at least 2 inches from the navel) or outer thigh and to rotate the injection site daily according to the Instructions for Use. Instruct patients and caregivers not to inject where the skin is tender, bruised, red, or hard and to avoid injecting into scars or stretch marks.
Inform patients and caregivers that injection site reactions such as erythema, pain, induration, pruritus, bruising, or urticaria can be treated with antihistamines or topical corticosteroids.
Advise the patient and caregiver to discontinue FORZINITY and seek immediate medical attention if any signs or symptoms of an immediate hypersensitivity reaction occur [see Warnings and Precautions (5.2)] .
-
SPL UNCLASSIFIED SECTION
FORZINITY manufactured for:
Stealth BioTherapeutics Inc.
Needham, MA 02494FORZINITY™ (elamipretide) is a trademark of Stealth BioTherapeutics Inc.
This product is covered by one or more of the following US Patents: US 7,576,061, US 7,550,439, US 9,687,519, US 11,083,771, US 11,083,772, US 11,771,734
-
PATIENT PACKAGE INSERT
INSTRUCTIONS FOR USE
FORZINITY™ [for zin' i tee]
(elamipretide)
injectionThis Instructions for Use contains information on how to inject FORZINITY. Read this Instructions for Use before you inject a dose of FORZINITY for the first time and each time you get a refill. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Ask your healthcare provider about any instructions you do not understand.
Important information you need to know before preparing and injecting FORZINITY:
- Your or your child's healthcare provider will show you how to prepare and inject FORZINITY before you use the vial for the first time. It is recommended that FORZINITY injections be prepared and given by adults or adult caregivers.
- FORZINITY is for injection under the skin (subcutaneous injection).
- FORZINITY is injected 1 time each day, at the same time each day.
- If a dose of FORZINITY is missed, skip that dose, and take the next dose of FORZINITY at the next scheduled time. Do not double a dose of FORZINITY.
- FORZINITY vial contains enough medicine for more than 1 dose.
- Do notmix FORZINITY with other medicines in the same syringe.
- Only use disposable syringes and needles prescribed by your healthcare provider or included with your medicine. Using the wrong syringe can lead to a mistake in your dose and you could inject too much or too little FORZINITY.
- Always use a new disposable syringe and needle for each injection.
- Do notreuse or share your needles with other people.
- Check the package containing the disposable syringe. If the package has been opened or damaged, do not use the syringe. Throw away the damaged syringe in an FDA-cleared sharps disposal container. See Step 8: "Throw away (dispose of) used needles and syringes."
Storing FORZINITY
- Store unused vials of FORZINITY in the refrigerator between 36°F to 46°F (2°C to 8°C).
- After the first dose, store opened vials in the refrigerator between 36°F to 46°F (2°C to 8°C) or at room temperature between 68°F to 77°F (20°C to 25°C).
- Do notfreeze FORZINITY vials.
- Throw away FORZINITY vials 8 days after first opening, even if some medicine is still left in the vial.
Keep FORZINITY, needles, disposable syringes, and all medicines out of the reach of children.
When using a new vial of FORZINITY:
- Remove the carton from the refrigerator.
- Make sure the name FORZINITY appears on the carton labeling.
- Make sure the tamper evident seal is on the carton.
- Remove the vial from the carton.
- Allow the vial to sit at room temperature for at least 10 minutes.
- Remove the "Instructions for Use" from the carton.
- Write the "Discard After" date on the vial. Repeat this step for each new vial you open.
- Return the carton containing unused vials to the refrigerator.
Step 1: Gather the following supplies on a clean flat, work surface (See Figure 1-A) - 1 vial of FORZINITY (See Figure 1-B). If using an opened vial stored in a refrigerator, allow the opened vial to sit at room temperature for at least 10 minutes.
- 1 new disposable syringe with the needle attached for each injection (See Figure 1-C)
- 2 alcohol wipes
- cotton ball or gauze pad
- sharps disposal container

Step 2: Check your FORZINITY vial - Make sure the name FORZINITY (elamipretide) appears on the vial label.
- Check the expiration date (EXP) on the vial label (See Figure 2). Do notuse after the expiration date (EXP) on label.
- Do notuse beyond the "Discard After" date written on the vial.
- Make sure that the liquid in the vial is clear, colorless to yellow. Do notuse if the liquid is discolored or cloudy, or if the liquid has lumps, flakes, or particles.
- Do notuse if the glass vial has cracks.
- Do notuse if the plastic cap on a new vial is broken or missing.
Step 3: Prepare the vial - Wash your hands well with soap and water (See Figure 3-A)
- Remove the plastic cap on the vial (if using a new vial) (See Figure 3-B). Throw away the plastic cap in the household trash.
- Clean the top of the vial stopper with an alcohol wipe before each injection (See Figure 3-C).
- Throw away the alcohol wipe in the household trash.
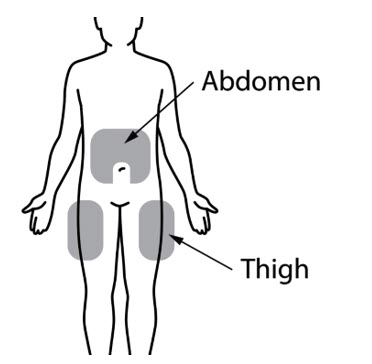
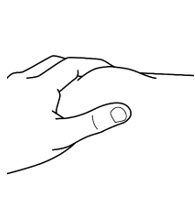
Step 4: Prepare the injection site FORZINITY is injected directly into a layer of fat under your skin (subcutaneous injection). - Choose 1 of the following injection sites (See
Figure 4).
- stomach-area (abdomen), 2 inches away from belly button
- outer area of the thigh
- Change (rotate) the injection site daily with each injection to reduce the risk of skin problems. You may want to use a calendar or diary to record your injection sites.
- Do notinject or give injections in the same location sites as other medicines.
- Do notinject or give FORZINITY injections into areas where the skin is tender, red, bruised, hard, scarred or has stretch marks.
Clean the injection site with the second alcohol wipe. - Do nottouch, fan, or blow on the cleaned area.
- Wait for about 10 seconds for the area to dry.
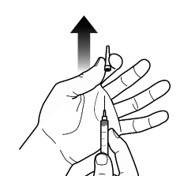
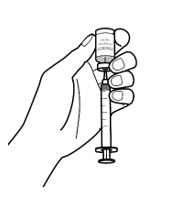
Step 5: Prepare syringe - Hold the syringe by the barrel with the needle cap pointing up and carefully pull the needle cap straight off and away from your body (See Figure 5-A).
- Throw the needle cap into the sharps disposal container (See Figure 5-B).
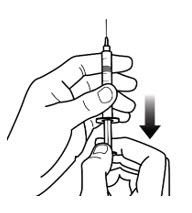
- Fill the syringe with air by pulling back on the plunger to the prescribed dose in milliliters (mL) (See Figure 5-C).
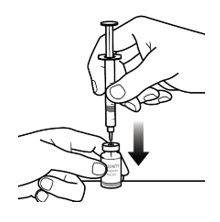
- With the vial on a flat work surface and in the upright position, insert the needle straight down into the center of the vial rubber stopper (See Figure 5-D).
- Slowly inject all the air from the syringe into the vial (See Figure 5-E).
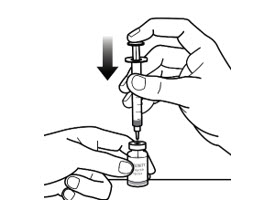
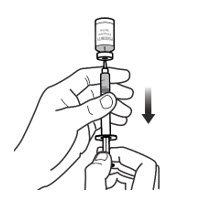
Step 6: Withdraw the dose - Keep the needle inside the vial. Turn the vial and syringe upside down with 1 hand. Keep needle tip in the liquid (See Figure 6-A).
- Slowly pull back on the plunger to withdraw your prescribed dose into the syringe (See Figure 6-B). Be careful not to pull the plunger out of the syringe.
- Keep the needle inside the vial. Check for air bubble(s). Small air bubble(s) are normal. Large amounts of air bubble(s) may give you the wrong dose of FORZINITY.
- If there are large air bubble(s), then gently tap the side of the syringe with your fingers until the large air bubble(s) rises to the top of the syringe (See Figure 6-C).
- Slowlypush the plunger up to push the large air bubble(s) back into the vial (See Figure 6-C).
- If no large air bubbles are present, gently pull back on the plunger again more slowly to fill the syringe to your prescribed dose.
- Return the vial to an upright position and place it on the clean, flat work surface.
- While holding the vial with 1 hand and the barrel of the syringe between the fingertips of your other hand, pull the needle straight out of the vial (See Figure 6-D).
- Keep the syringe in your hand. Do notset it down.

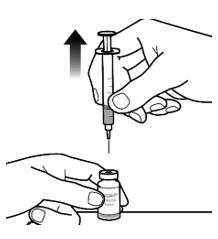
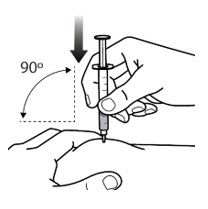
Step 7: Take or give the injection - With 1 hand, pinch about 2 inches of skin at the injection site between your thumb and index (pointer) finger (See Figure 7-A). Pinching the skin helps to make sure that you inject the medicine under the skin (subcutaneous).
- With the other hand, hold the middle of the syringe at a 90º degree angle to your body and insert the needle into the folded skin between the thumb and index (pointer) finger (See Figure 7-B).
- Maintaining the 90º degree angle to your body, push the needle straight into the injection site (See Figure 7-B).
- Make sure that you push the needle all the way into the skin. Do nothold or push on the plunger while inserting the needle.
- After the needle is inserted, the skin no longer needs to be pinched.
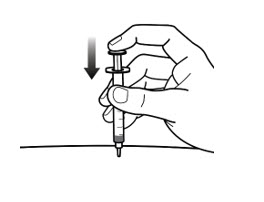
- Inject the solution by gently pushing the syringe plunger all the way until the syringe is empty (See Figure 7-C).
- Take the needle out of your skin at the same angle you inserted it. Keep the plunger pushed in while removing the needle.
- If there is bleeding, lightly press a cotton ball or gauze pad over the injection site.
- Throw away used alcohol wipes in the household trash.
- Do not put the needle cover back on the needle or replace the needle.
- Do not throw away (dispose of) needles and syringes in your household trash.
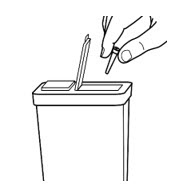
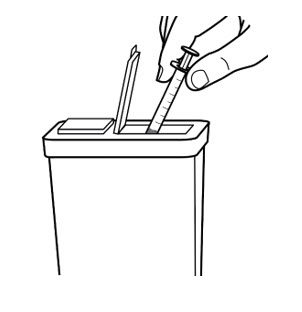
- Throw away used needles and syringes in an FDA-cleared sharps disposal container right away after use (See Figure 8).
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at:
http://www.fda.gov/safesharpsdisposal Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
FORZINITY manufactured for:
Stealth BioTherapeutics Inc.
Needham, MA 02494This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: 9/2025
-
PRINCIPAL DISPLAY PANEL - 3.5 mL Vial Carton
NDC: 72507-800-04
Rx OnlyFORZINITY™
(elamipretide) Injection280 mg/3.5 mL (80 mg/mL)
For Subcutaneous Use Only
Contains Preservative. Not for use in neonates.
Contains 4 Single-Patient-Use Vials

-
INGREDIENTS AND APPEARANCE
FORZINITY
elamipretide hydrochloride injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72507-800 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ELAMIPRETIDE HYDROCHLORIDE (UNII: E40WZ3BK2D) (ELAMIPRETIDE - UNII:87GWG91S09) ELAMIPRETIDE HYDROCHLORIDE 280 mg in 3.5 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) BENZYL ALCOHOL (UNII: LKG8494WBH) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Product Characteristics Color yellow (Clear, colorless to yellow aqueous) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72507-800-04 4 in 1 CARTON 10/30/2025 1 NDC: 72507-800-30 3.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215244 10/30/2025 Labeler - Stealth Biotherapeutics Inc. (963920215) Establishment Name Address ID/FEI Business Operations PCI San Diego, Inc. 023050730 manufacture(72507-800) , analysis(72507-800) Establishment Name Address ID/FEI Business Operations Sharp Clinical Services, Inc. 079209266 pack(72507-800) , label(72507-800) Establishment Name Address ID/FEI Business Operations AMPAC Fine Chemicals LLC 841185296 analysis(72507-800) Establishment Name Address ID/FEI Business Operations AMPAC Fine Chemicals LLC 073903937 analysis(72507-800)
Trademark Results [FORZINITY]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FORZINITY 90860355 not registered Live/Pending |
STEALTH BIOTHERAPEUTICS INC. 2021-08-02 |
 FORZINITY 87815982 not registered Live/Pending |
Stealth Biotherapeutics Corp 2018-03-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.