CABENUVA- cabotegravir and rilpivirine kit
Cabenuva by
Drug Labeling and Warnings
Cabenuva by is a Prescription medication manufactured, distributed, or labeled by ViiV Healthcare Company, Glaxo Wellcome Manufacturing Pte. Ltd, Janssen Pharmaceutica NV. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CABENUVA safely and effectively. See full prescribing information for CABENUVA.
CABENUVA (cabotegravir extended-release injectable suspension; rilpivirine extended-release injectable suspension), co-packaged for intramuscular use
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
Warnings and Precautions, Hypersensitivity Reactions (5.1)
4/2025
INDICATIONS AND USAGE
CABENUVA, a 2-drug co-packaged product of cabotegravir, an HIV-1 integrase strand transfer inhibitor (INSTI), and rilpivirine, an HIV-1 non‑nucleoside reverse transcriptase inhibitor (NNRTI), is indicated as a complete regimen for the treatment of HIV-1 infection in adults and adolescents 12 years of age and older and weighing at least 35 kg to replace the current antiretroviral regimen in those who are virologically suppressed (HIV‑1 RNA <50 copies/mL) on a stable antiretroviral regimen with no history of treatment failure and with no known or suspected resistance to either cabotegravir or rilpivirine. (1)
DOSAGE AND ADMINISTRATION
- Refer to full prescribing information for detailed information on dosage and administration recommendations. (2)
- Prior to initiating treatment with CABENUVA, oral lead-in dosing may be considered to assess the tolerability of cabotegravir and rilpivirine with the recommended dosage used for approximately 1 month. (2.3)
- For gluteal intramuscular injection only. (2.4, 2.5, 2.9)
- Recommended Monthly Dosing Schedule: Initiate injections of CABENUVA (600 mg of cabotegravir and 900 mg of rilpivirine) on the last day of current antiretroviral therapy or oral lead-in and continue with injections of CABENUVA (400 mg of cabotegravir and 600 mg of rilpivirine) every month thereafter. (2.4)
- Recommended Every-2-Month Dosing Schedule: Initiate injections of CABENUVA (600 mg of cabotegravir and 900 mg of rilpivirine) on the last day of current antiretroviral therapy or oral lead-in for 2 consecutive months and continue with injections of CABENUVA every 2 months thereafter. (2.5)
DOSAGE FORMS AND STRENGTHS
Injection:
- Kit of single-dose vials of 400 mg/2 mL (200 mg/mL) of cabotegravir extended-release injectable suspension and 600 mg/2 mL (300 mg/mL) of rilpivirine extended-release injectable suspension. (3)
- Kit of single-dose vials of 600 mg/3 mL (200 mg/mL) of cabotegravir extended-release injectable suspension and 900 mg/3 mL (300 mg/mL) of rilpivirine extended-release injectable suspension. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Serious or severe hypersensitivity reactions have been reported with cabotegravir- and rilpivirine-containing regimens. Reactions associated with cabotegravir include Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). Reactions associated with rilpivirine-containing regimens include cases of drug reaction with eosinophilia and systemic symptoms (DRESS). Discontinue CABENUVA immediately if signs or symptoms of hypersensitivity reactions develop. (5.1)
- Serious post-injection reactions with rilpivirine were reported. Monitor and treat as clinically indicated. (5.2)
- Hepatotoxicity has been reported in patients receiving cabotegravir or rilpivirine. Monitoring of liver chemistries is recommended. Discontinue CABENUVA if hepatotoxicity is suspected. (5.3)
- Depressive disorders have been reported with CABENUVA. Prompt evaluation is recommended for depressive symptoms. (5.4)
- Residual concentrations of cabotegravir and rilpivirine may remain in the systemic circulation of patients up to 12 months or longer. It is essential to initiate an alternative, fully suppressive antiretroviral regimen no later than 1 month after the final injections of CABENUVA when dosed monthly and no later than 2 months after the final injections when dosed every 2 months. If virologic failure is suspected, prescribe an alternative regimen as soon as possible. (5.6)
ADVERSE REACTIONS
The most common adverse reactions (Grades 1 to 4) observed in ≥2% of participants receiving CABENUVA were injection site reactions, pyrexia, fatigue, headache, musculoskeletal pain, nausea, sleep disorders, dizziness, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact ViiV Healthcare at 1-877-844-8872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Refer to the full prescribing information for important drug interactions with CABENUVA. (4, 5.5, 7)
- Because CABENUVA is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended. (7.1)
- Drugs that induce uridine diphosphate glucuronosyltransferase (UGT)1A1 or cytochrome P450 (CYP)3A4 may decrease the plasma concentrations of the components of CABENUVA. (4, 7.3, 7.4)
- CABENUVA should be used with caution in combination with drugs with a known risk of Torsade de Pointes. (7.3, 7.4)
USE IN SPECIFIC POPULATIONS
- Pregnancy: After oral use of rilpivirine, exposures were generally lower during pregnancy compared with the postpartum period. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
2.2 Adherence to CABENUVA
2.3 Optional Oral Lead-In Dosing to Assess Tolerability of CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
2.4 Recommended Monthly Gluteal Intramuscular Injection Dosing with CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
2.5 Recommended Every-2-Month Gluteal Intramuscular Injection Dosing with CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
2.6 Dosing Recommendations When Switching from Monthly to Every-2-Month Intramuscular Injections
2.7 Dosing Recommendations When Switching from Every-2-Month to Monthly Intramuscular Injections
2.8 Recommended Dosing Schedule for Missed Injections
2.9 Administration Instructions for Injections
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Post-Injection Reactions
5.3 Hepatotoxicity
5.4 Depressive Disorders
5.5 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
5.6 Long-Acting Properties and Potential Associated Risks with CABENUVA
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Use with Other Antiretroviral Medicines
7.2 Use of Other Antiretroviral Drugs after Discontinuation of CABENUVA
7.3 Potential for Other Drugs to Affect CABENUVA
7.4 Established and Other Potentially Significant Drug Interactions
7.5 Drugs without Clinically Significant Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Trials in Adults
14.2 Clinical Trial in Adolescents
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
CABENUVA is indicated as a complete regimen for the treatment of HIV-1 infection in adults and adolescents 12 years of age and older and weighing at least 35 kg to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA <50 copies/mL) on a stable antiretroviral regimen with no history of treatment failure and with no known or suspected resistance to either cabotegravir or rilpivirine [see Microbiology (12.4), Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
- CABENUVA contains cabotegravir extended-release injectable suspension in a single-dose vial and rilpivirine extended-release injectable suspension in a single-dose vial [see Dosage Forms and Strengths (3)].
- CABENUVA must be administered by a healthcare provider by gluteal intramuscular injection [see Dosage and Administration (2.9)].
- CABENUVA may be initiated with oral cabotegravir and oral rilpivirine prior to the intramuscular injections or the patient may proceed directly to injection of CABENUVA without an oral lead-in [see Dosage and Administration (2.3)].
- CABENUVA can be injected monthly or every 2 months [see Dosage and Administration (2.4, 2.5)]. Healthcare providers should discuss these 2 dosing options with the patient prior to starting CABENUVA and decide which injection dosing frequency would be the most appropriate option for the patient [see Adverse Reactions (6.1), Microbiology (12.4), Clinical Studies (14.1)].
2.2 Adherence to CABENUVA
Prior to starting CABENUVA, healthcare providers should carefully select patients who agree to the required monthly or every‑2-month injection dosing schedule and counsel patients about the importance of adherence to scheduled dosing visits to help maintain viral suppression and reduce the risk of viral rebound and potential development of resistance with missed doses [see Dosage and Administration (2.1), Warnings and Precautions (5.6), Microbiology (12.4)].
2.3 Optional Oral Lead-In Dosing to Assess Tolerability of CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
The healthcare provider and patient may decide to use an oral lead-in with oral cabotegravir and oral rilpivirine prior to the initiation of CABENUVA to assess the tolerability of cabotegravir and rilpivirine, or the healthcare provider and patient may proceed directly to injection of CABENUVA without the use of an oral lead-in.
If oral lead-in is used, the recommended oral lead-in daily dose is one 30-mg tablet of VOCABRIA (cabotegravir) and one 25-mg tablet of EDURANT (rilpivirine) taken with a meal for approximately 1 month (at least 28 days), followed by intramuscular initiation injections of CABENUVA. See Tables 1 and 2 for recommended oral lead-in and monthly or every-2-month intramuscular injection dosing schedule for CABENUVA [see Dosage and Administration (2.4, 2.5)].
2.4 Recommended Monthly Gluteal Intramuscular Injection Dosing with CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
Initiation Injections (CABENUVA 600-mg/900-mg Kit)
Initiate injections on the last day of current antiretroviral therapy or oral lead-in, if used [see Dosage and Administration (2.3)]. The recommended initiation injection doses of CABENUVA are a single 600-mg (3-mL) intramuscular injection of cabotegravir and a single 900-mg (3-mL) intramuscular injection of rilpivirine. Administer cabotegravir and rilpivirine at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)]. Continuation injections should be initiated a month after the initiation injections.
Continuation Injections (CABENUVA 400-mg/600-mg Kit)
After the initiation injections, the recommended monthly continuation injection doses of CABENUVA are a single 400-mg (2-mL) intramuscular injection of cabotegravir and a single 600-mg (2-mL) intramuscular injection of rilpivirine at each visit (Table 1). Administer cabotegravir and rilpivirine at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)]. Patients may be given CABENUVA up to 7 days before or after the date the patient is scheduled to receive monthly injections.
Table 1. Recommended Dosing Schedule with Optional Oral Lead-In or Direct to Injection for Monthly Injection a The optional oral therapy should be continued until the day the first injection is administered. b Given on the last day of current antiretroviral therapy or oral lead-in if used. Drug
Optional Oral Lead-Ina
(at Least 28 Days)Intramuscular (Gluteal)
Initiation Injections
(One-Time Dosing)Intramuscular (Gluteal)
Continuation Injections
(Once-Monthly Dosing)Month (at Least 28 Days) Prior to Starting Injections
Initiate Injections at Month 1b
One Month after Initiation Injection and Monthly Onwards
Cabotegravir
30 mg once daily
with a meal600 mg (3 mL)
400 mg (2 mL)
Rilpivirine
25 mg once daily
with a meal900 mg (3 mL)
600 mg (2 mL)
2.5 Recommended Every-2-Month Gluteal Intramuscular Injection Dosing with CABENUVA in Adults and Adolescents 12 Years of Age and Older and Weighing at Least 35 kg
Initiation Injections (CABENUVA 600-mg/900-mg Kit)
Initiate injections on the last day of current antiretroviral therapy or oral lead-in, if used [see Dosage and Administration (2.3)]. The recommended initiation injection doses of CABENUVA are a single 600-mg (3-mL) intramuscular injection of cabotegravir and a single 900-mg (3-mL) intramuscular injection of rilpivirine 1 month apart for 2 consecutive months (Table 2). Administer cabotegravir and rilpivirine at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)]. Patients may be given CABENUVA up to 7 days before or after the date the patient is scheduled to receive the second initiation injections.
Continuation Injections (CABENUVA 600-mg/900-mg Kit)
After the 2 initiation doses given consecutively 1 month apart (Months 1 and 2), the recommended continuation injection doses (Month 4 onwards) of CABENUVA are a single 600-mg (3-mL) intramuscular injection of cabotegravir and a single 900-mg (3-mL) intramuscular injection of rilpivirine administered every 2 months (Table 2). Administer cabotegravir and rilpivirine at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)]. Patients may be given CABENUVA up to 7 days before or after the date the patient is scheduled to receive the injections.
Table 2. Recommended Dosing Schedule with Optional Oral Lead-In or Direct to Injection for Every-2-Month Injection a The optional oral therapy should be continued until the day the first injection is administered.
b For the every-2-month injection dosing schedule in adults, Initiation Injections are injections administered at Month 1 and Month 2 and Continuation Injections are injections administered every 2 months onwards (starting Month 4).
c Given on the last day of current antiretroviral therapy or oral lead-in if used.Drug
Optional Oral Lead-Ina
(at Least 28 Days)
Intramuscular (Gluteal) Injectionsb
Month (at Least 28 Days) Prior to Starting Injections
Initiate Injectionsc at Month 1, Month 2, and then Every 2 Months Onwards
(Starting at Month 4)Cabotegravir
30 mg once daily with a meal
600 mg (3 mL)
Rilpivirine
25 mg once daily with a meal
900 mg (3 mL)
2.6 Dosing Recommendations When Switching from Monthly to Every-2-Month Intramuscular Injections
Patients switching from a monthly continuation injection schedule (a single 400-mg [2-mL] gluteal intramuscular injection of cabotegravir and a single 600-mg [2-mL] intramuscular injection of rilpivirine) to an every-2-month continuation injection dosing schedule should receive a single 600-mg (3-mL) intramuscular injection of cabotegravir and a single 900-mg (3-mL) intramuscular injection of rilpivirine administered 1 month after the last monthly continuation injections and then every 2 months thereafter. Cabotegravir and rilpivirine injections should be administered at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)].
2.7 Dosing Recommendations When Switching from Every-2-Month to Monthly Intramuscular Injections
Patients switching from an every-2-month continuation injection schedule (a single 600-mg [3-mL] intramuscular injection of cabotegravir and a single 900-mg [3-mL] intramuscular injection of rilpivirine) to a monthly continuation dosing schedule should receive a single 400-mg (2-mL) intramuscular injection of cabotegravir and a single 600-mg (2-mL) intramuscular injection of rilpivirine 2 months after the last every-2‑month continuation injection and then monthly thereafter. Cabotegravir and rilpivirine injections should be administered at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit [see Dosage and Administration (2.9)].
2.8 Recommended Dosing Schedule for Missed Injections
Adherence to the injection dosing schedule is strongly recommended [see Dosage and Administration (2.2)]. Patients who miss a scheduled injection visit should be clinically reassessed to ensure resumption of therapy remains appropriate. Refer to Table 3 for dosing recommendations after missed injections.
Planned Missed Injections for Patients on the Monthly Dosing Schedule
If a patient plans to miss a scheduled injection visit by more than 7 days, VOCABRIA in combination with EDURANT once daily may be used for up to 2 months to replace missed injection visits, or any other fully suppressive oral antiretroviral regimen may be used until injections are resumed. The recommended oral daily dose is one 30-mg tablet of VOCABRIA (cabotegravir) and one 25-mg tablet of EDURANT (rilpivirine). Take VOCABRIA with EDURANT at approximately the same time each day with a meal.
The first dose of oral therapy should be taken 1 month (+/-7 days) after the last injection dose of CABENUVA and continued until the day injection dosing is restarted. Refer to Table 3 for injection dosing recommendations. For oral therapy with VOCABRIA and EDURANT of durations greater than 2 months, an alternative oral regimen is recommended.
Unplanned Missed Injections for Patients on the Monthly Dosing Schedule
If monthly injections are missed or delayed by more than 7 days and oral therapy has not been taken in the interim, clinically reassess the patient to determine if resumption of injection dosing remains appropriate [see Warnings and Precautions (5.6)]. If injection dosing will be continued, see Table 3 for dosing recommendations.
Table 3. Injection Dosing Recommendations after Missed Injections for Patients on the Monthly Dosing Schedulea a Refer to oral dosing recommendations if a patient plans to miss a scheduled injection visit. Time since Last Injection
Recommendation
Less than or equal to 2 months
Resume with 400-mg (2-mL) cabotegravir and 600-mg (2-mL) rilpivirine gluteal intramuscular monthly injections as soon as possible.
Greater than 2 months
Re-initiate the patient with 600-mg (3-mL) cabotegravir and 900-mg (3‑mL) rilpivirine gluteal intramuscular injections then continue to follow the 400‑mg (2‑mL) cabotegravir and 600-mg (2-mL) rilpivirine gluteal intramuscular monthly injection dosing schedule.
Planned Missed Injections for Patients on the Every-2-Month Dosing Schedule
If a patient plans to miss a scheduled injection visit by more than 7 days, VOCABRIA in combination with EDURANT once daily may be used for up to 2 months to replace 1 missed injection visit, or any other fully suppressive oral antiretroviral regimen may be used until injections are resumed. The recommended oral daily dose is one 30‑mg tablet of VOCABRIA (cabotegravir) and one 25‑mg tablet of EDURANT (rilpivirine). Take VOCABRIA with EDURANT at approximately the same time each day with a meal.
The first dose of oral therapy should be taken approximately 2 months after the last injection dose of CABENUVA and continued until the day injection dosing is restarted. Refer to Table 4 for injection dosing recommendations. For oral therapy with VOCABRIA and EDURANT of durations greater than 2 months, an alternative oral regimen is recommended.
Unplanned Missed Injections for Patients on the Every-2-Month Dosing Schedule
If a scheduled every-2-month injection visit is missed or delayed by more than 7 days and oral therapy has not been taken in the interim, clinically reassess the patient to determine if resumption of injection dosing remains appropriate [see Warnings and Precautions (5.6)]. If the every-2‑month dosing schedule will be continued, see Table 4 for dosing recommendations.
Table 4. Injection Dosing Recommendations after Missed Injections for Patients on the Every-2-Month Dosing Schedule Missed Injection Visit
Time since Last Injection
Recommendation
Injection 2 (Month 2)
Less than or equal to 2 months
Resume with 600-mg (3-mL) cabotegravir and 900‑mg (3‑mL) rilpivirine intramuscular injections as soon as possible, then continue to follow the every-2-month injection dosing schedule.
Greater than 2 months
Re-initiate the patient with 600-mg (3-mL) cabotegravir and 900-mg (3‑mL) rilpivirine intramuscular injections, followed by the second initiation injection dose 1 month later. Then continue to follow the every-2-month injection dosing schedule thereafter.
Injection 3 or later (Month 4 onwards)
Less than or equal to 3 months
Resume with 600-mg (3-mL) cabotegravir and 900‑mg (3‑mL) rilpivirine intramuscular injections as soon as possible and continue with the every‑2‑month injection dosing schedule.
Greater than 3 months
Re-initiate the patient with 600-mg (3-mL) cabotegravir and 900-mg (3‑mL) rilpivirine intramuscular injections, followed by the second initiation injection dose 1 month later. Then continue with the every-2-month injection dosing schedule thereafter.
2.9 Administration Instructions for Injections
Refer to the Instructions for Use for complete administration instructions with illustrations. Carefully follow these instructions and ensure that the vial adaptor is used correctly when preparing the suspension for injection to avoid leakage.
A complete dose requires 2 injections: 1 injection of cabotegravir and 1 injection of rilpivirine [see Dosage and Administration (2.4, 2.5)].
Cabotegravir and rilpivirine are suspensions for gluteal intramuscular injection that do not need further dilution or reconstitution.
Administer each injection at separate gluteal injection sites (on opposite sides or at least 2 cm apart) during the same visit. The ventrogluteal site is recommended. A dorsogluteal approach (upper outer quadrant) is acceptable, if preferred by the healthcare professional. Do not administer by any other route or anatomical site. Consider the body mass index (BMI) of the patient to ensure that the needle length is sufficient to reach the gluteus muscle. Longer needle lengths (not included in the dosing kit) may be required for patients with higher BMI (example: >30 kg/m2) to ensure that injections are administered intramuscularly as opposed to subcutaneously. The administration order of cabotegravir and rilpivirine injections is not important.
Before preparing the injections, remove CABENUVA from the refrigerator and wait at least 15 minutes to allow the medicines to come to room temperature. The vials may remain in the carton at room temperature for up to 6 hours; do not put back into the refrigerator. If not used within 6 hours, the medication must be discarded.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The cabotegravir vial has a brown tint to the glass that may limit visual inspection. Discard CABENUVA if either medicine exhibits particulate matter or discoloration.
Shake each vial of CABENUVA vigorously so that the suspensions look uniform before injecting. Small air bubbles are expected and acceptable.
Once the suspensions have been drawn into the respective syringes, the injections should be administered as soon as possible, but may remain in the syringes for up to 2 hours. The filled syringes should not be placed in the refrigerator. If the medicine remains in the syringes for more than 2 hours, the filled syringes and needles must be discarded [see How Supplied/Storage and Handling (16)].
-
3 DOSAGE FORMS AND STRENGTHS
Injection:
- Kit of single-dose vials of 400 mg/2 mL (200 mg/mL) of cabotegravir extended-release injectable suspension and 600 mg/2 mL (300 mg/mL) of rilpivirine extended-release injectable suspension. (3)
- Kit of single-dose vials of 600 mg/3 mL (200 mg/mL) of cabotegravir extended-release injectable suspension and 900 mg/3 mL (300 mg/mL) of rilpivirine extended-release injectable suspension. (3)
-
4 CONTRAINDICATIONS
CABENUVA is contraindicated in patients:
- with previous hypersensitivity reaction to cabotegravir or rilpivirine [see Warnings and Precautions (5.1)].
-
receiving the following coadministered drugs for which significant decreases in cabotegravir and/or rilpivirine plasma concentrations may occur due to uridine diphosphate glucuronosyltransferase (UGT)1A1 and/or cytochrome P450 (CYP)3A enzyme induction, which may result in loss of virologic response [see Drug Interactions (7), Clinical Pharmacology (12.3)]:
- o Anticonvulsants: Carbamazepine, oxcarbazepine, phenobarbital, phenytoin
- o Antimycobacterials: Rifabutin, rifampin, rifapentine
- o Glucocorticoid (systemic): Dexamethasone (more than a single-dose treatment)
- o Herbal product: St John’s wort (Hypericum perforatum)
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious or severe hypersensitivity reactions have been reported with cabotegravir- and rilpivirine-containing regimens. Reactions associated with cabotegravir include Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) [see Adverse Reactions (6.2)]. Reactions associated with rilpivirine-containing regimens include cases of drug reaction with eosinophilia and systemic symptoms (DRESS) [see Adverse Reactions (6.1, 6.2)]. While some skin reactions were accompanied by constitutional symptoms such as fever, other skin reactions were associated with organ dysfunctions, including elevations in hepatic serum biochemistries. Administration of cabotegravir and rilpivirine oral lead-in dosing was used in clinical studies to help identify patients who may be at risk of a hypersensitivity reaction [see Dosage and Administration (2.3), Contraindications (4)]. Remain vigilant and discontinue CABENUVA if a hypersensitivity reaction is suspected [see Contraindications (4), Adverse Reactions (6)].
Discontinue CABENUVA immediately if signs or symptoms of hypersensitivity reactions develop (including, but not limited to, severe rash, or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, mucosal involvement [oral blisters or lesions], conjunctivitis, facial edema, hepatitis, eosinophilia, angioedema, difficulty breathing). Clinical status, including liver transaminases, should be monitored and appropriate therapy initiated. For information regarding the long-acting properties of CABENUVA, [see Warnings and Precautions (5.6)]. Administer oral lead-in dosing prior to administration of CABENUVA to help identify patients who may be at risk of a hypersensitivity reaction [see Dosage and Administration (2.3), Contraindications (4)].
5.2 Post-Injection Reactions
In clinical trials, serious post-injection reactions were reported within minutes after the injection of rilpivirine. These events included symptoms such as dyspnea, bronchospasm, agitation, abdominal cramping, rash/urticaria, dizziness, flushing, sweating, oral numbness, changes in blood pressure, and pain (e.g., back and chest). These events were reported in <1% of participants and began to resolve within minutes after the injection, with some patients receiving supportive care. These events may have been associated with accidental intravenous administration during the intramuscular injection procedure [see Adverse Reactions (6.1)].
Carefully follow the Instructions for Use when preparing and administering CABENUVA. The suspensions should be injected slowly via intramuscular injection, and care should be taken to avoid accidental intravenous administration [see Dosage and Administration (2.9)]. Observe patients briefly (approximately 10 minutes) after the injection. If a patient experiences a post-injection reaction, monitor and treat as clinically indicated.
5.3 Hepatotoxicity
Hepatotoxicity has been reported in patients receiving cabotegravir or rilpivirine with or without known pre-existing hepatic disease or identifiable risk factors [see Adverse Reactions (6.1)].
Patients with underlying liver disease or marked elevations in transaminases prior to treatment may be at increased risk for worsening or development of transaminase elevations.
Monitoring of liver chemistries is recommended and treatment with CABENUVA should be discontinued if hepatotoxicity is suspected. For information regarding the long-acting properties of CABENUVA, [see Warnings and Precautions (5.6)].
5.4 Depressive Disorders
Depressive disorders (including depressed mood, depression, major depression, mood altered, mood swings, dysphoria, negative thoughts, suicidal ideation, suicide attempt) have been reported with CABENUVA or the individual drug products [see Adverse Reactions (6.1)]. Promptly evaluate patients with depressive symptoms to assess whether the symptoms are related to CABENUVA and to determine whether the risks of continued therapy outweigh the benefits.
5.5 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of CABENUVA and other drugs may result in known or potentially significant drug interactions, some of which may lead to adverse events, loss of virologic response of CABENUVA, and possible development of viral resistance [see Contraindications (4), Drug Interactions (7.4)].
Rilpivirine 75-mg and 300-mg once-daily oral doses (3 and 12 times, respectively, the recommended oral dosage) in healthy adults prolonged the QTc interval with mean steady-state Cmax values 4.4- and 11.6-fold, respectively, higher than Cmax values associated with the recommended 600-mg monthly dose of rilpivirine extended-release injectable suspension and 4.1- and 10.7-fold, respectively, higher than Cmax values associated with the recommended 900-mg every-2-month dose of rilpivirine extended‑release injectable suspension [see Clinical Pharmacology (12.2)]. CABENUVA should be used with caution in combination with drugs with a known risk of Torsade de Pointes [see Drug Interactions (7.3, 7.4)].
See Table 8 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during therapy with, and after discontinuation of, CABENUVA; review concomitant medications during therapy with CABENUVA [see Drug Interactions (7.4)].
5.6 Long-Acting Properties and Potential Associated Risks with CABENUVA
Residual concentrations of both cabotegravir and rilpivirine may remain in the systemic circulation of patients for prolonged periods (up to 12 months or longer). It is important to carefully select patients who agree to the required monthly or every-2-month injection dosing schedule because non-adherence to monthly or every-2-month injections or missed doses could lead to loss of virologic response and development of resistance [see Dosage and Administration (2.2), Adverse Reactions (6.1), Drug Interactions (7.4)].
To minimize the potential risk of developing viral resistance, it is essential to initiate an alternative, fully suppressive antiretroviral regimen no later than 1 month after the final injections of CABENUVA when dosed monthly and no later than 2 months after the final injections of CABENUVA when dosed every 2 months. If virologic failure is suspected, switch the patient to an alternative regimen as soon as possible.
-
6 ADVERSE REACTIONS
The following adverse reactions are described below and in other sections of the labeling:
- Hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Post-injection reactions [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Depressive disorders [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect rates observed in practice.
Clinical Trials Experience in Adults
The safety assessment of CABENUVA is based on the analysis of pooled 48-week data from 1,182 virologically suppressed participants with HIV-1 infection in 2 international, multicenter, open-label pivotal trials, FLAIR and ATLAS, and 1,045 virologically suppressed participants from the ATLAS-2M trial [see Clinical Studies (14.1)]. Additional safety information from other ongoing or earlier clinical trials in the cabotegravir and rilpivirine program have been considered in assessing the overall safety profile of CABENUVA.
Adverse reactions were reported following exposure to CABENUVA extended-release injectable suspensions (median time exposure at the time of analysis: 54 weeks in FLAIR and ATLAS, and 64 weeks in ATLAS-2M) and data from VOCABRIA (cabotegravir) tablets and EDURANT (rilpivirine) tablets administered in combination as oral lead-in therapy (median time exposure: 5.3 weeks in FLAIR and ATLAS, and 5.6 weeks in ATLAS-2M). Adverse reactions included those attributable to both the oral and injectable formulations of cabotegravir and rilpivirine administered as a combination regimen. Refer to the prescribing information for EDURANT (rilpivirine) for other adverse reactions associated with oral rilpivirine.
The most common adverse reactions regardless of severity reported in ≥2% of adult participants from FLAIR and ATLAS at Week 48 are presented in Table 5. Selected laboratory abnormalities are included in Table 7. At Week 96, the overall safety profile for FLAIR was consistent with that observed at Week 48, with no new safety findings identified.
In the extension phase of the FLAIR study at Week 124, the overall safety profile was consistent with that observed at Week 48 and when injection therapy with CABENUVA was initiated directly without the oral lead-in phase.
Overall, 4% of participants in the group receiving CABENUVA and 2% of participants in the control group in FLAIR and ATLAS discontinued due to adverse events. Non-injection-site-related adverse events leading to discontinuation and occurring in more than 1 participant were headache, diarrhea, hepatitis A, and acute hepatitis B (all with an incidence <1%). In ATLAS-2M, 2% of participants in both treatment groups discontinued due to adverse events. Non-injection-site–related adverse events leading to discontinuation and occurring in more than 1 participant in ATLAS-2M were fatigue, pyrexia, headache, presyncope, acute hepatitis B, hyperhidrosis, and abnormal dreams that occurred with an incidence of ≤1% in either treatment group.
Table 5. Adverse Reactionsa (Grades 1 to 4) Reported in at Least 2% of Participants with HIV-1 Infection in FLAIR and ATLAS Trials (Week 48 Pooled Analyses) a Adverse reactions defined as “treatment-related” as assessed by the investigator.
b See Injection-Associated Adverse Reactions for additional information.
c Pyrexia: includes pyrexia, feeling hot, chills, influenza-like illness, body temperature increased.
d Fatigue: includes fatigue, malaise, asthenia.
e Musculoskeletal pain: includes musculoskeletal pain, musculoskeletal discomfort, back pain, myalgia, pain in extremity.
f Sleep disorders: includes insomnia, poor quality sleep, somnolence.
g Rash: includes erythema, pruritus, pruritus generalized, purpura, rash, rash- erythematous, generalized, macular.Adverse Reactions
Cabotegravir plus Rilpivirine
Once Monthly
(n = 591)
Current Antiretroviral
Regimen(n = 591)
All Grades
At Least Grade 2
All Grades
At Least Grade 2
Injection site reactionsb
83%
37%
0
0
Pyrexiac
8%
2%
0
0
Fatigued
5%
1%
<1%
<1%
Headache
4%
<1%
<1%
<1%
Musculoskeletal paine
3%
1%
<1%
0
Nausea
3%
<1%
1%
<1%
Sleep disordersf
2%
<1%
<1%
0
Dizziness
2%
<1%
<1%
0
Rashg
2%
<1%
0
0
In ATLAS-2M, the type and frequency of adverse reactions reported in participants receiving CABENUVA once monthly or CABENUVA once every 2 months for 48 weeks were similar. Differences between treatment arms were reported for the types of injection-associated adverse reactions (see Injection-Associated Adverse Reactions for additional information).
Injection-Associated Adverse Reactions: Local Injection Site Reactions (ISRs): In the 3 Phase 3 studies, FLAIR, ATLAS, and ATLAS‑2M, the most frequent adverse reactions associated with the intramuscular administration of CABENUVA were ISRs.
In the pooled analysis of FLAIR and ATLAS, 83% of participants reported any injection site reaction with the monthly dosing regimen, with 1% of participants who discontinued treatment with CABENUVA because of ISRs. After 14,682 injections, 3,663 ISRs were reported. The severity of ISRs was generally mild (Grade 1: 75% of participants) or moderate (Grade 2: 36% of participants). Four percent (4%) of participants experienced severe (Grade 3) ISRs, and no participant experienced Grade 4 ISRs. The median duration of overall ISR events was 3 days. The most commonly reported ISR in FLAIR and ATLAS was pain/discomfort, with 79% reported in the group receiving CABENUVA.
In ATLAS-2M, 75% of participants reported any injection site reaction in both the monthly and every‑2‑month dosing regimens, with <1% of participants who discontinued treatment with CABENUVA because of ISRs. When dosing monthly, after 15,711 injections, 3,152 ISRs were reported. When dosing every 2 months, after 8,470 injections, 2,507 ISRs were reported. The severity of ISRs was generally mild (Grade 1: 70% and 71% of participants) or moderate (Grade 2: 28% and 27% of participants) in monthly and every‑2‑month dosing regimens, respectively. Four percent (4%) of participants in the monthly group and 3% of participants in the every‑2‑month group experienced severe (Grade 3) ISRs, and no participant experienced Grade 4 ISRs. The median duration of overall ISR events was 3 days for both dosing regimens. The most commonly reported ISR in ATLAS‑2M was pain/discomfort, with 71% and 73% reported in the monthly and every‑2‑month dosing regimens, respectively. The severity and duration of ISRs, including pain/discomfort, were similar for both dosing regimens and in participants without prior exposure to CABENUVA.
The most commonly reported ISR (Grades 1 to 3) in at least 1% of adult participants in the pooled analyses from FLAIR and ATLAS, and from ATLAS‑2M, are presented in Table 6. The side‑by‑side tabulation is to simplify presentation; direct comparison across trials should not be made due to differing trials.
Table 6. Injection Site Reactions (Grades 1 to 3) Reported in at Least 1% of Participants in FLAIR and ATLAS (Pooled Analysis) and ATLAS-2M Trials (Week 48 Analysis) Anesthesia, abscess, cellulitis, and hemorrhage at the injection site were each reported in <1% of participants. Injection Site Reactions
FLAIR and ATLAS
ATLAS-2M
Cabotegravir plus Rilpivirine
Once Monthly
(n = 591)
Cabotegravir plus Rilpivirine
Once Every 2 Months
(n = 522)
Cabotegravir plus Rilpivirine
Once Monthly
(n = 523)
Any injection site reaction
83%
75%
75%
Pain/discomfort
79%
73%
71%
Nodules
14%
10%
17%
Induration
12%
8%
7%
Swelling
8%
6%
5%
Erythema
4%
2%
3%
Pruritus
4%
5%
5%
Bruising/discoloration
3%
2%
2%
Warmth
2%
1%
2%
Hematoma
2%
<1%
3%
Other Injection-Associated Adverse Reactions: In the ATLAS and FLAIR clinical trials, an increased incidence of pyrexia (8%) was reported by participants receiving cabotegravir plus rilpivirine injections compared with no events among participants receiving current antiretroviral regimen. In ATLAS and FLAIR, no cases were serious or led to withdrawal and the occurrences of pyrexia may represent a response to administration of CABENUVA via intramuscular injection. In ATLAS-2M, 1 participant in each arm reported pyrexia that led to withdrawal.
In ATLAS and FLAIR, reports of musculoskeletal pain (3%) and less frequently, sciatica, were also more common in participants receiving cabotegravir plus rilpivirine compared with the current antiretroviral regimen and some events had a temporal association with injection.
Vasovagal or pre-syncopal reactions were reported in <1% of participants after injection with rilpivirine or cabotegravir.
Less Common Adverse Reactions: The following select adverse reactions (regardless of severity) occurred in <2% of participants receiving cabotegravir plus rilpivirine.
Gastrointestinal Disorders: Abdominal pain (including upper abdominal pain), gastritis, dyspepsia, vomiting, diarrhea, and flatulence.
Hepatobiliary Disorders: Hepatotoxicity.
Investigations: Weight increase (see below).
Psychiatric Disorders: Anxiety (including anxiety and irritability), depression, abnormal dreams, suicidal ideation, and suicide attempt (these events were observed primarily in participants with a pre-existing history of depression or other psychiatric illness).
Skin and Hypersensitivity Reactions: Hypersensitivity reactions.
Weight Increase: At Week 48, participants in FLAIR and ATLAS who received cabotegravir plus rilpivirine had a median weight gain of 1.5 kg; those in the current antiretroviral regimen group had a median weight gain of 1.0 kg (pooled analysis). In the FLAIR trial, the median weight gain in participants receiving cabotegravir plus rilpivirine or a dolutegravir-containing regimen was 1.3 kg and 1.5 kg, respectively, compared with 1.8 kg and 0.3 kg, respectively, in the ATLAS trial in participants receiving either cabotegravir plus rilpivirine or a protease inhibitor-, non-nucleoside reverse transcriptase inhibitor (NNRTI)-, or integrase strand transfer inhibitor (INSTI)-containing regimen, respectively. At Week 48, participants in ATLAS-2M who received cabotegravir plus rilpivirine in both the monthly and every-2-month treatment arms had a median weight gain of 1.0 kg.
Laboratory Abnormalities: Selected laboratory abnormalities with a worsening grade from baseline and representing the worst-grade toxicity are presented in Table 7. The side-by-side tabulation is to simplify presentation; direct comparison across trials should not be made due to differing trials.
Table 7. Selected Laboratory Abnormalities (Grades 3 to 4, Week 48) in FLAIR and ATLAS (Pooled Analyses) and ATLAS-2M Trials ALT = Alanine Aminotransferase, ULN = Upper limit of normal, AST = Aspartate Transaminase. Laboratory Parameter
FLAIR and ATLAS
ATLAS-2M
Cabotegravir
plus Rilpivirine
Once Monthly(n = 591)
Current
Antiretroviral
Regimen(n = 591)
Cabotegravir
plus Rilpivirine
Once Every
2 Months(n = 522)
Cabotegravir
plus Rilpivirine
Once Monthly(n = 523)
ALT (≥5.0 x ULN)
2%
<1%
<1%
<1%
AST (≥5.0 x ULN)
2%
<1%
<1%
1%
Total bilirubin (≥2.6 x ULN)
<1%
<1%
<1%
<1%
Creatine phosphokinase (≥10.0 x ULN)
8%
4%
3%
4%
Lipase (≥3.0 x ULN)
5%
3%
3%
2%
Changes in Total Bilirubin: Small, non-progressive increases in total bilirubin (without clinical jaundice) were observed with cabotegravir plus rilpivirine. These changes are not considered clinically relevant as they likely reflect competition between cabotegravir and unconjugated bilirubin for a common clearance pathway (UGT1A1) [see Clinical Pharmacology (12.3)].
Serum Cortisol: In pooled Phase 3 trials of EDURANT (rilpivirine), the overall mean change from baseline in basal cortisol was -0.69 (-1.12, 0.27) mcg/dL in the group receiving EDURANT compared with -0.02 (-0.48, 0.44) mcg/dL in the control group. Abnormal responses to ACTH stimulation tests were also higher in the group receiving EDURANT. The clinical significance of the higher abnormal rate of ACTH stimulation tests in the group receiving EDURANT is not known. Refer to the prescribing information for EDURANT for additional information.
Clinical Trial Experience in Adolescents
Based on data from the Week 16 (cohort 1) and Week 24 (cohort 2) analyses of the MOCHA study, the safety profile in adolescents (aged 12 to younger than 18 years and weighing ≥35 kg) was consistent with the safety profile established with cabotegravir plus rilpivirine in adults [see Clinical Studies (14.2)].
In cohort 1, 55 virologically suppressed adolescents with HIV-1 received background antiretroviral therapy in addition to either oral cabotegravir (n = 30) followed by injectable cabotegravir (n = 29), or oral rilpivirine (n = 25) followed by injectable rilpivirine (n = 23). Adverse reactions were reported in 38% of adolescents receiving either cabotegravir or rilpivirine. Thirty-three percent of participants reported at least 1 ISR. All ISRs were Grade 1 or Grade 2. Two participants had Grade 3 adverse reactions of hypersensitivity (n = 1, oral rilpivirine) and insomnia (n = 1, injectable cabotegravir). The Grade 3 adverse reaction of drug hypersensitivity led to discontinuation of rilpivirine during oral lead-in. The adverse reactions reported by more than one participant (regardless of severity) were injection site pain (n = 18), headache (n = 2), hypersensitivity (n = 2), insomnia (n = 2), and nausea (n = 2).
In cohort 2, 144 virologically suppressed adolescents with HIV-1 received oral cabotegravir plus oral rilpivirine followed by injectable cabotegravir plus injectable rilpivirine. Adverse reactions were reported in 35% of adolescents receiving cabotegravir plus rilpivirine. Thirty-four percent of participants reported at least 1 ISR. The majority of these participants experienced Grade 1 or Grade 2 ISRs. Two participants had Grade 3 ISRs consisting of injection site abscess (n = 2) and injection site pain in one of these two participants (symptoms resolved in both participants). All non-ISR adverse reactions were ≤ Grade 2. Non-injection-site associated adverse reactions reported by more than one participant (regardless of severity) were headache (n = 3), nausea (n = 2), rash (n = 2) and rash pruritic (n = 2).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing experience in patients receiving cabotegravir- or oral-rilpivirine-containing regimens. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders
Hypersensitivity reactions (including angioedema and urticaria) [see Warnings and Precautions (5.1)].
Renal and Genitourinary Disorders
Nephrotic syndrome.
Skin and Subcutaneous Tissue Disorders
Severe skin and hypersensitivity reactions, including DRESS, Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) [see Warnings and Precautions (5.1)].
-
7 DRUG INTERACTIONS
7.1 Concomitant Use with Other Antiretroviral Medicines
Because CABENUVA is a complete regimen, coadministration with other antiretroviral medications for the treatment of HIV-1 infection is not recommended [see Indications and Usage (1)].
7.2 Use of Other Antiretroviral Drugs after Discontinuation of CABENUVA
Residual concentrations of cabotegravir and rilpivirine may remain in the systemic circulation of patients for prolonged periods (up to 12 months or longer). These residual concentrations are not expected to affect the exposures of antiretroviral drugs that are initiated after discontinuation of CABENUVA [see Warnings and Precautions (5.6), Drug Interactions (7.4), Clinical Pharmacology (12.3)].
7.3 Potential for Other Drugs to Affect CABENUVA
Refer to the prescribing information for VOCABRIA (cabotegravir) and EDURANT (rilpivirine) for additional drug interaction information related to oral cabotegravir and oral rilpivirine, respectively.
Cabotegravir
Cabotegravir is primarily metabolized by UGT1A1 with some contribution from UGT1A9. Drugs that are strong inducers of UGT1A1 or UGT1A9 are expected to decrease cabotegravir plasma concentrations and may result in loss of virologic response; therefore, coadministration of CABENUVA with these drugs is contraindicated [see Contraindications (4)].
Rilpivirine
Rilpivirine is primarily metabolized by CYP3A. Coadministration of CABENUVA and drugs that induce CYP3A may result in decreased plasma concentrations of rilpivirine and loss of virologic response and possible resistance to rilpivirine or to the class of NNRTIs [see Contraindications (4), Drug Interactions (7.4)]. Coadministration of CABENUVA and drugs that inhibit CYP3A may result in increased plasma concentrations of rilpivirine [see Drug Interactions (7.4), Clinical Pharmacology (12.3)].
QT-Prolonging Drugs: At mean steady-state Cmax values 4.4- and 11.6-fold higher than those with the recommended 600‑mg dose of rilpivirine extended-release injectable suspension, rilpivirine may prolong the QTc interval [see Clinical Pharmacology (12.2)]. CABENUVA should be used with caution in combination with drugs with a known risk of Torsade de Pointes [see Warnings and Precautions (5.5), Drug Interactions (7.4)].
7.4 Established and Other Potentially Significant Drug Interactions
Refer to the prescribing information for VOCABRIA (cabotegravir) and EDURANT (rilpivirine) for additional drug interaction information related to oral cabotegravir and oral rilpivirine, respectively.
Information regarding potential drug interactions with cabotegravir and rilpivirine is provided in Table 8. These recommendations are based on either drug interaction trials following oral administration of cabotegravir or rilpivirine or predicted interactions due to the expected magnitude of the interaction and potential for loss of virologic response [see Contraindications (4), Warnings and Precautions (5.5), Clinical Pharmacology (12.3)]. Table 8 includes potentially significant interactions but is not all inclusive.
Table 8. Drug Interactions with CABENUVA ↑ = Increase, ↓ = Decrease, ↔ = No change.
a See Clinical Pharmacology (12.3) for magnitude of interaction.Concomitant Drug Class:
Drug Name
Effect on Concentration
Clinical Comment
Anticonvulsants:
Carbamazepine
Oxcarbazepine
Phenobarbital
Phenytoin
↓Cabotegravir
↓Rilpivirine
Coadministration is contraindicated with CABENUVA due to potential for loss of virologic response and development of resistance [see Contraindications (4)].
Antimycobacterials:
Rifampina
Rifapentine
↓Cabotegravir
↓Rilpivirine
Antimycobacterial:
Rifabutina
↓Cabotegravir
↔Rifabutin
↓Rilpivirine
Glucocorticoid (systemic):
Dexamethasone
(more than a single-dose treatment)
↓Rilpivirine
Herbal product:
St John’s wort (Hypericum perforatum)
↓Rilpivirine
Macrolide or ketolide antibiotics:
Azithromycin
Clarithromycin
Erythromycin
↔Cabotegravir
↑Rilpivirine
Macrolides are expected to increase concentrations of rilpivirine and are associated with a risk of Torsade de Pointes [see Warnings and Precautions (5.5)]. Where possible, consider alternatives, such as azithromycin, which increases rilpivirine concentrations less than other macrolides.
Narcotic analgesic:
Methadonea
↔Cabotegravir
↓Methadone
↔Rilpivirine
No dose adjustment of methadone is required when starting coadministration of methadone with CABENUVA. However, clinical monitoring is recommended as methadone maintenance therapy may need to be adjusted in some patients.
7.5 Drugs without Clinically Significant Interactions
Cabotegravir
Based on drug interaction study results, the following drugs can be coadministered with cabotegravir (non-antiretrovirals and rilpivirine) or given after discontinuation of cabotegravir (antiretrovirals and non-antiretrovirals) without a dose adjustment: etravirine, midazolam, oral contraceptives containing levonorgestrel and ethinyl estradiol, and rilpivirine [see Clinical Pharmacology (12.3)].
Rilpivirine
Based on drug interaction study results, the following drugs can be coadministered with rilpivirine (non-antiretrovirals and cabotegravir) or given after discontinuation of rilpivirine (antiretrovirals and non-antiretrovirals): acetaminophen, atorvastatin, cabotegravir, chlorzoxazone, dolutegravir, ethinyl estradiol, norethindrone, raltegravir, ritonavir-boosted atazanavir, ritonavir-boosted darunavir, sildenafil, tenofovir alafenamide, and tenofovir disoproxil fumarate [see Clinical Pharmacology (12.3)]. Rilpivirine did not have a clinically significant effect on the pharmacokinetics of digoxin or metformin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to CABENUVA during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
There are insufficient human data on the use of CABENUVA during pregnancy to adequately assess a drug-associated risk of birth defects and miscarriage. Discuss the benefit-risk of using CABENUVA with individuals of childbearing potential or during pregnancy.
Cabotegravir and rilpivirine are detected in systemic circulation for up to 12 months or longer after discontinuing injections of CABENUVA; therefore, consideration should be given to the potential for fetal exposure during pregnancy [see Warnings and Precautions (5.6), Drug Interactions (7.2)].
Cabotegravir use in pregnant individuals has not been evaluated. Available data from the APR show no difference in the overall risk of birth defects for rilpivirine compared with the background rate for major birth defects of 2.7% in a United States (U.S.) reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data).
The rate of miscarriage is not reported in the APR. The background risk for major birth defects and miscarriage for the indicated population is unknown. The background rate for major birth defects in a U.S. reference population of the MACDP is 2.7%. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15% to 20%. The APR uses the MACDP as the U.S. reference population for birth defects in the general population. The MACDP evaluates mothers and infants from a limited geographic area and does not include outcomes for births that occurred at <20 weeks’ gestation.
In animal reproduction studies with oral cabotegravir, a delay in the onset of parturition and increased stillbirths and neonatal deaths were observed in a rat pre- and postnatal development study at >28 times the exposure at the recommended human dose (RHD). No evidence of adverse developmental outcomes was observed with oral cabotegravir in rats or rabbits (>28 times or similar to the exposure at the RHD, respectively) given during organogenesis (see Data).
No adverse developmental outcomes were observed when rilpivirine was administered orally at exposures 15 (rats) and 70 (rabbits) times the exposure in humans at the RHD (see Data).
Clinical Considerations
Lower exposures with oral rilpivirine were observed during pregnancy. Viral load should be monitored closely if the patient remains on CABENUVA during pregnancy. Cabotegravir and rilpivirine are detected in systemic circulation for up to 12 months or longer after discontinuing injections of CABENUVA; therefore, consideration should be given to the potential for fetal exposure during pregnancy [see Warnings and Precautions (5.6), Drug Interactions (7.2)].
Human Data:
Rilpivirine: Based on prospective reports to the APR of over 580 exposures to oral rilpivirine-containing regimens during the first trimester of pregnancy and over 200 during second/third trimester of pregnancy, the prevalence of birth defects in live births was 1.5% (95% CI: 0.7% to 2.9%) and 1.5% (95% CI: 0.3% to 4.2%) following first and second/third trimester exposures, respectively, compared with the background birth defect rate of 2.7% in the U.S. reference population of the MACDP. In a clinical trial, total oral rilpivirine exposures were generally lower during pregnancy compared with the postpartum period. Refer to the prescribing information for EDURANT (rilpivirine) for additional information on rilpivirine.
Animal Data: Cabotegravir: Cabotegravir was administered orally to pregnant rats at 0, 0.5, 5, or 1,000 mg/kg/day from 15 days before cohabitation, during cohabitation, and from Gestation Days 0 to 17. There were no effects on fetal viability when fetuses were delivered by caesarean, although a minor decrease in fetal body weight was observed at 1,000 mg/kg/day (>28 times the exposure in humans at the RHD). No drug-related fetal toxicities were observed at 5 mg/kg/day (approximately 13 times the exposure in humans at the RHD), and no drug-related fetal malformations were observed at any dose.
Cabotegravir was administered orally to pregnant rabbits at 0, 30, 500, or 2,000 mg/kg/day from Gestation Days 7 to 19. No drug-related fetal toxicities were observed at 2,000 mg/kg/day (approximately 0.7 times the exposure in humans at the RHD).
In a rat pre- and postnatal development study, cabotegravir was administered orally to pregnant rats at 0, 0.5, 5, or 1,000 mg/kg/day from Gestation Day 6 to Lactation Day 21. A delay in the onset of parturition and increases in the number of stillbirths and neonatal deaths by Lactation Day 4 were observed at 1,000 mg/kg/day (>28 times the exposure in humans at the RHD); there were no alterations to growth and development of surviving offspring. In a cross-fostering study, similar incidences of stillbirths and early postnatal deaths were observed when rat pups born to cabotegravir-treated mothers were nursed from birth by control mothers. There was no effect on neonatal survival of control pups nursed from birth by cabotegravir-treated mothers. A lower dose of 5 mg/kg/day (13 times the exposure at the RHD) was not associated with delayed parturition or neonatal mortality in rats. Studies in pregnant rats showed that cabotegravir crosses the placenta and can be detected in fetal tissue.
Rilpivirine: Rilpivirine was administered orally to pregnant rats (40, 120, or 400 mg/kg/day) and rabbits (5, 10, or 20 mg/kg/day) through organogenesis (on Gestation Days 6 through 17 and 6 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with rilpivirine in rats and rabbits at exposures 15 (rats) and 70 (rabbits) times the exposure in humans at the RHD. In a pre- and postnatal development study, rilpivirine was administered orally up to 400 mg/kg/day through lactation. No adverse effects were noted in the offspring at maternal exposures up to 63 times the exposure in humans at the RHD.
8.2 Lactation
Risk Summary
Based on limited data after oral administration, rilpivirine is present in human breast milk. The data do not allow determination of the amount of rilpivirine that is transferred to milk. There are no data on the presence of cabotegravir in human milk. Cabotegravir is present in animal milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk. It is not known if the components of CABENUVA affect human milk production or have effects on the breastfed infant. Residual exposures in human milk of cabotegravir (if present) and rilpivirine may remain for 12 months or longer after the last injections have been administered [see Warnings and Precautions (5.6)].
Potential risks of breastfeeding include: (1) HIV‑1 transmission (in HIV-1–negative infants), (2) developing viral resistance (in HIV-1–positive infants), and (3) adverse reactions in a breastfed infant similar to those seen in adults.
Cabotegravir: Animal lactation studies with cabotegravir have not been conducted. However, cabotegravir was detected in the plasma of nursing pups on Lactation Day 10 in the rat pre- and postnatal development study.
8.4 Pediatric Use
The safety and effectiveness of CABENUVA have been established in adolescents aged 12 to younger than 18 years and weighing at least 35 kg, which is supported by the following:
- Trials in adults [see Clinical Studies (14.1)]
- MOCHA (NCT03497676) trial in adolescents, in which virologically suppressed adolescents with HIV-1 (aged 12 to younger than 18 years and weighing at least 35 kg) received either cabotegravir or rilpivirine in addition to their background antiretroviral regimen (cohort 1), or cabotegravir plus rilpivirine as a complete regimen (cohort 2) [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.2)].
The safety and efficacy in adolescents (aged 12 to younger than 18 years and weighing at least 35 kg) were similar to that in adults and there was no clinically significant change in exposure for the components of CABENUVA [see Adverse Reactions (6.1)]. The efficacy of CABENUVA in adolescents is extrapolated from adults with support from pharmacokinetic analyses showing similar drug exposure and additional supportive efficacy data from the MOCHA study [see Clinical Pharmacology (12.3), Clinical Studies (14.2)].
The safety, efficacy, and pharmacokinetics of CABENUVA have not been established in pediatric patients younger than 12 years of age or weighing <35 kg.
8.5 Geriatric Use
Clinical trials of CABENUVA did not include sufficient numbers of participants aged 65 years and older to determine whether they respond differently from younger participants. In general, caution should be exercised in administration of CABENUVA in elderly patients, reflecting greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Based on studies with oral cabotegravir and population pharmacokinetic analyses of oral rilpivirine, no dosage adjustment of CABENUVA is necessary for patients with mild (creatinine clearance ≥60 to <90 mL/min) or moderate renal impairment (creatinine clearance ≥30 to <60 mL/min). In patients with severe renal impairment (creatinine clearance 15 to <30 mL/min) or end-stage renal disease (creatinine clearance <15 mL/min), increased monitoring for adverse effects is recommended [see Clinical Pharmacology (12.3)]. In patients with end-stage renal disease not on dialysis, effects on the pharmacokinetics of cabotegravir or rilpivirine are unknown. As cabotegravir and rilpivirine are >99% protein bound, dialysis is not expected to alter exposures of cabotegravir or rilpivirine.
8.7 Hepatic Impairment
Based on separate studies with oral cabotegravir and oral rilpivirine, no dosage adjustment of CABENUVA is necessary for patients with mild or moderate hepatic impairment (Child-Pugh A or B). The effect of severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of cabotegravir or rilpivirine is unknown [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no known specific treatment for overdose with cabotegravir or rilpivirine. If overdose occurs, monitor the patient and apply standard supportive treatment as required, including monitoring of vital signs and ECG (QT interval) as well as observation of the clinical status of the patient. As both cabotegravir and rilpivirine are highly bound to plasma proteins, it is unlikely that either would be significantly removed by dialysis. Consider the prolonged exposure to cabotegravir and rilpivirine (components of CABENUVA) following an injection when assessing treatment needs and recovery [see Warnings and Precautions (5.6)].
-
11 DESCRIPTION
CABENUVA contains cabotegravir extended-release injectable suspension, an HIV INSTI, co-packaged with rilpivirine extended-release injectable suspension, an HIV NNRTI.
Cabotegravir
The chemical name for cabotegravir is (3S,11aR)-N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. The empirical formula is C19H17F2N3O5 and the molecular weight is 405.35 g/mol. It has the following structural formula:
Cabotegravir extended-release injectable suspension is a white to light pink free-flowing suspension for intramuscular injection in a sterile single-dose vial. Each vial contains 2 mL or 3 mL of the following: cabotegravir 200 mg/mL and the inactive ingredients mannitol (35 mg/mL), polyethylene glycol (PEG) 3350 (20 mg/mL), polysorbate 20 (20 mg/mL), and Water for Injection.
The vial stoppers are not made with natural rubber latex.
Rilpivirine
The chemical name for rilpivirine is 4-[[4-[[4-[(E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile. Its molecular formula is C22H18N6 and its molecular weight is 366.42. Rilpivirine has the following structural formula:
Rilpivirine extended-release injectable suspension is a white to off-white suspension for intramuscular injection. Each sterile single-dose vial contains 2 mL or 3 mL of the following: rilpivirine 300 mg/mL and the following inactive ingredients: citric acid monohydrate (1 mg/mL), dextrose to ensure isotonicity, monobasic sodium phosphate (1.74 mg/mL), poloxamer 338 (50 mg/mL), sodium hydroxide to adjust pH, and Water for Injection.
The vial stoppers are not made with natural rubber latex.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CABENUVA contains 2 long-acting HIV-1 antiretroviral drugs, cabotegravir and rilpivirine [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
At a dose of cabotegravir 150 mg orally every 12 hours (10 times the recommended total daily oral lead-in dosage of CABENUVA), the QT interval is not prolonged to any clinically relevant extent. Administration of 3 doses of cabotegravir 150 mg orally every 12 hours resulted in a geometric mean Cmax approximately 2.8-, 5.4-, and 5.6-fold above the geometric mean steady-state Cmax associated with the recommended 30‑mg dose of oral cabotegravir, the recommended 400‑mg dose given monthly, and the recommended 600-mg dose given every 2 months of cabotegravir extended-release injectable suspension, respectively.
At the recommended dose of rilpivirine 25 mg orally once daily, the QT interval is not prolonged to any clinically relevant extent. The rilpivirine 25-mg once-daily mean steady-state Cmax was 247 ng/mL, which is 1.7-fold higher than the mean steady-state Cmax observed with the recommended 600-mg dose of rilpivirine extended-release injectable suspension given monthly and 1.6-fold higher than the mean steady-state Cmax observed with the recommended 900-mg dose of rilpivirine extended-release injectable suspension given every 2 months.
When rilpivirine 75-mg and 300-mg once-daily oral doses (3 and 12 times, respectively, the recommended oral lead-in dosage) were studied in healthy adults, the maximum mean time-matched (95% upper confidence bound) differences in QTcF interval were 10.7 (15.3) and 23.3 (28.4) msec, respectively, after baseline and placebo adjustment. Steady-state administration of rilpivirine 75 mg once daily and 300 mg once daily resulted in a mean steady-state Cmax approximately 4.4- and 11.6-fold, respectively, higher than the mean steady-state Cmax observed with the recommended 600-mg dose of rilpivirine extended-release injectable suspension given monthly and approximately 4.1- and 10.7-fold, respectively, higher than the mean steady-state Cmax observed with the recommended 900-mg dose of rilpivirine extended-release injectable suspension given every 2 months. The corresponding Cmax ratios are 2.6 and 6.7 when compared with the recommended oral rilpivirine dosage [see Warnings and Precautions (5.5)].
12.3 Pharmacokinetics
Absorption, Distribution, and Elimination
The pharmacokinetic properties of the components of CABENUVA are provided in Table 9. The multiple-dose pharmacokinetic parameters are provided in Table 10. For the pharmacokinetic properties of oral cabotegravir and oral rilpivirine, refer to the full prescribing information for VOCABRIA (cabotegravir) and EDURANT (rilpivirine), respectively.
Table 9. Pharmacokinetic Properties of the Components of CABENUVA CSF = Cerebrospinal Fluid, BLQ = Below Limit of Quantification. a When taken orally with a high-fat meal versus fasted, the AUC(0-inf) (geometric mean ratio [90% CI] of cabotegravir and rilpivirine are 1.14 [1.02, 1.28] and 1.72 [1.36, 2.16]), respectively.
b The clinical relevance of CSF-to-plasma concentration ratios is unknown. Concentrations were measured at steady-state 1 week after intramuscular administration of cabotegravir and rilpivirine extended-release injectable suspensions given monthly or every 2 months.
c Elimination half-life driven by slow absorption rate from the intramuscular injection site.
d Dosing in mass balance studies: single-dose oral administration of [14C] cabotegravir; single-dose oral administration of [14C] rilpivirine.Cabotegravir
Rilpivirine
Absorptiona
Tmax (days), median
7
3 to 4
Distribution
% Bound to human plasma proteins
>99.8
99.7
Blood-to-plasma ratio
0.52
0.7
CSF-to-plasma concentration ratio (median [range])b
0.003
0.01
(0.002 to 0.004)
(BLQ to 0.02)
Elimination
t1/2 (weeks), meanc
5.6 to 11.5
13 to 28
Metabolism
Metabolic pathways
UGT1A1
CYP3A
UGT1A9 (minor)
Excretion
Major route of elimination
Metabolism
Metabolism
% of dose excreted as total 14C (unchanged drug) in urined
27 (0)
6 (<1)
% of dose excreted as total 14C (unchanged drug) in fecesd
59 (47)
85 (26)
Table 10. Pharmacokinetic Parameters following Once-Daily Oral Cabotegravir and Rilpivirine and following Initiation and Monthly and Every-2-Month Continuation Intramuscular Injections of the Components of CABENUVA in Adults IM = Intramuscular. a Pharmacokinetic parameter values were based on individual post-hoc estimates from separate cabotegravir and rilpivirine population pharmacokinetic models (cabotegravir: pooled FLAIR and ATLAS for the oral, initial, and monthly injection dosing schedule and ATLAS-2M [participants with no prior exposure to cabotegravir plus rilpivirine] for the every-2-month injection dosing schedule; rilpivirine: pooled FLAIR, ATLAS, and ATLAS-2M [participants with no prior exposure], except for initial injection (direct to injection) [see footnote e] and for oral rilpivirine [see footnote g]).
b tau is dosing interval: 24 hours for oral cabotegravir and rilpivirine, 1 month for cabotegravir and rilpivirine extended-release injectable suspensions given monthly, 2 months for cabotegravir and rilpivirine extended-release injectable suspensions given every 2 months.
c Oral lead-in pharmacokinetic parameter values represent steady-state.
d Initial injection Cmax values primarily reflect oral dosing because the initial injection was administered on the same day as the last oral dose; however, AUC(0-tau) and the Ctau values reflect the initial injections for cabotegravir and rilpivirine.
e Pharmacokinetic parameters for initial injection (direct to injection) based on observed data from FLAIR Extension Phase (n = 110), AUC not calculated based on observed data, Cmax = 1 week following initial injection, Ctau = 1 month following initial injection.
f Monthly and every-2-month injection pharmacokinetic parameter values represent Week 48 data.
g Oral rilpivirine: AUC(0-tau) based on population pharmacokinetic estimates of rilpivirine 25 mg once daily from pooled Phase 3 trials with EDURANT (rilpivirine); Ctau based on observed data from FLAIR, ATLAS, and ATLAS-2M; Cmax based on observed data for rilpivirine 25 mg once daily from a pharmacokinetic substudy in pooled Phase 3 trials with EDURANT (rilpivirine).Drug
Dosing
PhaseDosage
RegimenGeometric Mean (5th, 95th Percentile)a
AUC(0-tau)b
(mcgh/mL)Cmax
(mcg/mL)Ctaub
(mcg/mL)Cabotegravir
Oral lead-inc
30 mg
once daily145
(93.5, 224)
8.0
(5.3, 11.9)
4.6
(2.8, 7.5)
Initial injection (after oral lead-in)d
600 mg IM
initial dose1,591
(714; 3,245)
8.0
(5.3, 11.9)
1.5
(0.65, 2.9)
Initial injection (direct to injection)e
600 mg IM
initial dose—
1.89
(0.438, 5.69)1.43
(0.403, 3.90)Monthly injectionf
400 mg IM
monthly2,415
(1,494; 3,645)
4.2
(2.5, 6.5)
2.8
(1.7, 4.6)
Every-2-month injectionf
600 mg IM
every 2 months
3,764
(2,431; 5,857)
4.0
(2.3, 6.8)
1.6
(0.8, 3.0)
Drug
Dosing
PhaseDosage
RegimenGeometric Mean (5th, 95th Percentile)a
AUC(0-tau)b
(ngh/mL)Cmax
(ng/mL)Ctaub
(ng/mL)Rilpivirine
Oral lead-inc,g
25 mg
once daily2,083
(1,125; 3,748)
116
(48.6, 244)
79.4
(31.8, 177)
Initial injection (after oral lead-in)d
900 mg IM
initial dose44,842
(21,712; 87,575)
144
(93.9, 221)
41.9
(21.7, 78.9)
Initial injection (direct to injection)e
900 mg IM
initial dose—
68
(27.5, 220)48.9
(17.7, 138)Monthly injectionf
600 mg IM
monthly68,324
(39,042; 118,111)
121
(68.1, 210)
85.8
(49.6, 147)
Every-2-month injectionf
900 mg IM
every 2 months132,450
(76,638; 221,783)138
(80.6, 228)68.9
(38.0, 119)Specific Populations
No clinically significant differences in the pharmacokinetics of cabotegravir or rilpivirine were observed based on age, sex, race/ethnicity, BMI, or UGT1A1 polymorphisms.
Cabotegravir and rilpivirine concentrations in participants who were hepatitis C virus antibody positive at baseline were similar to those in the overall study population. The effect of hepatitis B virus co-infection on the pharmacokinetics of cabotegravir is unknown. No clinically relevant differences in the pharmacokinetics of oral rilpivirine have been observed with hepatitis B and/or C virus co-infection.
Patients with Renal Impairment: With oral cabotegravir, no clinically significant differences in the pharmacokinetics of cabotegravir are expected in patients with mild, moderate, or severe renal impairment. Cabotegravir has not been studied in patients with end-stage renal disease not on dialysis. As cabotegravir is >99% protein bound, dialysis is not expected to alter exposures of cabotegravir [see Use in Specific Populations (8.6)].
Population pharmacokinetic analyses indicated that mild renal impairment had no clinically relevant effect on the exposure of oral rilpivirine. There is limited or no information regarding the pharmacokinetics of rilpivirine in patients with moderate or severe renal impairment or end-stage renal disease not on dialysis. As rilpivirine is >99% protein bound, dialysis is not expected to alter exposures of rilpivirine [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment: No clinically significant differences in the pharmacokinetics of cabotegravir are expected in mild to moderate (Child-Pugh A or B) hepatic impairment. The effect of severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of cabotegravir has not been studied [see Use in Specific Populations (8.7)].
No clinically significant differences in the pharmacokinetics of rilpivirine were observed in mild to moderate (Child-Pugh A or B) hepatic impairment. The effect of severe hepatic impairment (Child-Pugh C) has not been studied [see Use in Specific Populations (8.7)].
Geriatric Patients: The pharmacokinetics of cabotegravir (oral or injectable) and of injectable rilpivirine have not been studied and data are limited in participants aged 65 years or older [see Use in Specific Populations (8.5)].
Pediatric Patients: Population pharmacokinetic analyses revealed no clinically relevant differences in exposure between adolescent participants (weighing ≥35 kg and aged at least 12 years) and adult participants with and without HIV-1 from the cabotegravir or rilpivirine development program.
Table 11. Pharmacokinetic Parameters following Cabotegravir and Rilpivirine Orally Once Daily, and Initiation, Monthly, and Every-2-Months Continuation Intramuscular Injections in Adolescents Aged 12 to Younger than 18 Years (≥35 kg) IM = Intramuscular, PO = By Mouth. a Pharmacokinetic parameter values for cabotegravir were based on individual post-hoc estimates from population pharmacokinetic models in both adolescents with HIV-1 (n = 147) weighing 35.2 to 98.5 kg and adolescents without HIV-1 (n = 62) weighing 39.9 to 167 kg. Pharmacokinetic parameter values for rilpivirine were based on individual post-hoc estimates from a population pharmacokinetic model in adolescents with HIV-1 (n = 148) weighing 35.2 to 98.5 kg. b tau is dosing interval: 24 hours for oral administration, 1 month for the initial injection and monthly intramuscular injections, and 2 months for every-2-months intramuscular injections of extended‑release injectable suspension. c Oral lead-in pharmacokinetic parameter values represent steady-state. d Initial injection Cmax values primarily reflect oral dosing because the initial injection was administered on the same day as the last oral dose; however, the AUC(0-tau) and Ctau values reflect the initial injection. e Monthly and every-2-month injection pharmacokinetic parameter values represent Week 48 data. Drug
Dosing
PhaseDosage
RegimenGeometric Mean (5th, 95th Percentile)a
AUC(0-tau)b
(mcgh/mL)Cmax
(mcg/mL)Ctaub
(mcg/mL)Cabotegravir
Oral lead-inc
30 mg
once daily203
(136, 320)10.7
(7.36, 16.6)6.43
(4.15, 10.5)Initial injectiond
600 mg IM
initial dose2,085
(1,056; 4,259)10.8
(7.42, 16.6)1.88
(0.801, 3.71)Every-1-month injectione
400 mg IM every 1 month
3,416
(2,303; 5,109)
5.73
(3.76, 8.90)
4.24
(2.74, 6.45)
Every-2-months injectione
600 mg IM
every 2 months5,184
(3,511; 7,677)5.10
(3.06, 8.24)2.54
(1.25, 4.19)Drug
Dosing
PhaseDosage
RegimenGeometric Mean (5th, 95th Percentile)a
AUC(0-tau)b
(ngh/mL)Cmax
(ng/mL)Ctaub
(ng/mL)Rilpivirine
Oral lead-inc
25 mg PO
once daily2,389
(1,259; 4,414)144
(80.8, 234)76.1
(27.9, 184)Initial injectiond
900 mg IM
initial dose35,259
(20,301; 63,047)135
(85.8, 211)36.5
(22.4, 59.4)Every-1-month injectione
600 mg IM
every month84,280
(49,444; 156,987)146
(84.8, 269)109
(64.8, 202)Every-2-months injectione
900 mg IM
every 2 months110,686
(78,480; 151,744)108
(68.0, 164)61.8
(44.5, 88.0)Drug Interaction Studies
Cabotegravir is not a clinically relevant inhibitor of the following enzymes and transporters: CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4; UGT1A1, 1A3, 1A4, 1A6, 1A9, 2B4, 2B7, 2B15, and 2B17; P-glycoprotein (P‑gp); breast cancer resistance protein (BCRP); bile salt export pump (BSEP); organic cation transporter (OCT)1, OCT2; organic anion transporter polypeptide (OATP)1B1, OATP1B3; multidrug and toxin extrusion transporter (MATE) 1, MATE 2-K; and multidrug resistance protein (MRP)2 or MRP4.
In vitro, cabotegravir inhibited renal OAT1 (IC50 = 0.81 microM) and OAT3 (IC50 = 0.41 microM). Based on physiologically based pharmacokinetic (PBPK) modeling, cabotegravir may increase the AUC of OAT1/3 substrates up to approximately 80%.
In vitro, cabotegravir did not induce CYP1A2, CYP2B6, or CYP3A4.
Simulations using PBPK modeling show that no clinically significant interaction is expected during coadministration of cabotegravir with drugs that inhibit UGT1A1.
In vitro, cabotegravir was not a substrate of OATP1B1, OATP1B3, OATP2B1, or OCT1.
Cabotegravir is a substrate of P-gp and BCRP in vitro; however, because of its high permeability, no alteration in cabotegravir absorption is expected with coadministration of P-gp or BCRP inhibitors.
Rilpivirine is not likely to have a clinically relevant effect on the exposure of drugs metabolized by CYP enzymes.
Drug interaction studies were not conducted with injectable cabotegravir or injectable rilpivirine. Drug interaction studies with oral cabotegravir or oral rilpivirine are summarized in Tables 12, 13, 14, and 15.
Table 12. Effect of Coadministered Drugs on the Pharmacokinetics of Cabotegravir n = Maximum number of participants with data, CI = Confidence Interval. Coadministered
Drug(s)
and Dose(s)Dose of
Cabotegravirn
Geometric Mean Ratio (90% CI) of
Cabotegravir Pharmacokinetic Parameters
with/without Coadministered Drugs
No Effect = 1.00Cmax
AUC
Ctau or C24
Etravirine
30 mg
12
1.04
1.01
1.00
200 mg twice daily
once daily
(0.99, 1.09)
(0.96, 1.06)
(0.94, 1.06)
Rifabutin
30 mg
12
0.83
0.79
0.74
300 mg once daily
once daily
(0.76, 0.90)
(0.74, 0.83)
(0.70, 0.78)
Rifampin
30-mg
15
0.94
0.41
0.50
600 mg once daily
single dose
(0.87, 1.02)
(0.36, 0.46)
(0.44, 0.57)
Rilpivirine
30 mg
11
1.05
1.12
1.14
25 mg once daily
once daily
(0.96, 1.15)
(1.05, 1.19)
(1.04, 1.24)
Table 13. Effect of Coadministered Drugs on the Pharmacokinetics of Rilpivirine N = Maximum number of participants with data, CI = Confidence Interval, ↔ = No Change.
a This interaction study has been performed with a dose higher than the recommended dose for rilpivirine (25 mg once daily) assessing the maximal effect on the coadministered drug.
b Comparison based on historic controls.Coadministered
Drug(s)
and Dose(s)Dose of
Rilpivirinen
Geometric Mean Ratio (90% CI) of
Rilpivirine Pharmacokinetic Parameters
with/without Coadministered Drugs
No Effect = 1.00Cmax
AUC
Cmin
Acetaminophen
150 mg
16
1.09
1.16
1.26
500-mg single dose
once dailya
(1.01 to 1.18)
(1.10 to 1.22)
(1.16 to 1.38)
Atorvastatin
150 mg
16
0.91
0.90
0.90
40 mg once daily
once dailya
(0.79 to 1.06)
(0.81 to 0.99)
(0.84 to 0.96)
Chlorzoxazone
150 mg
16
1.17
1.25
1.18
500-mg single dose taken 2 hours after rilpivirine
once dailya
(1.08 to 1.27)
(1.16 to 1.35)
(1.09 to 1.28)
Darunavir/ritonavir
150 mg
14
1.79
2.30
2.78
800/100 mg once daily
once dailya
(1.56 to 2.06)
(1.98 to 2.67)
(2.39 to 3.24)
Didanosine
150 mg
21
1.00
1.00
1.00
400 mg once daily delayed-release capsules taken 2 hours before rilpivirine
once dailya
(0.90 to 1.10)
(0.95 to 1.06)
(0.92 to 1.09)
Ethinyl estradiol/
norethindrone25 mg
once daily15
↔b
↔b
↔b
0.035 mg once daily/1 mg once daily
Ketoconazole
150 mg
15
1.30
1.49
1.76
400 mg once daily
once dailyb
(1.13 to 1.48)
(1.31 to 1.70)
(1.57 to 1.97)
Lopinavir/ritonavir
150 mg
15
1.29
1.52
1.74
400/100 mg twice daily (soft gel capsule)
once dailya
(1.18 to 1.40)
(1.36 to 1.70)
(1.46 to 2.08)
Methadone
25 mg
12
↔b
↔b
↔b
60 to 100 mg once daily, individualized dose
once daily
Raltegravir
25 mg
23
1.12
1.12
1.03
400 mg twice daily
once daily
(1.04 to 1.20)
(1.05 to 1.19)
(0.96 to 1.12)
Rifabutin
25 mg
18
0.69
0.58
0.52
300 mg once daily
once daily
(0.62 to 0.76)
(0.52 to 0.65)
(0.46 to 0.59)
Rifabutin
300 mg once daily
50 mg
once daily
18
1.43
(1.30 to 1.56)
1.16
(1.06 to 1.26)
0.93
(0.85 to 1.01)
(reference arm for comparison was 25 mg once-daily rilpivirine administered alone)
Rifampin
150 mg
16
0.31
0.20
0.11
600 mg once daily
once dailya
(0.27 to 0.36)
(0.18 to 0.23)
(0.10 to 0.13)
Sildenafil
75 mg
16
0.92
0.98
1.04
50-mg single dose
once dailya
(0.85 to 0.99)
(0.92 to 1.05)
(0.98 to 1.09)
Tenofovir disoproxil fumarate
150 mg
once dailya
16
0.96
1.01
0.99
300 mg once daily
(0.81 to 1.13)
(0.87 to 1.18)
(0.83 to 1.16)
Table 14. Effect of Cabotegravir on the Pharmacokinetics of Coadministered Drugs n = Maximum number of participants with data, CI = Confidence Interval, NA = Not Available. Coadministered
Drug(s)
and Dose(s)Dose of
Cabotegravirn
Geometric Mean Ratio (90% CI) of
Pharmacokinetic Parameters of
Coadministered Drug
with/without CabotegravirNo Effect = 1.00
Cmax
AUC
Ctau or C24
Ethinyl estradiol
30 mg
19
0.92
1.02
1.00
0.03 mg once daily
once daily
(0.83, 1.03)
(0.97, 1.08)
(0.92, 1.10)
Levonorgestrel
30 mg
19
1.05
1.12
1.07
0.15 mg once daily
once daily
(0.96, 1.15)
(1.07, 1.18)
(1.01, 1.15)
Midazolam
30 mg
12
1.09
1.10
NA
3 mg
once daily
(0.94, 1.26)
(0.95, 1.26)
Rilpivirine
30 mg
11
0.96
0.99
0.92
25 mg once daily
once daily
(0.85, 1.09)
(0.89, 1.09)
(0.79, 1.07)
Table 15. Effect of Rilpivirine on the Pharmacokinetics of Coadministered Drugs n = Maximum number of participants with data, CI = Confidence Interval, NA = Not Available.
a This interaction study has been performed with a dose higher than the recommended dose for rilpivirine (25 mg once daily) assessing the maximal effect on the coadministered drug.
b AUC(0-last)
c n = (maximum number of participants with data) for AUC(0-∞) = 15.Coadministered
Drug(s)
and Dose(s)Dose of
Rilpivirinen
Geometric Mean Ratio (90% CI)
of Coadministered Drug Pharmacokinetic
Parameters with/without EDURANT
No Effect = 1.00Cmax
AUC
Cmin
Acetaminophen
150 mg
16
0.97
0.91
NA
500-mg single dose
once dailya
(0.86 to 1.10)
(0.86 to 0.97)
Atorvastatin
150 mg
16
1.35
1.04
0.85
40 mg once daily
once dailya
(1.08 to 1.68)
(0.97 to 1.12)
(0.69 to 1.03)
2-hydroxy-atorvastatin
1.58
1.39
1.32
(1.33 to 1.87)
(1.29 to 1.50)
(1.10 to 1.58)
4-hydroxy-atorvastatin
1.28
1.23
NA
(1.15 to 1.43)
(1.13 to 1.33)
Chlorzoxazone
150 mg
16
0.98
1.03
NA
500-mg single dose taken 2 hours after rilpivirine
once dailya
(0.85 to 1.13)
(0.95 to 1.13)
Darunavir/ritonavir
150 mg
15
0.90
0.89
0.89
800/100 mg once daily
once dailya
(0.81 to 1.00)
(0.81 to 0.99)
(0.68 to 1.16)
Didanosine
150 mg
13
0.96
1.12
NA
400 mg once daily delayed-release capsules taken 2 hours before rilpivirine
once dailya
(0.80 to 1.14)
(0.99 to 1.27)
Digoxin
25 mg
22
1.06
0.98
NA
0.5-mg single dose
once daily
(0.97 to 1.17)
(0.93 to 1.04)b
Ethinyl estradiol
25 mg
17
1.17
1.14
1.09
0.035 mg once daily
once daily
(1.06 to 1.30)
(1.10 to 1.19)
(1.03 to 1.16)
Norethindrone
0.94
0.89
0.99
1 mg once daily
(0.83 to 1.06)
(0.84 to 0.94)
(0.90 to 1.08)
Ketoconazole
150 mg
14
0.85
0.76
0.34
400 mg once daily
once dailya
(0.80 to 0.90)
(0.70 to 0.82)
(0.25 to 0.46)
Lopinavir/ritonavir
150 mg
15
0.96
0.99
0.89
400/100 mg twice daily (soft gel capsule)
once dailya
(0.88 to 1.05)
(0.89 to 1.10)
(0.73 to 1.08)
Methadone
25 mg
13
60 to 100 mg once daily, individualized dose
once daily
R(‑) methadone
0.86
0.84
0.78
(0.78 to 0.95)
(0.74 to 0.95)
(0.67 to 0.91)
S(+) methadone
0.87
0.84
0.79
(0.78 to 0.97)
(0.74 to 0.96)
(0.67 to 0.92)
Metformin
25 mg
20
1.02
0.97
NA
850-mg single dose
once daily
(0.95 to -1.10)
(0.90 to 1.06)c
Raltegravir
25 mg
23
1.10
1.09
1.27
400 mg twice daily
once daily
(0.77 to 1.58)
(0.81 to 1.47)
(1.01 to 1.60)
Rifampin
150 mg
16
1.02
0.99
NA
600 mg once daily
once dailya
(0.93 to 1.12)
(0.92 to 1.07)
25-desacetylrifampin
1.00
0.91
NA
(0.87 to 1.15)
(0.77 to 1.07)
Sildenafil
75 mg
16
0.93
0.97
NA
50-mg single dose
once dailya
(0.80 to 1.08)
(0.87 to 1.08)
N-desmethyl-sildenafil
0.90
0.92
NA
(0.80 to 1.02)
(0.85 to 0.99)b
Tenofovir disoproxil
150 mg
16
1.19
1.23
1.24
fumarate
once dailya
(1.06 to 1.34)
(1.16 to 1.31)
(1.10 to 1.38)
300 mg once daily
12.4 Microbiology
Mechanism of Action
Cabotegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral deoxyribonucleic acid (DNA) integration that is essential for the HIV replication cycle. The mean 50% inhibitory concentration (IC50) value of cabotegravir in a strand transfer assay using purified recombinant HIV-1 integrase was 3.0 nM.
Rilpivirine is a diarylpyrimidine NNRTI of HIV-1 and inhibits HIV-1 replication by non-competitive inhibition of HIV-1 reverse transcriptase (RT). Rilpivirine does not inhibit the human cellular DNA polymerases α, β, and γ.
Antiviral Activity in Cell Culture
Cabotegravir exhibited antiviral activity against laboratory strains of HIV-1 (subtype B, n = 4) with mean 50 percent effective concentration (EC50) values of 0.22 to 1.7 nM in peripheral blood mononuclear cells (PBMCs) and 293 cells. Cabotegravir demonstrated antiviral activity in PBMCs against a panel of 24 HIV-1 clinical isolates (3 in each of group M subtypes A, B, C, D, E, F, and G and 3 in group O) with a median EC50 value of 0.19 nM (range: 0.02 to 1.06 nM, n = 24). The median EC50 value against subtype B clinical isolates was 0.05 nM (range: 0.02 to 0.50 nM, n = 3). Against clinical HIV-2 isolates, the median EC50 value was 0.12 nM (range: 0.10 to 0.14 nM, n = 3).
Rilpivirine exhibited activity against laboratory strains of wild-type HIV-1 in an acutely infected T-cell line with a median EC50 value for HIV-1IIIB of 0.73 nM (0.27 ng/mL). Rilpivirine demonstrated antiviral activity against a broad panel of HIV-1 group M (subtypes A, B, C, D, F, G, and H) primary isolates with EC50 values ranging from 0.07 nM to 1.01 nM (0.03 to 0.37 ng/mL) and was less active against group O primary isolates with EC50 values ranging from 2.88 nM to 8.45 nM (1.06 to 3.10 ng/mL).
In cell culture, cabotegravir was not antagonistic in combination with the NNRTI rilpivirine, or the nucleoside reverse transcriptase inhibitors (NRTIs) emtricitabine (FTC), lamivudine (3TC), or tenofovir disoproxil fumarate (TDF).
The antiviral activity of rilpivirine was not antagonistic when combined with the NNRTIs efavirenz, etravirine, or nevirapine; the NRTIs abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, or zidovudine; the protease inhibitors amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, or tipranavir; the fusion inhibitor enfuvirtide; the CCR5 co-receptor antagonist maraviroc, or the INSTI raltegravir.
Resistance
Cell Culture: Cabotegravir-resistant viruses were selected during passage of HIV-1 strain IIIB in MT-2 cells in the presence of cabotegravir. Amino acid substitutions in integrase that emerged and conferred decreased susceptibility to cabotegravir included Q146L (fold change: 1.3 to 4.6), S153Y (fold change: 2.8 to 8.4), and I162M (fold change: 2.8). The integrase substitution T124A also emerged alone (fold change: 1.1 to 7.4 in cabotegravir susceptibility), in combination with S153Y (fold change: 3.6 to 6.6 in cabotegravir susceptibility) or I162M (2.8-fold change in cabotegravir susceptibility). Cell culture passage of virus harboring integrase substitutions Q148H, Q148K, or Q148R selected for additional substitutions (C56S, V72I, L74M, V75A, T122N, E138K, G140S, G149A, and M154I), with substituted viruses having reduced susceptibility to cabotegravir of 2.0- to 410-fold change. The combinations of E138K+Q148K and V72I+E138K+Q148K conferred the greatest reductions of 53- to 260‑fold change and 410-fold change, respectively.
Rilpivirine-resistant strains were selected in cell culture starting from wild-type HIV-1 of different origins and subtypes as well as NNRTI-resistant HIV-1. The frequently observed amino acid substitutions that emerged and conferred decreased phenotypic susceptibility to rilpivirine included L100I; K101E; V106I and A; V108I; E138K and G, Q, R; V179F and I; Y181C and I; V189I; G190E; H221Y; F227C; and M230I and L.
Clinical Trials: In the pooled Phase 3 FLAIR (Week 124) and ATLAS (Week 96) analysis, there were 8 confirmed virologic failures (2 consecutive HIV-1 RNA ≥200 copies/mL) on cabotegravir plus rilpivirine (8/591, 1.4%) and 8 confirmed virologic failures on current antiretroviral regimen (8/591, 1.4%). Of the 8 confirmed virologic failures in the cabotegravir plus rilpivirine arm, 7 (88%) had treatment-emergent NNRTI resistance-associated substitutions K101E, V106V/A, V108I, E138A, E138G, E138K, H221H/L, or M230L in reverse transcriptase, and 6 of them showed reduced phenotypic susceptibility to rilpivirine (range: 2- to 27-fold). One additional participant in the ATLAS Extension Switch on cabotegravir plus rilpivirine had emergent NNRTI resistance substitution E138A at Week 80 with HIV-1 RNA >50 copies/mL and <200 copies/mL.
Additionally, 5 of the 8 (63%) cabotegravir plus rilpivirine confirmed virologic failures had treatment-emergent INSTI resistance-associated substitutions and reduced phenotypic susceptibility to cabotegravir: Q148R (n = 2; 5- and 9-fold decreased susceptibility to cabotegravir), G140R (n = 1; 7-fold decreased susceptibility to cabotegravir), N155H (n = 1; 3‑fold decreased susceptibility to cabotegravir), or N155H+R263K (n = 1; 9-fold decreased susceptibility to cabotegravir).
There was another confirmed virologic failure participant at Week 112 in FLAIR who had switched to cabotegravir plus rilpivirine direct to injection at Week 100; there were no INSTI resistance‑associated substitutions detected at failure.
In comparison, in the current antiretroviral regimen arm with 8 confirmed virologic failures, 2 of 7 (29%) who had post-baseline resistance data had treatment-emergent resistance substitutions and phenotypic resistance to their antiretroviral drugs; both had treatment-emergent NRTI substitutions, M184V or I, which conferred resistance to emtricitabine or lamivudine in their regimen, and one of them also had the treatment-emergent NNRTI resistance substitution G190S, which conferred resistance to efavirenz in their regimen.
In the ATLAS-2M trial, there were 11 confirmed virologic failures (2 consecutive HIV-1 RNA ≥200 copies/mL) through Week 48: 9 participants (1.7%) in the every-2-month treatment arm and 2 participants (0.4%) in the monthly treatment arm. Of note, 8 of the 11 (73%) participants met confirmed virologic failure criteria at or before the Week 24 injection visit.
Four of the 9 confirmed virologic failure participants in the every-2-month arm transitioned from the oral current antiretroviral regimen arm of ATLAS into this trial.
In the every-2-month treatment arm, 8 of 9 (89%) confirmed virologic failure participants had NNRTI resistance-associated substitutions (E138E/K+V179V/I, K101E+E138A, A98G+K103N, E138K, V179I+Y188L+P225H, Y188L, K103N+E138A, or K101E) at virologic failure. Decreases in rilpivirine susceptibility for these 8 participant isolates ranged from 2- to 30-fold. Six of the 8 participants with NNRTI resistance-associated substitutions also had INSTI resistance-associated substitutions (L74I+Q148Q/R+N155N/H (n = 2), L74I+T97A+N155H, L74I+N155H, N155H, or L74I+Q148R) at failure with cabotegravir fold changes ranging from 1- to 9-fold. The INSTI polymorphism L74I was detected at baseline in 5 of the virologic failure participants by a HIV-1 proviral DNA assay.
In the monthly treatment arm, both of the confirmed virologic failure participants had NNRTI resistance-associated substitutions (K101E+M230L or Y188F+G190Q) at virologic failure with decreased susceptibility to rilpivirine of 17- and >119.2-fold, respectively. Both participants also had INSTI resistance-associated substitutions (N155N/H or Q148R+E138E/K) at failure with decreased susceptibility to cabotegravir of 2- and 5-fold, respectively. Neither participant had the L74I integrase polymorphism at baseline.
Genotypic Baseline Factors Associated with Virologic Failure
An increased risk of cabotegravir plus rilpivirine confirmed virologic failure is associated with baseline virological factors: HIV-1 subtype A1, the presence of baseline integrase L74I polymorphism, and archived NNRTI resistance-associated substitutions.
Association of Subtype A1 and Baseline L74I Polymorphism in Integrase with Cabotegravir plus Rilpivirine Virologic Failure
Eight of the 18 (44%) cabotegravir plus rilpivirine confirmed virologic failures in FLAIR, ATLAS, and ATLAS-2M had HIV‑1 subtype A1 with 7 of the 8 subtype A1 failures having the integrase polymorphism L74I detected at baseline and failure timepoints (Table 16). There was no detectable phenotypic resistance to cabotegravir conferred by the presence of L74I at baseline. Subtype A1 is uncommon in the U.S.
The presence of the integrase polymorphism L74I in subtype B commonly seen in the U.S. was not associated with virologic failure. In contrast to FLAIR and ATLAS, where all virologic failures were subtype A, A1, or AG, subtypes of the cabotegravir plus rilpivirine virologic failures in ATLAS-2M included A (n = 1), A1 (n = 3), B (n = 4), C (n = 2), and complex/A1 (n = 1).
Association of Archived Baseline NNRTI Substitutions with Cabotegravir plus Rilpivirine Virologic Failure
The presence of archived NNRTI resistance-associated substitutions at baseline detected using an exploratory HIV-1 proviral DNA assay was associated with a higher virologic failure rate in the every-2-month arm of ATLAS-2M and in the cabotegravir plus rilpivirine arm (monthly) of ATLAS compared to without archived NNRTI resistance-associated substitutions (Table 16). However, in clinical practice, it is unlikely baseline resistance testing will be performed on virologically suppressed patients (HIV-1 RNA <50 copies/mL). Thus, in patients with an incomplete or uncertain NNRTI treatment history, consideration should be given before starting cabotegravir plus rilpivirine treatment.
Table 16. Rate of Confirmed Virologic Failure in FLAIR, ATLAS, and ATLAS-2M: Baselinea Analysis (Subtype Al, Presence of lntegrase Polymorphism L74I, and Presence of Archived NNRTI Resistance-Associated Substitutions) NNRTI = Non-Nucleoside Reverse Transcriptase Inhibitor, CAB+RPV = Cabotegravir + Rilpivirine, CAR = Current Antiretroviral Regimen, RAS = Resistance-Associated Substitutions, NA = Not Available.
a Baseline and/or Screening result used.
b Per Standard Monogram Nomenclature Reports. Based on June 2020 Los Alamos National Library panel, the majority of HIV-1 subtype A1 was reclassified as HIV-1 subtype A6.
c L74I and L74L/I mixture.
d Baseline/Screening NNRTI substitutions at L100, K101, K103, V106, V108, E138, V179, Y181, Y188, G190, H221, P225, M230.FLAIR
CAB+RPV
N = 283
FLAIR
CAR
N = 283
ATLAS
CAB+RPV
N = 308
ATLAS
CAR
N = 308
ATLAS-2M
Q4W
N = 523
ATLAS-2M
Q8W
N = 522
Total confirmed virologic failures
5
4
3
4
2
9
Subtype A1b
4/8 (50%)
1/4 (25%)
1/17 (6%)
0/21 (0%)
0/30 (0%)
4/31 (13%)
- +L74Ic
4/5 (80%)
1/3 (33%)
1/16 (6%)
0/19 (0%)
2/28 (0%)
3/26 (12%)
- -L74I
0/3 (0%)
0/1 (0%)
0/1 (0%)
0/2 (0%)
0/2 (0%)
1/5 (20%)
Other subtypes
2/268
(0.7%)3/272
(1%)2/240
(0.8%)4/252
(1.6%)2/409
(0.5%)5/415
(1.2%)- +L74Ic
0/49 (0%)
1/43
(2.3%)1/29 (3%)
1/39 (2.6%)
0/42 (0%)
2/48 (4%)
- -L74I
0/219
(2.5%)2/229
(0.9%)1/211
(0.5%)3/213
(1.4%)2/367
(0.5%)3/367
(0.8%)Missing data
7
7
51
35
84
76
With NNRTI RASd
NA
NA
3/78
(4%)2/83
(2%)1/128
(0.8%)7/117
(6%)Without NNRTI RAS
NA
NA
0/179
(0%)2/190
(1%)1/310
(0.3%)2/327
(0.6%)Missing data
NA
NA
51
35
84
76
Cross-Resistance
Virologic failure isolates from cabotegravir plus rilpivirine treatment in FLAIR, ATLAS, and ATLAS-2M exhibited cross-resistance to INSTIs and NNRTIs. All confirmed virologic isolates with genotypic evidence of cabotegravir resistance had cross-resistance to elvitegravir and raltegravir but retained phenotypic susceptibility to dolutegravir and when tested bictegravir. Virologic failure isolates with rilpivirine resistance had cross-resistance with NNRTIs delavirdine, doravirine, efavirenz, etravirine, and nevirapine.
Cabotegravir: Cross-resistance has been observed among INSTIs. Cabotegravir had reduced susceptibility (>5-fold change) to recombinant HIV-1 strain NL432 viruses harboring the following integrase amino acid substitutions: G118R, Q148K, Q148R, T66K+L74M, E92Q+N155H, E138A+Q148R, E138K+Q148K/R, G140C+Q148R, G140S+Q148H/K/R, Y143H+N155H, and Q148R+N155H (range: 5.1- to 81-fold). The substitutions E138K+Q148K and Q148R+N155H conferred the greatest reductions in susceptibility of 81- and 61-fold, respectively.
Cabotegravir was active against viruses harboring the NNRTI substitutions K103N or Y188L, or the NRTI substitutions M184V, D67N/K70R/T215Y, or V75I/F77L/F116Y/Q151M.
Rilpivirine: Cross-resistance has been observed among NNRTIs. The single NNRTI substitutions K101P, Y181I, and Y181V conferred 52-, 15-, and 12-times fold change to rilpivirine, respectively. The K103N substitution did not show reduced susceptibility to rilpivirine by itself. Combinations of 2 or 3 NNRTI resistance-associated substitutions gave 3.7- to 554-fold change to rilpivirine in 38% and 66% of substitutions, respectively. Considering all available cell culture and clinical data, any of the following amino acid substitutions, when present at baseline, are likely to decrease the antiviral activity of rilpivirine: K101E and P; E138A, G, K, R, and Q; V179L; Y181C, I, and V; Y188L; H221Y; F227C; M230I and L, and the combination of L100I/K103N.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies in mice and rats were conducted with cabotegravir. In mice, no drug-related increases in tumor incidence were observed at cabotegravir exposures (AUC) up to approximately 8 times (males) and 7 times (females) higher than those in humans at the RHD. In rats, no drug-related increases in tumor incidence were observed at cabotegravir exposures up to approximately 26 times higher than those in humans at the RHD.
Two-year carcinogenicity studies in mice and rats were conducted with rilpivirine. In mice, rilpivirine was positive for hepatocellular neoplasms in both males and females. The observed hepatocellular findings in mice may be rodent-specific. At the lowest tested dose in the mouse carcinogenicity study, the systemic exposure to rilpivirine was 21 times that observed in humans at the RHD. In rats, no drug-related neoplasms were observed at exposures 3 times those observed in humans at the RHD.
Mutagenesis
Cabotegravir and rilpivirine were not genotoxic in the bacterial reverse mutation assay, mouse lymphoma assay, or in the in vivo rodent micronucleus assay.
Impairment of Fertility
In rats, no effects on fertility were observed at cabotegravir exposures (AUC) >20 times (male) and 28 times (female) the exposure in humans at the RHD. Similarly, no effects on fertility were observed in rats at rilpivirine exposures (AUC) >36 times (male) and 40 times (female) the exposure in humans at the RHD.
-
14 CLINICAL STUDIES
14.1 Clinical Trials in Adults
Monthly Dosing Trials
The efficacy of CABENUVA has been evaluated in 2 Phase 3 randomized, multicenter, active-controlled, parallel-arm, open-label, non-inferiority trials:
- Trial 201584 (FLAIR [NCT02938520]), (n = 629): antiretroviral treatment (ART)‑naive participants with HIV-1 received a dolutegravir INSTI-containing regimen for 20 weeks (either dolutegravir/abacavir/lamivudine or dolutegravir plus 2 other NRTIs if participants were HLA‑B*5701 positive). Participants who were virologically suppressed (HIV-1 RNA <50 copies/mL, n = 566) were then randomized (1:1) to receive either a cabotegravir plus rilpivirine regimen or remain on the current antiretroviral regimen. Participants randomized to receive cabotegravir plus rilpivirine initiated treatment with daily oral lead-in dosing with one 30-mg VOCABRIA (cabotegravir) tablet plus one 25-mg EDURANT (rilpivirine) tablet for at least 4 weeks followed by monthly injections with CABENUVA for an additional 44 weeks [see Dosage and Administration (2.2, 2.3)].
- Trial 201585 (ATLAS [NCT02951052]), (n = 616): ART-experienced, virologically-suppressed (for at least 6 months; median prior treatment duration was 4.3 years) participants (HIV-1 RNA <50 copies/mL) with HIV-1 were randomized and received either a cabotegravir plus rilpivirine regimen or remained on their current antiretroviral regimen. Participants randomized to receive cabotegravir plus rilpivirine initiated treatment with daily oral lead-in dosing with one 30-mg VOCABRIA (cabotegravir) tablet plus one 25-mg EDURANT (rilpivirine) tablet for at least 4 weeks followed by monthly injections with CABENUVA for an additional 44 weeks [see Dosage and Administration (2.2, 2.3)].
The primary analysis was conducted after all participants completed their Week 48 visit or discontinued the trial prematurely.
At baseline, in FLAIR and ATLAS the median age was 34 years and 40 years, 22% and 32% were female, and 24% and 31% were non-White, respectively. In both studies, 7% had CD4+ cell count <350 cells/mm3; these characteristics were similar between treatment arms. In ATLAS, participants received an NNRTI (50%), integrase inhibitor (33%), or protease inhibitor (17%) as their baseline third-agent class prior to randomization; this was similar between treatment arms. Participants with hepatitis B co-infection were excluded from the trial.
The primary endpoint of FLAIR and ATLAS was the proportion of participants with plasma HIV-1 RNA ≥50 copies/mL at Week 48.
The primary endpoint and other Week 48 outcomes, including outcomes by key baseline factors, for FLAIR and ATLAS are shown in Tables 17 and 18.
Table 17. Virologic Outcomes of Randomized Treatment in FLAIR and ATLAS Trials at Week 48 CAB = Cabotegravir, RPV = Rilpivirine, CAR = Current Antiretroviral Regimen, n = Number of participants in each treatment group, CI = Confidence Interval.
a Includes participants who discontinued for lack of efficacy and discontinued while not suppressed.Virologic Outcomes
FLAIR
Monthly DosingATLAS
Monthly DosingCAB plus RPV
(n = 283)
CAR
(n = 283)
CAB plus RPV
(n = 308)
CAR
(n = 308)
HIV-1 RNA ≥50 copies/mLa
2%
2%
2%
1%
Treatment difference
-0.4%
0.7%
(95% CI: -2.8%, 2.1%)
(95% CI: -1.2%, 2.5%)
HIV-1 RNA <50 copies/mL
94%
93%
93%
95%
No virologic data at Week 48 window
4%
4%
6%
4%
Discontinued due to adverse event or death
3%
<1%
4%
2%
Discontinued for other reasons
1%
4%
2%
2%
Missing data during window but on study
0
0
0
0
Adjusted for study and randomization stratification factors, treatment difference of HIV-1 RNA ≥50 copies/mL for the pooled data was 0.2% with 95% CI (-1.4%, 1.7%).
Table 18. Proportion of Participants in FLAIR and ATLAS Trials with Plasma HIV-1 RNA ≥50 copies/mL at Week 48 for Key Baseline Factors CAB = Cabotegravir, RPV = Rilpivirine, CAR = Current Antiretroviral Regimen. Baseline Factors
FLAIR
Monthly DosingATLAS
Monthly DosingCAB plus RPV
(N = 283)
n/N (%)
CAR
(N = 283)
n/N (%)
CAB plus RPV
(N = 308)
n/N (%)
CAR
(N = 308)
n/N (%)
Baseline CD4+ (cells/mm3)
<350
0/19
1/27 (4%)
0/23
1/27 (4%)
≥350 to <500
3/64 (5%)
0/60
2/56 (4%)
0/60
≥500
3/200 (2%)
6/196 (3%)
3/299 (1%)
2/224 (<1%)
Gender
Male
3/220 (1%)
6/219 (3%)
3/209 (1%)
3/204 (1%)
Female
3/63 (5%)
1/64 (2%)
2/99 (2%)
0/104
Race
White
6/216 (3%)
5/201 (2%)
3/214 (1%)
2/207 (<1%)
African American/African Heritage
0/47
2/56 (4%)
2/62 (3%)
1/77 (1%)
Asian/Other
0/20
0/24
0/32
0/24
Body mass index
<30 kg/m2
3/243 (1%)
7/246 (3%)
3/248 (1%)
1/242 (<1%)
≥30 kg/m2
3/40 (8%)
0/37
2/60 (3%)
2/66 (3%)
Age (years)
<50
5/250 (2%)
6/254 (2%)
4/242 (2%)
2/212 (<1%)
≥50
1/33 (3%)
1/29 (3%)
1/66 (2%)
1/96 (1%)
Baseline antiviral therapy at randomization
Protease inhibitor-containing regimen
0
0
1/51 (2%)
0/54
Integrase inhibitor-containing regimen
6/283 (2%)
7/283 (2%)
0/102
2/99 (2%)
Non-nucleoside reverse transcriptase inhibitor-containing regimen
0
0
4/155 (3%)
1/155 (<1%)
Participants in both the FLAIR and ATLAS trials were virologically suppressed prior to Day 1 or at study entry, respectively, and no clinically relevant change from baseline in CD4+ cell counts was observed.
In FLAIR at Week 96, the proportion of participants with HIV-1 RNA ≥50 copies/mL was 3.2 % for both the cabotegravir plus rilpivirine (n = 283) and current antiretroviral regimen (n = 283) treatment arms; adjusted treatment difference was 0.0% with 95% CI (-2.9%, 2.9%). The proportion of participants with HIV-1 RNA <50 copies/mL was 87% and 89% for the cabotegravir plus rilpivirine and the current antiretroviral regimen arms, respectively; adjusted treatment difference was -2.8% with 95% CI (-8.2%, 2.5%).
Optional Oral Lead-In: FLAIR Extension Phase
In the FLAIR study during the Extension Phase (Week 100 to Week 124), the efficacy of CABENUVA was evaluated in patients who switched (at Week 100) from their current antiretroviral regimen to CABENUVA, with and without an oral lead-in phase. A total of 121 participants chose to start the treatment with oral lead-in and 111 participants chose direct to injection. Participants were not randomized during the Extension Phase. At Week 124, the proportion of participants with HIV-1 RNA ≥50 copies/mL was 0.8% and 0.9% for the oral lead-in and direct to injection groups, respectively. The rates of virologic suppression (HIV-1 RNA <50 copies/mL) were similar in both the oral lead-in (93%) and direct to injection (99%) groups.
Every-2-Month Dosing Trial
The efficacy of CABENUVA dosed every 2 months has been evaluated in 1 Phase 3b randomized, multicenter, parallel-arm, open-label, non-inferiority trial:
- Trial 207966 (ATLAS-2M [NCT03299049]), (n = 1,045): ART‑experienced, virologically suppressed participants with HIV-1, including 504 participants from the ATLAS trial (randomized to CAB plus RPV [n = 253] or CAR [n = 251]; prior exposure to cabotegravir plus rilpivirine [n = 391]), were randomized and received a cabotegravir plus rilpivirine regimen administered as injection doses of cabotegravir 400 mg plus rilpivirine 600 mg either monthly or cabotegravir 600 mg plus rilpivirine 900 mg every 2 months. Participants without prior exposure to cabotegravir plus rilpivirine initiated treatment with daily oral lead-in dosing with one 30-mg VOCABRIA (cabotegravir) tablet plus one 25-mg EDURANT (rilpivirine) tablet for at least 4 weeks followed by monthly or every-2-month injections with CABENUVA for an additional 44 weeks.
The primary analysis was conducted after all participants completed their Week 48 visit or discontinued the study prematurely.
At baseline, the median age was 42 years, 27% were female, 27% were non-White, and 6% had a CD4+ cell count <350 cells per mm3; these characteristics were similar between the treatment arms. Participants received either an NNRTI (29%), an integrase inhibitor besides cabotegravir plus rilpivirine (26%), a protease inhibitor (7%), or cabotegravir plus rilpivirine (37%) as their baseline third-agent class prior to randomization.
The primary endpoint of ATLAS-2M was the proportion of participants with a plasma HIV-1 RNA ≥50 copies/mL at Week 48.
The primary endpoint and other Week 48 outcomes, including outcomes by key baseline factors, for ATLAS-2M are shown in Tables 19 and 20.
Table 19. Virologic Outcomes of Randomized Treatment in ATLAS 2-M Trial at Week 48 a Includes participants who discontinued for lack of efficacy, discontinued while not suppressed.
n = Number of participants in each treatment group, CI = Confidence Interval.Virologic Outcomes
Cabotegravir plus Rilpivirine
Every-2-Month Dosing
n = 522
Monthly Dosing
n = 523
HIV-1 RNA ≥50 copies/mLa
2%
1%
Treatment difference
0.8
(95% CI: -0.6%, 2.2%)
HIV-1 RNA <50 copies/mL
94%
94%
No virologic data at Week 48 window
4%
6%
Discontinued study due to adverse event or death
2%
3%
Discontinued for other reasons
2%
3%
Missing data during window but on study
0
0
Table 20. Proportion of Participants in ATLAS 2-M Trial with Plasma HIV-1 RNA ≥50 copies/mL at Week 48 for Key Baseline Factors Baseline Factors
Cabotegravir plus Rilpivirine
Every-2-Month Dosing
(N = 522)
n/N (%)
Monthly Dosing
(N = 523)
n/N (%)
Baseline CD4+ (cells/mm3)
<350
1/35 (3%)
1/27 (4%)
≥350 to <500
1/96 (1%)
0/89
≥500
7/391 (2%)
4/407 (1%)
Gender
Male
4/385 (1%)
5/380 (1%)
Female
5/137 (4%)
0/143
Race
White
5/370 (1%)
5/393 (1%)
Black/African American
Asian/Other
4/101 (4%)
0/51
0/90
0/40
Body mass index
<30 kg/m2
3/409 (1%)
3/425 (1%)
≥30 kg/m2
6/113 (5%)
2/98 (2%)
Age (years)
<35
4/137 (3%)
1/145 (1%)
35 to <50
3/242 (1%)
2/239 (1%)
≥50
2/143 (1%)
2/139 (1%)
Prior exposure to cabotegravir plus rilpivirine
None
5/327 (2%)
5/327 (2%)
1 to 24 Weeks
3/69 (4%)
0/68
>24 Weeks
1/126 (1%)
0/128
Baseline third-agent class
Protease inhibitor-containing regimen
1/40 (3%)
1/30 (3%)
Integrase inhibitor-containing regimen
3/136 (2%)
2/141 (1%)
NNRTI-containing regimen
1/151 (1%)
2/156 (1%)
Cabotegravir plus rilpivirine
4/195 (2%)
0/196
14.2 Clinical Trial in Adolescents
Trial 208580 (MOCHA, [NCT03497676])
The safety, tolerability, and pharmacokinetics of oral and injectable cabotegravir and oral and injectable rilpivirine were assessed in an ongoing Phase 1/2 multicenter, open-label, non‑comparative study, MOCHA (IMPAACT 2017) [see Adverse Reactions (6.1), Clinical Pharmacology (12.3)].
Cohort 1
Fifty-five virologically suppressed adolescents with HIV-1 aged 12 to younger than 18 years and weighing at least 35 kg were enrolled to 1 of 4 subgroups, 1C (Q4W): cabotegravir monthly dosing, 1C (Q8W): cabotegravir every‑2‑month dosing, 1R (Q4W): rilpivirine monthly dosing or 1R (Q8W): rilpivirine every‑2‑month dosing.
In cohort 1C, participants (n = 30) received one 30-mg cabotegravir tablet daily for at least 4 weeks followed by monthly cabotegravir injections for 3 months (Month 1: 600-mg injection, Months 2 and 3: 400-mg injection), or every-2-month cabotegravir injections for 2 months (Months 1 and 2: 600-mg injection), while continuing background antiretroviral therapy. In cohort 1R, participants (n = 25) received one 25-mg rilpivirine tablet daily for at least 4 weeks followed by monthly rilpivirine injections for 3 months (Month 1: 900-mg injection, Months 2 and 3: 600-mg injection), or every-2-month rilpivirine injections for 2 months (Months 1 and 2: 900-mg injection), while continuing background antiretroviral therapy.
At baseline, in cohort 1 (n = 55), the median age of participants was 15.0 years, the median weight was 50.0 kg (range: 37.4, 98.5), 47% were female, 76% were Black/African American, 16% were Asian, 7% were White; and no participant had a CD4+ cell count <350 cells per mm3. At baseline, median CD4+ cell count was 725 cells per mm3 (range: 397 to 1808).
The primary objectives at Week 16, which were to confirm the use of the adult dose through the evaluation of safety and pharmacokinetics in virologically suppressed adolescents with HIV-1, were met enabling the progression of participants to cohort 2 [see Adverse Reactions (6.1), Clinical Pharmacology (12.3)].
Cohort 2
Cohort 2 enrolled eligible participants who had completed cohort 1 as well as eligible participants who had not been previously enrolled in the study. Cohort 2 participants (n = 144) discontinued their background antiretroviral therapy and received one 30-mg cabotegravir tablet plus one 25-mg rilpivirine tablet daily for at least 4 weeks followed by every-2-month cabotegravir injections (Months 1 and 2: 600-mg injection, then 600-mg injection once every 2 months) and rilpivirine injections (Months 1 and 2: 900-mg injection, then 900-mg injection once every 2 months).
At baseline, in cohort 2, the median age of participants was 15.0 years, the median weight was 48.5 kg (range: 35.2, 100.9), 51% were female, 74% were Black/African American, 25% were Asian, 1% was White; and 4 participants had a CD4+ cell count less than 350 cells per mm3. At baseline, median CD4+ cell count was 739.5 cells per mm3 (range: 81 to 1925).
The primary objective at Week 24, which was to confirm the safety of injectable cabotegravir plus injectable rilpivirine in virologically suppressed adolescents with HIV-1, was met [see Adverse Reactions (6.1)]. Antiviral activity was assessed as a secondary objective: 139 of the 141 (98.6%) participants with available data remained virologically suppressed (HIV-1 RNA <50 copies/mL) at Week 24. The median change from baseline in CD4+ cell count at Week 24 was -36.0 cells per mm3.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
CABENUVA is supplied in 2 dosing kits. Each kit contains 1 vial of cabotegravir extended‑release injectable suspension and 1 vial of rilpivirine extended-release injectable suspension, co‑packaged as follows:
CABENUVA 400-mg/600-mg Kit (NDC: 49702-253-15) containing:
- One single-dose vial of cabotegravir extended-release injectable suspension containing 400 mg/2 mL (200 mg/mL) of cabotegravir.
- One single-dose vial of rilpivirine extended-release injectable suspension containing 600 mg/2 mL (300 mg/mL) of rilpivirine
CABENUVA 600-mg/900-mg Kit (NDC: 49702-240-15) containing:
- One single-dose vial of cabotegravir extended-release injectable suspension containing 600 mg/3 mL (200 mg/mL) of cabotegravir.
- One single-dose vial of rilpivirine extended-release injectable suspension containing 900 mg/3 mL (300 mg/mL) of rilpivirine.
Each dosing kit also contains 2 syringes, 2 syringe labels, 2 vial adapters, and 2 needles for intramuscular injection (23-gauge, 1½ inch). The vial stoppers are not made with natural rubber latex.
Storage and Handling
Store CABENUVA in the refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton until ready to use. Do not freeze. Do not mix with any other product or diluent.
Prior to administration, vials should be brought to room temperature (not to exceed 25°C [77°F]). Vials may remain in the carton at room temperature for up to 6 hours; do not put back into the refrigerator. If not used within 6 hours, they must be discarded.
Once the suspensions have been drawn into the respective syringes, the injections should be administered as soon as possible, but may remain in the syringes for up to 2 hours. The filled syringes should not be placed in the refrigerator. If the medicines remain in the syringes for more than 2 hours, the filled syringes and needles must be discarded [see Dosage and Administration (2.9)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Advise patients to immediately contact their healthcare provider if they develop a rash. Instruct patients to immediately stop taking CABENUVA and seek medical attention if they develop a rash associated with any of the following symptoms, as it may be a sign of a more serious reaction such as Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) or drug reaction with eosinophilia and systemic symptoms (DRESS): fever; generally ill feeling; extreme tiredness; muscle or joint aches; blisters; oral blisters or lesions; eye inflammation; facial swelling; swelling of the eyes, lips, tongue, or mouth; difficulty breathing; and/or signs and symptoms of liver problems (e.g., yellowing of the skin or whites of the eyes; dark or tea-colored urine; pale-colored stools or bowel movements; nausea; vomiting; loss of appetite; or pain, aching, or sensitivity on the right side below the ribs). Advise patients that if hypersensitivity occurs, they will be closely monitored, laboratory tests will be ordered, and appropriate therapy will be initiated [see Warnings and Precautions (5.1)].
Adverse Reactions Following Injections
Advise patients that injection site reactions have been reported in the majority of patients receiving CABENUVA. These local reactions typically consist of one or more of the following: pain, erythema, tenderness, pruritus, and local swelling. Systemic reactions have also been reported, such as fever, musculoskeletal pain, and sciatica pain [see Adverse Reactions (6.1)]. Serious post-injection reactions were reported within minutes after the injection of rilpivirine, including dyspnea, bronchospasm, agitation, abdominal cramping, rash/urticaria, dizziness, flushing, sweating, oral numbness, changes in blood pressure, and back pain (e.g., back and chest). These events began to resolve within minutes after the injection. Advise patients that they will be observed briefly (approximately 10 minutes) after the injection. If they experience a post-injection reaction, they will be monitored and appropriate treatment administered [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients that hepatotoxicity has been reported with cabotegravir and rilpivirine, components of CABENUVA [see Warnings and Precautions (5.3), Adverse Reactions (6.1)]. Inform patients that monitoring for liver transaminases is recommended.
Depressive Disorders
Inform patients that depressive disorders (including depressed mood, depression, major depression, mood altered, mood swings, unusual mood, negative thoughts, suicidal ideation, suicide attempt) have been reported with at least one of the components of CABENUVA. Advise patients to seek prompt medical evaluation if they experience depressive symptoms [see Warnings and Precautions (5.4), Adverse Reactions (6.1)].
Drug Interactions
Inform patients that CABENUVA may interact with other drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John’s wort. CABENUVA is an extended-release injectable that may be systemically present for 12 months or longer. These residual concentrations are not expected to affect the exposures of antiretroviral drugs that are initiated after discontinuation of CABENUVA [see Contraindications (4), Drug Interactions (7)].
Adherence to CABENUVA
Counsel patients about the importance of continued medication adherence and scheduled visits to help maintain viral suppression and to reduce risk of loss of virologic response and development of resistance [see Dosage and Administration (2.1), Warnings and Precautions (5.6)].
Two Options for Dosing Frequency
Advise the patient that CABENUVA can be injected monthly or every 2 months after oral lead‑in with cabotegravir and rilpivirine to assess tolerability. Discuss the two injection dosing frequency options with the patient prior to starting CABENUVA and decide which injection dosing frequency would be the most appropriate option for the patient [see Dosage and Administration (2.1)].
Missed Dose
Inform patients that CABENUVA can remain in the body for up to 12 months or longer after receiving their last injection. Advise patients that they should contact their healthcare provider if they miss or plan to miss a scheduled injection visit and that oral therapy with VOCABRIA and EDURANT may be used up to 2 months to replace missed injection visits, or any other fully suppressive oral antiretroviral regimen may be used until injections are resumed. Advise patients that if they stop treatment with CABENUVA, they will need to take other medicines to treat their HIV-1 infection [see Dosage and Administration (2.2, 2.8), Warnings and Precautions (5.6)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes in those exposed to CABENUVA during pregnancy. Patients who are of reproductive potential should be informed of the long duration of exposure of CABENUVA and that there is very limited clinical experience in human pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Lactation
Inform individuals with HIV-1 infection that the potential risks of breastfeeding include: (1) HIV-1 transmission (in HIV-1–negative infants), (2) developing viral resistance (in HIV-1–positive infants), and (3) adverse reactions in a breastfed infant similar to those seen in adults [see Use in Specific Populations (8.2)].
CABENUVA and VOCABRIA are trademarks owned by or licensed to the ViiV Healthcare group of companies.
The other brand listed is a trademark owned by or licensed to its respective owner and is not a trademark owned by or licensed to the ViiV Healthcare group of companies. The maker of this brand is not affiliated with and does not endorse the ViiV Healthcare group of companies or its products.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
©2025 ViiV Healthcare group of companies or its licensor.
CBN:11PI
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
CABENUVA [KAB-en-ew-vah]
(cabotegravir extended-release injectable suspension;
rilpivirine extended-release injectable suspension)co-packaged for intramuscular use
What is CABENUVA?
CABENUVA is a prescription medicine that is used without other human immunodeficiency virus-1 (HIV-1) medicines to treat HIV-1 infection in people 12 years of age and older who weigh at least 77 pounds (35 kg) to replace their current HIV-1 medicines when their healthcare provider determines that they meet certain requirements.
HIV-1 is the virus that causes acquired immune deficiency syndrome (AIDS).
CABENUVA contains 2 different medicines:
- cabotegravir
- rilpivirine
It is not known if CABENUVA is safe and effective in children younger than 12 years of age or weighing less than 77 pounds (35 kg).
Do not receive CABENUVA if you:
- have ever had an allergic reaction to cabotegravir or rilpivirine.
- are taking any of the following medicines:
- carbamazepine
- oxcarbazepine
- phenobarbital
- phenytoin
- rifabutin
- rifampin
- rifapentine
- dexamethasone (more than a single-dose treatment)
- St John’s wort (Hypericum perforatum)
Before you receive CABENUVA, tell your healthcare provider about all your medical conditions, including if you:
- have ever had a skin rash or an allergic reaction to medicines that contain cabotegravir or rilpivirine.
- have or have had liver problems, including hepatitis B or C infection.
- have or ever had kidney problems.
- have ever had mental health problems.
- are pregnant or plan to become pregnant. It is not known if CABENUVA will harm your unborn baby. CABENUVA can remain in your body for up to 12 months or longer after the last injection. Tell your healthcare provider if you become pregnant during or after treatment with CABENUVA.
- Pregnancy Registry. There is a pregnancy registry for those who take CABENUVA during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
-
are breastfeeding or plan to breastfeed. CABENUVA may pass into your breast milk. Talk to your healthcare provider about the following risks to your baby from breastfeeding during or after treatment with CABENUVA:
- o the HIV-1 virus may pass to your baby if your baby does not have HIV-1 infection.
- o the HIV-1 virus may become harder to treat if your baby has HIV-1 infection.
- o your baby may get side effects from CABENUVA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Some medicines interact with CABENUVA. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
You can ask your healthcare provider or pharmacist for a list of medicines that interact with CABENUVA.
Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take CABENUVA with other medicines.
How will I receive CABENUVA?
- Your healthcare provider will inject CABENUVA into the muscle of each side of your buttocks or on the same side of your buttocks at least 2 cm apart.
- You will receive CABENUVA as 2 injections (cabotegravir and rilpivirine), either one time every month or one time every 2 months.
- Before receiving your first injection doses of CABENUVA, your healthcare provider may have you take 1 VOCABRIA (cabotegravir) tablet and 1 EDURANT (rilpivirine) tablet 1 time a day for 1 month (at least 28 days). This will allow your healthcare provider to assess how well you tolerate these medicines.
- CABENUVA is a long-acting medicine and may stay in your system for 12 months or longer after your last injection.
- Stay under the care of a healthcare provider during treatment with CABENUVA. It is important that you attend your planned appointments to receive your injection doses of CABENUVA.
- If you miss or plan to miss a scheduled monthly or every-2-month injection of CABENUVA by more than 7 days, call your healthcare provider right away to discuss your treatment options.
- If you stop treatment with CABENUVA you will need to take other medicines to treat your HIV-1 infection and reduce the risk of developing viral resistance. Call your healthcare provider right away to discuss your treatment options.
What are the possible side effects of CABENUVA?
CABENUVA may cause serious side effects including:
- Allergic reactions. Call your healthcare provider right away if you develop a rash with CABENUVA. Stop receiving CABENUVA and get medical help right away if you develop a rash with any of the following signs or symptoms:
- fever
- generally ill feeling
- tiredness
- muscle or joint aches
- trouble breathing
- blisters or sores in mouth
- blisters
- redness or swelling of the eyes
- swelling of the mouth, face, lips, or tongue
- Post-injection reactions. Post-injection reaction symptoms have happened within minutes in some people after receiving their rilpivirine injection. Most symptoms resolved within minutes after the injection. Symptoms of post-injection reactions may include:
- trouble breathing
- narrowing of airways
- stomach cramps
- sweating
- numbness of your mouth
- pain (e.g., back and chest)
- feeling anxious
- feeling warm
- rash
- feeling lightheaded or feeling like you are going to pass out (faint)
- blood pressure changes
- Liver problems. Liver problems have happened in people with or without history of liver problems or other risk factors. Your healthcare provider may do blood tests to check your liver function. People with a history of liver problems or people who have certain liver function test changes may have an increased risk of developing new or worsening changes in certain liver tests during treatment with CABENUVA.
- Call your healthcare provider right away if you develop any of the following signs or symptoms of liver problems:
- your skin or the white part of your eyes turns yellow (jaundice)
- dark or “tea-colored” urine
- light-colored stools (bowel movements)
- nausea or vomiting
- loss of appetite
- pain, aching, or tenderness on the right side of your stomach area
- itching
-
Depression or mood changes. Call your healthcare provider or get emergency medical help right away if you develop any of the following symptoms:
- o feeling sad or hopeless
- o feeling anxious or restless
- o have thoughts of hurting yourself (suicide) or have tried to hurt yourself
The most common side effects of CABENUVA include:
- pain, tenderness, hardened mass or lump, swelling, redness, itching, bruising, and warmth at the injection site
- fever
- tiredness
- headache
- muscle or bone pain
- nausea
- sleep problems
- dizziness
- rash
These are not all the possible side effects of CABENUVA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088.
General information about the safe and effective use of CABENUVA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about CABENUVA that is written for health professionals.
What are the ingredients in CABENUVA?
Cabotegravir extended-release injectable suspension:
Active ingredient: cabotegravir
Inactive ingredients: mannitol, polyethylene glycol (PEG) 3350, polysorbate 20, and Water for Injection.
Rilpivirine extended-release injectable suspension:
Active ingredient: rilpivirine
Inactive ingredients: citric acid monohydrate, dextrose to ensure isotonicity, monobasic sodium phosphate, poloxamer 338, sodium hydroxide to adjust pH, and Water for Injection.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
CABENUVA and VOCABRIA are trademarks owned by or licensed to the ViiV Healthcare group of companies.
The other brand listed is a trademark owned by or licensed to its respective owner and is not a trademark owned by or licensed to the ViiV Healthcare group of companies. The maker of this brand is not affiliated with and does not endorse the ViiV Healthcare group of companies or its products.
©2025 ViiV Healthcare group of companies or its licensor.
CBN:8PIL
For more information, go to www.cabenuva.com or call 1-877-844-8872.
- This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 11/2025
-
INSTRUCTIONS FOR USE
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: 9/2024
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: 9/2024
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 49702-253-15
CABENUVA
Cabotegravir extended-release
injectable suspension
400 mg/2 mL
(200 mg/mL)
co-packaged with
Rilpivirine extended-release
injectable suspension
600 mg/2 mL
(300 mg/mL)
Rx Only
ViiV Healthcare
For gluteal intramuscular use only.
Healthcare Professional administration only.
Contents:
- 1 Cabotegravir single-dose vial
- 1 Rilpivirine single-dose vial
- 2 Vial adapters
- 2 Syringes
- 2 Injection needles (23 gauge, 1 ½ inch)
- 2 Syringe labels
- Prescribing Information
- Patient Information
- Instructions for Use
Store in refrigerator at 2°C to 8°C (36°F to 46°F).
Do not freeze.
Discard unused portion.
Prior to administration, bring vials to room temperature (not to exceed 25°C (77°F). Vials may remain at room temperature for up to 6 hours. If not used within 6 hours, they must be discarded. Ensure vial adapter is used correctly.
400 mg/600 mg Kit
Cabotegravir – Made in Singapore and Belgium
Rilpivirine – Made in Belgium and Ireland
©2025 ViiV Healthcare group of companies or its licensor.
- Rev. 9/25
- 620000000101118
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 49702-240-15
CABENUVA
Cabotegravir extended-release
injectable suspension
600 mg/3 mL
(200 mg/mL)
co-packaged with
Rilpivirine extended-release
injectable suspension
900 mg/3 mL
(300 mg/mL)
Rx Only
ViiV Healthcare
For gluteal intramuscular use only.
Healthcare Professional administration only.
Contents:
- 1 Cabotegravir single-dose vial
- 1 Rilpivirine single-dose vial
- 2 Vial adapters
- 2 Syringes
- 2 Injection needles (23 gauge, 1 ½ inch)
- 2 Syringe labels
- Prescribing Information
- Patient Information
- Instructions for Use
Store in refrigerator at 2°C to 8°C (36°F to 46°F).
Do not freeze.
Discard unused portion.
Prior to administration, bring vials to room temperature (not to exceed 25°C (77°F). Vials may remain at room temperature for up to 6 hours. If not used within 6 hours, they must be discarded. Ensure vial adapter is used correctly.
600 mg/900 mg Kit
Cabotegravir – Made in Singapore and Belgium
Rilpivirine – Made in Belgium and Ireland
©2025 ViiV Healthcare group of companies or its licensor.
- Rev. 9/25
- 620000000101176
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 49702-266-63
CABENUVA
Cabotegravir extended-release
injectable suspension
600 mg/3 mL
(200 mg/mL)
co-packaged with
Rilpivirine extended-release
injectable suspension
900 mg/3 mL
(300 mg/mL)
Rx Only
Sample-Not for Sale
ViiV Healthcare
For gluteal intramuscular use only.
Healthcare Professional administration only.
Contents:
- 1 Cabotegravir single-dose vial
- 1 Rilpivirine single-dose vial
- 2 Vial adapters
- 2 Syringes
- 2 Injection needles (23 gauge, 1 ½ inch)
- 2 Syringe labels
- Prescribing Information
- Patient Information
- Instructions for Use
Store in refrigerator at 2°C to 8°C (36°F to 46°F).
Do not freeze.
Discard unused portion.
Prior to administration, bring vials to room temperature (not to exceed 25°C (77°F). Vials may remain at room temperature for up to 6 hours. If not used within 6 hours, they must be discarded. Ensure vial adapter is used correctly.
600 mg/900 mg Kit
Cabotegravir – Made in Singapore and Belgium
Rilpivirine – Made in Belgium and Ireland
©2025 ViiV Healthcare group of companies or its licensor.
- Rev. 9/25
62000000010112
-
INGREDIENTS AND APPEARANCE
CABENUVA
cabotegravir and rilpivirine kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 49702-253 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-253-15 1 in 1 CARTON; Type 1: Convenience Kit of Co-Package 01/21/2021 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 1 mL Part 2 1 VIAL 1 mL Part 1 of 2 CABOTEGRAVIR
cabotegravir injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-245 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CABOTEGRAVIR (UNII: HMH0132Z1Q) (CABOTEGRAVIR - UNII:HMH0132Z1Q) CABOTEGRAVIR 200 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) POLYSORBATE 20 (UNII: 7T1F30V5YH) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE (white to light pink) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-245-01 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 Part 2 of 2 RILPIVIRINE
rilpivirine injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-249 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RILPIVIRINE (UNII: FI96A8X663) (RILPIVIRINE - UNII:FI96A8X663) RILPIVIRINE 300 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLOXAMER 338 (UNII: F75JV2T505) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) DEXTROSE MONOHYDRATE (UNII: LX22YL083G) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-249-02 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 CABENUVA
cabotegravir and rilpivirine kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 49702-240 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-240-15 1 in 1 CARTON; Type 1: Convenience Kit of Co-Package 01/21/2021 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 0 VIAL 1 mL Part 2 0 VIAL 1 mL Part 1 of 2 CABOTEGRAVIR
cabotegravir injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-238 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CABOTEGRAVIR (UNII: HMH0132Z1Q) (CABOTEGRAVIR - UNII:HMH0132Z1Q) CABOTEGRAVIR 200 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) POLYSORBATE 20 (UNII: 7T1F30V5YH) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE (white to light pink) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-238-01 3 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 Part 2 of 2 RILPIVIRINE
rilpivirine injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-243 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RILPIVIRINE (UNII: FI96A8X663) (RILPIVIRINE - UNII:FI96A8X663) RILPIVIRINE 300 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLOXAMER 338 (UNII: F75JV2T505) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) DEXTROSE MONOHYDRATE (UNII: LX22YL083G) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-243-02 3 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2021 CABENUVA
cabotegravir and rilpivirine kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 49702-266 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-266-63 1 in 1 CARTON; Type 1: Convenience Kit of Co-Package 01/21/2025 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 0 VIAL 1 mL Part 2 0 VIAL 1 mL Part 1 of 2 CABOTEGRAVIR
cabotegravir injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-238 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CABOTEGRAVIR (UNII: HMH0132Z1Q) (CABOTEGRAVIR - UNII:HMH0132Z1Q) CABOTEGRAVIR 200 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) POLYSORBATE 20 (UNII: 7T1F30V5YH) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE (white to light pink) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-238-64 3 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2025 Part 2 of 2 RILPIVIRINE
rilpivirine injection, suspension, extended releaseProduct Information Item Code (Source) NDC: 49702-243 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RILPIVIRINE (UNII: FI96A8X663) (RILPIVIRINE - UNII:FI96A8X663) RILPIVIRINE 300 mg in 1 mL Inactive Ingredients Ingredient Name Strength POLOXAMER 338 (UNII: F75JV2T505) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) DEXTROSE MONOHYDRATE (UNII: LX22YL083G) SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49702-243-61 3 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA212888 01/21/2025 Labeler - ViiV Healthcare Company (027295585) Establishment Name Address ID/FEI Business Operations Glaxo Wellcome Manufacturing Pte. Ltd 595295577 API MANUFACTURE(49702-253, 49702-240, 49702-245, 49702-238) , ANALYSIS(49702-253, 49702-240, 49702-245, 49702-238) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceutica NV 400345889 API MANUFACTURE(49702-253, 49702-240, 49702-249, 49702-243) , ANALYSIS(49702-253, 49702-240, 49702-249, 49702-243)
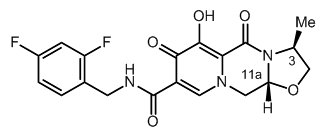
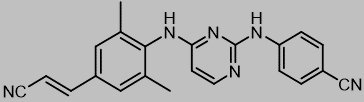

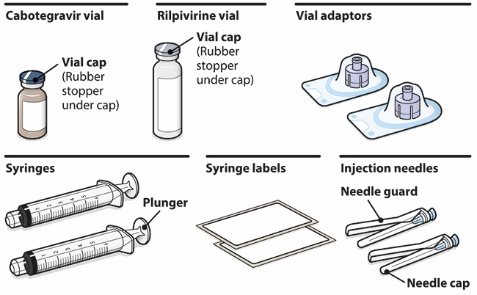
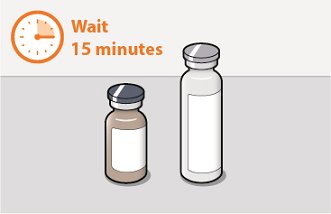
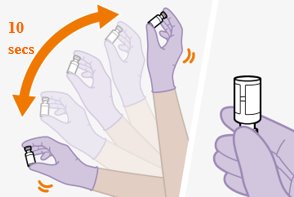
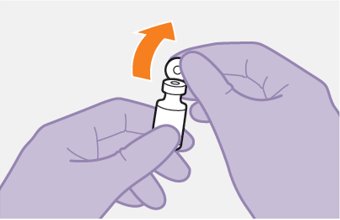
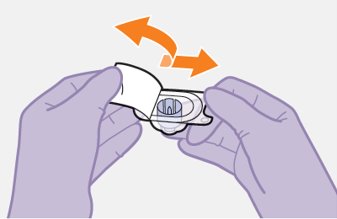
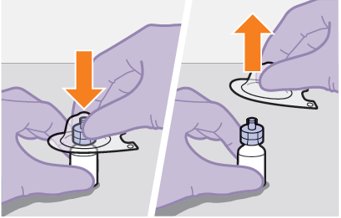
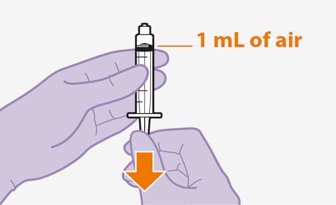
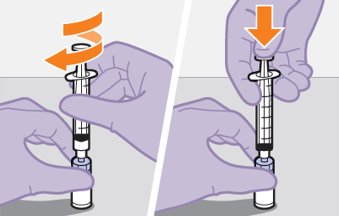
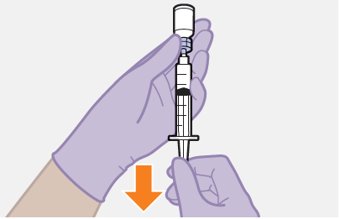
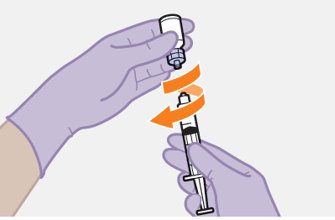
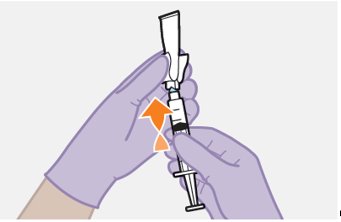
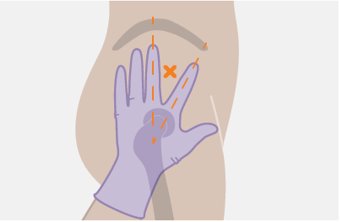
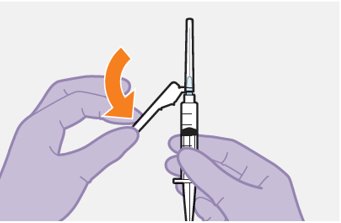
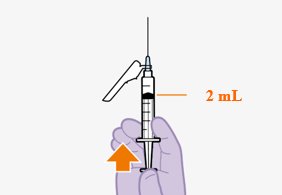
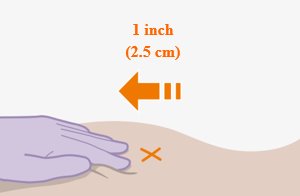
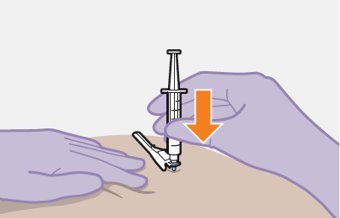
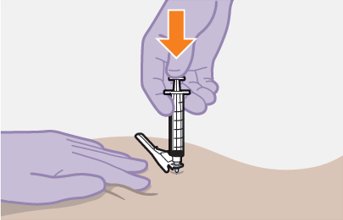
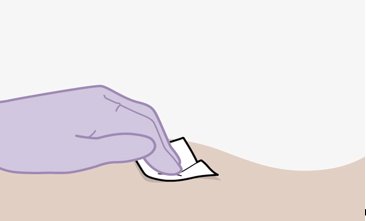
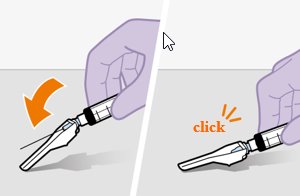
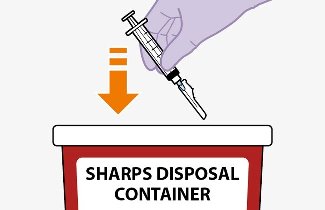


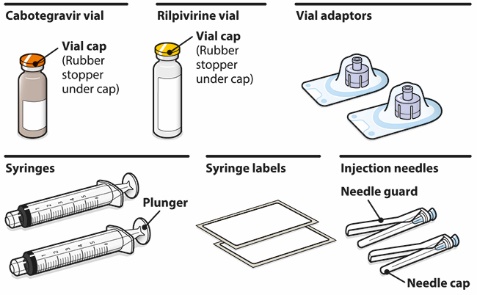
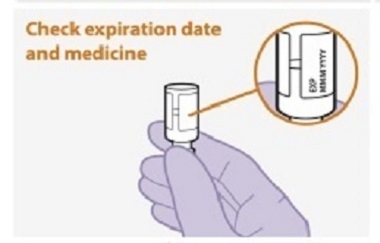
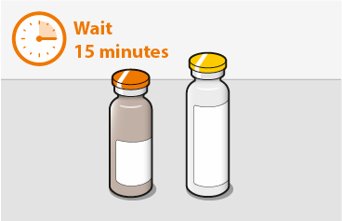
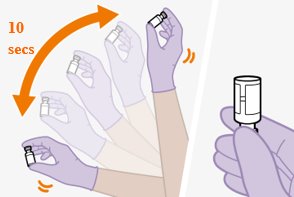
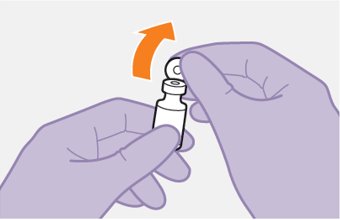
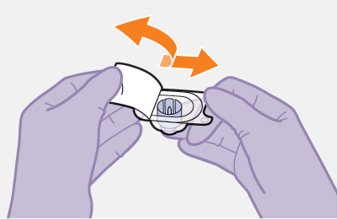
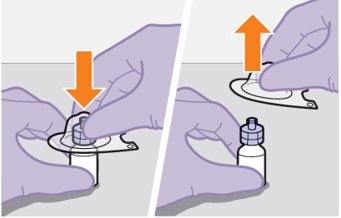
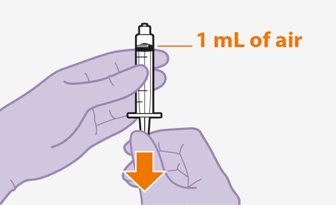
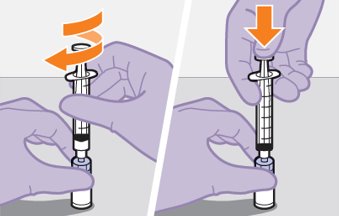
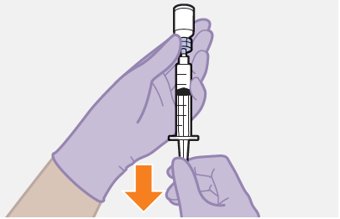
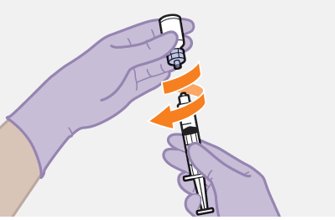
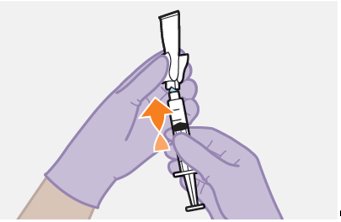
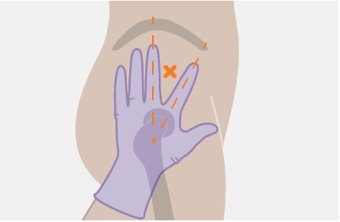
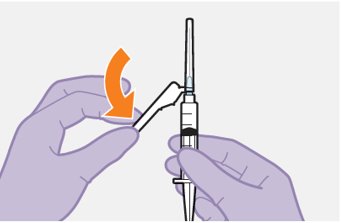
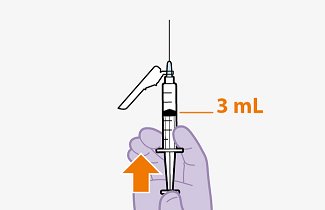
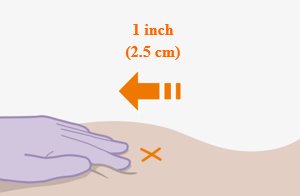
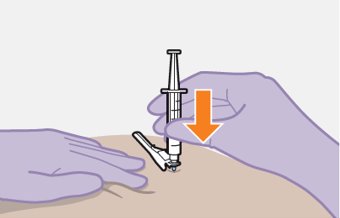
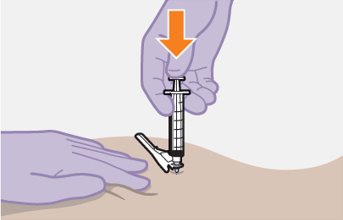
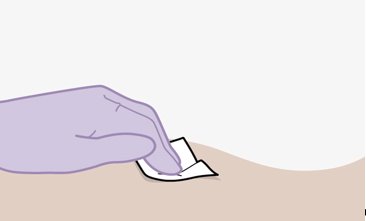
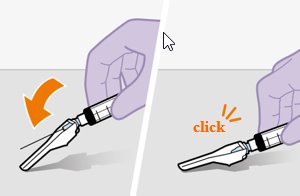
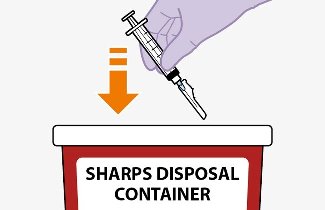



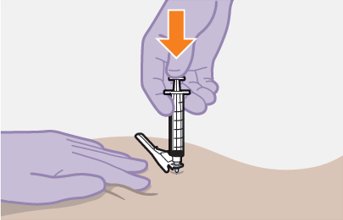
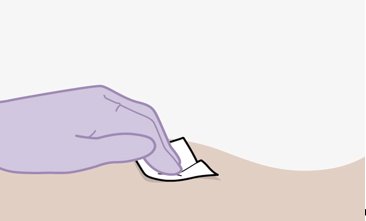
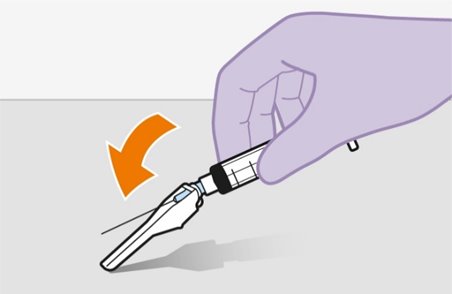

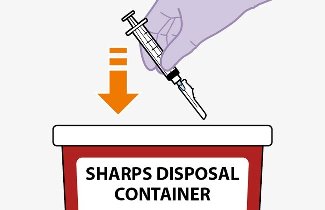




Trademark Results [Cabenuva]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CABENUVA 87420884 5335232 Live/Registered |
ViiV Healthcare UK (No.3) Limited 2017-04-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.