COMETRIQ- cabozantinib kit COMETRIQ- cabozantinib capsule
COMETRIQ by
Drug Labeling and Warnings
COMETRIQ by is a Prescription medication manufactured, distributed, or labeled by Exelixis, Inc., Catalent CTS, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use COMETRIQ safely and effectively. See full prescribing information for COMETRIQ.
COMETRIQ® (cabozantinib) capsules, for oral use
Initial U.S. Approval: 2012RECENT MAJOR CHANGES
INDICATIONS AND USAGE
COMETRIQ is a kinase inhibitor indicated for the treatment of patients with progressive, metastatic medullary thyroid cancer (MTC). (1)
DOSAGE AND ADMINISTRATION
- Recommended Dose: 140 mg orally, once daily. (2.1)
- Instruct patients not to eat for at least 2 hours before and at least 1 hour after taking COMETRIQ. (2.1)
- Do NOT substitute COMETRIQ capsules with cabozantinib tablets. (2.1)
- Hepatic Impairment: The recommended starting dose of COMETRIQ is 80 mg in patients with mild or moderate hepatic impairment. (2.1)
DOSAGE FORMS AND STRENGTHS
Capsules: 20 mg and 80 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Perforations and Fistulas: Monitor for symptoms. Discontinue COMETRIQ for Grade 4 fistula or perforation. (5.1)
- Hemorrhage: Do not administer COMETRIQ if recent history of hemorrhage. (5.2)
- Thrombotic Events: Discontinue COMETRIQ for myocardial infarction or serious arterial or venous thromboembolic events. (5.3)
- Impaired Wound Healing: Withhold COMETRIQ for at least 3 weeks before elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of COMETRIQ after resolution of wound healing complications has not been established. (5.4)
- Hypertension and hypertensive crisis: Monitor blood pressure regularly. Interrupt for hypertension that is not adequately controlled with anti- hypertensive therapy. Discontinue COMETRIQ for hypertensive crisis or severe hypertension that cannot be controlled with anti-hypertensive therapy. (5.5)
- Osteonecrosis of the Jaw (ONJ): Withhold COMETRIQ for at least 3 weeks prior to invasive dental procedure and for development of ONJ. (5.6).
- Diarrhea: May be severe. Interrupt COMETRIQ immediately until diarrhea resolves or decreases to Grade 1. Recommend standard antidiarrheal treatments. (5.7)
- Palmar-Plantar Erythrodysesthesia (PPE): Interrupt COMETRIQ until PPE resolves or decreases to Grade 1. (5.8)
- Proteinuria: Monitor urine protein. Discontinue for nephrotic syndrome. (5.9)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Discontinue COMETRIQ. (5.10)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.11, 8.1, 8.3)
ADVERSE REACTIONS
- The most common adverse reactions (≥ 25%) are diarrhea, stomatitis, palmar-plantar erythrodysesthesia (PPE), decreased weight, decreased appetite, nausea, fatigue, oral pain, hair color changes, dysgeusia, hypertension, abdominal pain, and constipation.
- The most common laboratory abnormalities (≥ 25%) are increased AST, increased ALT, lymphopenia, increased alkaline phosphatase, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Exelixis, Inc. at 1-855-500-3935 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
- Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications for Adverse Reactions
2.3 Dosage Modifications For Coadministration With Strong CYP3A4 Inhibitors
2.4 Dosage Modifications For Coadministration With Strong CYP3A4 Inducers
2.5 Dosage Modifications for Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Perforations and Fistulas
5.2 Hemorrhage
5.3 Thrombotic Events
5.4 Impaired Wound Healing
5.5 Hypertension and Hypertensive Crisis
5.6 Osteonecrosis of the Jaw
5.7 Diarrhea
5.8 Palmar-Plantar Erythrodysesthesia
5.9 Proteinuria
5.10 Reversible Posterior Leukoencephalopathy Syndrome
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of CYP3A4 Inhibitors
7.2 Effect of CYP3A4 Inducers
7.3 Effect of MRP2 Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Do NOT substitute COMETRIQ capsules with cabozantinib tablets.
The recommended daily dose of COMETRIQ is 140 mg once daily without food until disease progression or unacceptable toxicity. Instruct patients not to eat for at least 2 hours before and at least 1 hour after taking COMETRIQ.
Swallow COMETRIQ capsules whole. Do not open COMETRIQ capsules.
Do not take a missed dose within 12 hours of the next dose.
Do not ingest foods (e.g., grapefruit, grapefruit juice) or nutritional supplements that are known to inhibit cytochrome P450 while taking COMETRIQ.
2.2 Dosage Modifications for Adverse Reactions
Withhold COMETRIQ for NCI CTCAE Grade 4 hematologic adverse reactions, Grade 3 or greater non-hematologic adverse reactions, intolerable Grade 2 adverse reactions, or osteonecrosis of the jaw.
Upon resolution/improvement of the adverse reaction (i.e., return to baseline or resolution to Grade 1), reduce the dose as follows:
- If previously receiving 140 mg daily dose, resume treatment at 100 mg daily
- If previously receiving 100 mg daily dose, resume treatment at 60 mg daily
- If previously receiving 60 mg daily dose, resume at 60 mg if tolerated, otherwise, discontinue COMETRIQ
Permanently discontinue COMETRIQ for any of the following:
- development of gastrointestinal (GI) perforation or Grade 4 fistula
- severe hemorrhage
- acute myocardial infarction or arterial or venous thromboembolic events that require medical intervention
- nephrotic syndrome
- severe hypertension that cannot be controlled with anti-hypertensive therapy or hypertensive crisis
- reversible posterior leukoencephalopathy syndrome
2.3 Dosage Modifications For Coadministration With Strong CYP3A4 Inhibitors
Reduce the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 100 mg daily or from 100 mg to 60 mg daily). Resume the dose that was used prior to initiating the CYP3A4 inhibitor 2 to 3 days after discontinuation of the strong inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.4 Dosage Modifications For Coadministration With Strong CYP3A4 Inducers
Increase the daily COMETRIQ dose by 40 mg (for example, from 140 mg to 180 mg daily or from 100 mg to 140 mg daily) as tolerated. Resume the dose that was used prior to initiating the CYP3A4 inducer 2 to 3 days after discontinuation of the strong inducer. The daily dose of COMETRIQ should not exceed 180 mg [see Drug Interactions (7.2), Clinical Pharmacology (12.3)].
2.5 Dosage Modifications for Patients with Hepatic Impairment
The recommended starting dose of COMETRIQ for patients with mild to moderate hepatic impairment is 80 mg [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Perforations and Fistulas
Gastrointestinal (GI) perforations and fistulas, including fatal cases, were reported in 3% and 1% of COMETRIQ-treated patients (N=214), respectively. Non-GI fistulas including tracheal/esophageal, including fatal cases, were reported in 4% of COMETRIQ-treated patients.
Monitor patients for symptoms of perforations and fistulas, including abscess and sepsis. Discontinue COMETRIQ in patients who experience a Grade 4 fistula or a GI perforation [see Dosage and Administration (2.2)].
5.2 Hemorrhage
Severe and fatal hemorrhage occurred with COMETRIQ. The incidence of Grade ≥ 3 hemorrhagic events was higher in COMETRIQ-treated patients compared with placebo (3% vs. 1%).
Discontinue COMETRIQ for Grade 3 or 4 hemorrhage [see Dosage and Administration (2.2)]. Do not administer COMETRIQ to patients with a recent history of hemorrhage, including hemoptysis, hematemesis, or melena.
5.3 Thrombotic Events
COMETRIQ increased the incidence of thrombotic events (venous thromboembolism: 6% vs. 3% and arterial thromboembolism: 2% vs. 0% in COMETRIQ-treated and placebo-treated patients, respectively).
Discontinue COMETRIQ in patients who develop an acute myocardial infarction or arterial or venous thromboembolic events that require medical intervention [see Dosage and Administration (2.2)].
5.4 Impaired Wound Healing
Wound complications have been reported with COMETRIQ. Withhold COMETRIQ for at least 3 weeks prior to elective surgery. Do not administer COMETRIQ for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of COMETRIQ after resolution of wound healing complications has not been established.
5.5 Hypertension and Hypertensive Crisis
COMETRIQ can an increase the incidence of treatment-emergent hypertension with Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (modified JNC criteria) stage 1 or 2 hypertension identified in 61% in COMETRIQ-treated patients compared with 30% of placebo-treated patients in the randomized trial [see Adverse Reactions (6.1)].
Do not initiate COMETRIQ in patients with uncontrolled hypertension. Monitor blood pressure regularly during COMETRIQ treatment. Withhold COMETRIQ for hypertension that is not adequately controlled with medical management; when controlled, resume COMETRIQ at a reduced dose. Discontinue COMETRIQ for severe hypertension that cannot be controlled with anti-hypertensive therapy and for hypertensive crisis [see Dosage and Administration (2.2)].
5.6 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ) occurred in 1% of COMETRIQ-treated patients. ONJ can manifest as jaw pain, osteomyelitis, osteitis, bone erosion, tooth or periodontal infection, toothache, gingival ulceration or erosion, persistent jaw pain or slow healing of the mouth or jaw after dental surgery. Perform an oral examination prior to initiation of COMETRIQ and periodically during COMETRIQ therapy. Advise patients regarding good oral hygiene practices. Withhold COMETRIQ treatment for at least 3 weeks prior to scheduled dental surgery, or invasive dental procedures, if possible. Withhold COMETRIQ for development of ONJ until complete resolution [see Dosage and Administration (2.2)].
5.7 Diarrhea
Diarrhea occurred in 63% of patients treated with COMETRIQ. Grade 3-4 diarrhea occurred in 16% of patients treated with COMETRIQ [see Adverse Reactions (6.1)].
Withhold COMETRIQ until improvement to Grade 1 and resume COMETRIQ at a reduced dose for intolerable Grade 2 diarrhea, Grade 3 diarrhea that cannot be managed with standard antidiarrheal treatments, or Grade 4 diarrhea.
5.8 Palmar-Plantar Erythrodysesthesia
Palmar-plantar erythrodysesthesia (PPE) occurred in 50% of patients treated with COMETRIQ, including 13% Grade 3 [see Adverse Reactions (6.1)].
Withhold COMETRIQ until improvement to Grade 1 and resume COMETRIQ at a reduced dose for intolerable Grade 2 PPE or Grade 3 PPE.
5.9 Proteinuria
Proteinuria was observed in 2% of patients receiving COMETRIQ, including one with nephrotic syndrome. Monitor urine protein regularly during COMETRIQ treatment. Discontinue COMETRIQ in patients who develop nephrotic syndrome.
5.10 Reversible Posterior Leukoencephalopathy Syndrome
Reversible Posterior Leukoencephalopathy Syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI, occurred in one (<1%) patient. Perform an evaluation for RPLS in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. Discontinue COMETRIQ in patients who develop RPLS [see Dosage and Administration (2.2)].
5.11 Embryo-Fetal Toxicity
Based on data from animal studies and its mechanism of action, COMETRIQ can cause fetal harm when administered to a pregnant woman. Cabozantinib administration to pregnant animals during organogenesis resulted in embryolethality at exposures below those occurring clinically at the recommended dose, and in increased incidences of skeletal variations in rats and visceral variations and malformations in rabbits. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with COMETRIQ and for 4 months after the last dose [see Use in Specific Populations (8.1), (8.3), and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- Perforations and Fistula [see Warnings and Precautions (5.1)]
- Hemorrhage [see Warnings and Precautions (5.2)]
- Thromboembolic Events [see Warnings and Precautions (5.3)]
- Impaired Wound Healing [see Warnings and Precautions (5.4)]
- Hypertension and Hypertensive Crisis [see Warnings and Precautions (5.5)]
- Osteonecrosis of the Jaw [see Warnings and Precautions (5.6)]
- Diarrhea [see Warnings and Precautions (5.7)]
- Palmar-Plantar Erythrodysesthesia [see Warnings and Precautions (5.8)]
- Proteinuria [see Warnings and Precautions (5.9)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of COMETRIQ was evaluated in 330 patients with progressive metastatic medullary thyroid cancer randomized to receive 140 mg COMETRIQ (n = 214) or placebo (n = 109) administered daily until disease progression or intolerable toxicity in a randomized, doubleblind, controlled trial (Study 1) [see Clinical Studies (14)]. The data described below reflect a median exposure to COMETRIQ for 204 days. The population exposed to COMETRIQ was 70% male, 90% white, and had a median age of 55 years.
Fatal adverse reactions occurred in 6% of patients receiving COMETRIQ and resulted from hemorrhage, pneumonia, septicemia, fistulas, cardiac arrest, respiratory failure, and unspecified death. Fatal adverse reactions occurred in 5% of patients receiving placebo and resulted from septicemia, pneumonia, and general deterioration.
The COMETRIQ dose was reduced in 79% of patients receiving COMETRIQ and in 9% of patients receiving placebo. The median number of dosing delays was one in patients receiving COMETRIQ and in no patients receiving placebo. Adverse reactions led to study treatment discontinuation in 16% of patients receiving COMETRIQ. The most frequent adverse reactions leading to permanent discontinuation of COMETRIQ were: hypocalcemia, increased lipase, PPE, diarrhea, fatigue, hypertension, nausea, pancreatitis, tracheal fistula formation and vomiting.
Increased levels of thyroid stimulating hormone (TSH) were observed in 57% of patients receiving COMETRIQ after the first dose compared to 19% of patients receiving placebo (regardless of baseline value). Ninety-two percent (92%) of patients on the COMETRIQ arm had a prior thyroidectomy, and 89% were taking thyroid hormone replacement prior to the first dose.
Adverse reactions which occurred in ≥ 25% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥ 5% included, in order of decreasing frequency: diarrhea, stomatitis, palmar-plantar erythrodysesthesia (PPE), decreased weight, decreased appetite, nausea, fatigue, oral pain, hair color changes, dysgeusia, hypertension, abdominal pain, and constipation. The most common laboratory abnormalities (≥25%) were increased AST, increased ALT, lymphopenia, increased ALP, hypocalcemia, neutropenia, thrombocytopenia, hypophosphatemia, and hyperbilirubinemia. Grade 3-4 adverse reactions and laboratory abnormalities which occurred in ≥ 5% of COMETRIQ-treated patients occurring more frequently in the COMETRIQ arm with a between-arm difference of ≥ 2% included, in order of decreasing frequency; diarrhea, PPES, lymphopenia, hypocalcemia, fatigue, hypertension, asthenia, increased ALT, decreased weight, stomatitis, and decreased appetite (Table 1 and Table 2 summarize the adverse reactions and laboratory abnormalities reported in Study 1).
Table 1. Selected Adverse Reactions Occurring at a Higher Incidence in COMETRIQ-Treated Patients (Study 1) [Between Arm Difference of ≥ 5% (All Grades)* or ≥ 2% (Grades 3-4)] System Organ Class/Preferred Terms COMETRIQ
(n=214)Placebo
(n=109)All
Grades
(%)Grades
3-4
(%)All
Grades
(%)Grades
3-4
(%)- * National Cancer Institute Common Terminology Criteria for Adverse Events Version 3.0
- † Includes the following terms: stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation
- ‡ Includes the following terms: oral pain, oropharyngeal pain, glossitis, burning mouth syndrome, glossodynia
- § Includes the following terms: abdominal pain, abdominal pain lower, abdominal pain upper, abdominal rigidity, abdominal tenderness, esophageal pain
- ¶ Palmar-plantar erythrodysesthesia
GASTROINTESTINAL DISORDERS Diarrhea 63 16 33 2 Stomatitis† 51 5 6 0 Nausea 43 1 21 0 Oral pain‡ 36 2 6 0 Constipation 27 0 6 0 Abdominal pain§ 27 3 13 1 Vomiting 24 2 2 1 Dysphagia 13 4 6 1 Dyspepsia 11 0 0 0 Hemorrhoids 9 0 3 0 SKIN AND SUBCUTANEOUS TISSUE DISORDERS PPE¶ 50 13 2 0 Hair color changes/ depigmentation, graying 34 0 1 0 Rash 19 1 10 0 Dry skin 19 0 3 0 Alopecia 16 0 2 0 Erythema 11 1 2 0 Hyperkeratosis 7 0 0 0 INVESTIGATIONS Decreased weight 48 5 10 0 METABOLISM AND NUTRITION DISORDERS Decreased appetite 46 5 16 1 Dehydration 7 2 2 1 GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS Fatigue 41 9 28 3 Asthenia 21 6 15 1 NERVOUS SYSTEM DISORDERS Dysgeusia 34 0 6 0 Headache 18 0 8 0 Dizziness 14 0 7 0 Paresthesia 7 0 2 0 Peripheral sensory neuropathy 7 0 0 0 Peripheral neuropathy 5 0 0 0 VASCULAR DISORDERS Hypertension 33 8 4 0 Hypotension 7 1 0 0 RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS Dysphonia 20 0 9 0 MUSCULOSKELETAL AND CONNECTIVE
TISSUE DISORDERSArthralgia 14 1 7 0 Muscle spasms 12 0 5 0 Musculoskeletal chest pain 9 1 4 0 PSYCHIATRIC DISORDERS Anxiety 9 0 2 0 Table 2 Laboratory Abnormalities Occurring at a Higher Incidence in COMETRIQ-Treated Patients (Study 1) [Between Arm Difference of ≥ 5% (All Grades) or ≥ 2% (Grades 3-4)] Test COMETRIQ
(n=214)Placebo
(N=109)All Grades
(%)Grade 3-4
(%)All Grades
(%)Grade 3-4
(%)Chemistries Increased AST 86 3 35 2 Increased ALT 86 6 41 2 Increased ALP 52 3 35 3 Hypocalcemia 52 12 27 3 Hypophosphatemia 28 3 10 1 Hyperbilirubinemia 25 2 14 5 Hypomagnesemia 19 1 4 0 Hypokalemia 18 4 9 3 Hyponatremia 10 2 5 0 Hematologic Lymphopenia 53 16 51 11 Neutropenia 35 3 15 2 Thrombocytopenia 35 0 4 3 ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase Nearly all COMETRIQ-treated patients (96% vs. 84% placebo) experienced elevated blood pressure and there was a doubling in the incidence of overt hypertension in COMETRIQ-treated patients over placebo-treated patients (61% vs. 30%) according to modified Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC) staging criteria. No patients developed malignant hypertension.
Table 3 Per-Patient Incidence of Hypertension (Study 1) - * Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, JAMA 2003: 289:2560. Criteria applied were modified, as multiple readings were not available per timepoint, and therefore not averaged.
- † Patients classified by highest category based on all recorded blood pressure readings beginning after the first dose through 30 days after last dose.
- ‡ Patients with at least two blood pressure measurements after the first dose
Hypertension, JNC* Stage† COMETRIQ
N=211‡(%)Placebo
N=107‡(%)Normal: Grade 0: Systolic < 120 mmHg and Diastolic < 80 mmHg 4 15 Pre-hypertension: Systolic ≥ 120 mmHg or Diastolic ≥ 80 mmHg 34 54 Stage 1: Systolic ≥ 140 mmHg or Diastolic ≥ 90 mmHg 46 25 Stage 2: Systolic ≥ 160 mmHg or Diastolic ≥ 100 mmHg 15 5 Malignant: Diastolic ≥ 120 mmHg 0 0 Other clinically important adverse reactions (all grades) that were reported in clinical trials include: hepatitis cholestatic (<1%).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of COMETRIQ. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hematology: A case of supratherapeutic international normalized ratio (INR) and epistaxis during concomitant use of warfarin
-
7 DRUG INTERACTIONS
7.1 Effect of CYP3A4 Inhibitors
Administration of a strong CYP3A4 inhibitor, ketoconazole to healthy subjects increased single-dose plasma cabozantinib exposure by 38%. Avoid taking a strong CYP3A4 inhibitor (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole) while taking COMETRIQ or reduce the dosage of COMETRIQ if concomitant use with strong CYP3A4 inhibitors cannot be avoided [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
Avoid ingestion of foods (e.g., grapefruit, grapefruit juice) or nutritional supplements that are known to inhibit cytochrome P450 while taking COMETRIQ.
7.2 Effect of CYP3A4 Inducers
Administration of a strong CYP3A4 inducer, rifampin to healthy subjects decreased single-dose plasma cabozantinib exposure by 77%. Avoid chronic co-administration of strong CYP3A4 inducers (e.g., phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital, St. John’s Wort) with COMETRIQ or increase the dosage of COMETRIQ if concomitant use with strong CYP3A4 inducers cannot be avoided [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
7.3 Effect of MRP2 Inhibitors
Concomitant administration of MRP2 inhibitors may increase the exposure to cabozantinib. Monitor patients for increased toxicity when MRP2 inhibitors (e.g., abacavir, adefovir, cidofovir, furosemide, lamivudine, nevirapine, ritonavir, probenecid, saquinavir, and tenofovir) are co-administered with COMETRIQ [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, COMETRIQ can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal developmental and reproductive toxicology studies administration of cabozantinib to pregnant rats and rabbits during organogenesis resulted in embryofetal lethality and structural anomalies at exposures that were below those occurring clinically at the recommended dose (see Data). Advise pregnant women or women of childbearing potential of the potential hazard to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study in pregnant rats, daily oral administration of cabozantinib throughout organogenesis caused increased embryo-fetal lethality compared to controls at a dose of 0.03 mg/kg (less than 1% of the human exposure by AUC at the 140 mg dose). Findings included delayed ossifications and skeletal variations at a dose of 0.01 mg/kg/day (approximately 0.03% of the human exposure by AUC at the 140 mg dose).
In pregnant rabbits, daily oral administration of cabozantinib throughout organogenesis resulted in findings of visceral malformations and variations including reduced spleen size and missing lung lobe at 3 mg/kg (approximately 11% of the human exposure by AUC at the 140 mg dose).
In a pre- and postnatal study in rats, cabozantinib was administered orally from gestation day 10 through postnatal day 20. Cabozantinib did not produce adverse maternal toxicity or affect pregnancy, parturition or lactation of female rats, and did not affect the survival, growth or postnatal development of the offspring at doses up to 0.3 mg/kg/day (approximately 0.02 times the recommended clinical dose of 140 mg based on body surface area).
8.2 Lactation
Risk Summary
There is no information regarding the presence of cabozantinib or its metabolites in human milk, or their effects on the breastfed infant, or milk production. Because of the potential for serious adverse reactions in a breastfed infant from COMETRIQ, advise a lactating woman not to breastfeed during treatment with COMETRIQ and for 4 months after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating COMETRIQ [see Use in Specific Populations (8.1)].
Contraception
COMETRIQ can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with COMETRIQ and for 4 months after the final dose.
Infertility
Females and Males
Based on findings in animals, COMETRIQ may impair fertility in females and males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of COMETRIQ in pediatric patients have not been studied.
Juvenile Animal Toxicity Data
Juvenile rats were administered cabozantinib daily at doses of 1 or 2 mg/kg/day from Postnatal Day 12 (comparable to less than 2 years in humans) through Postnatal Day 35 or 70. Mortalities occurred at doses equal and greater than 1 mg/kg/day (approximately 0.07 times the clinical dose of 140 mg/day based on body surface area). Hypoactivity was observed at both doses tested on Postnatal Day 22. Targets were generally similar to those seen in adult animals, occurred at both doses, and included the kidney (nephropathy, glomerulonephritis), reproductive organs, gastrointestinal tract (cystic dilatation and hyperplasia in Brunner’s gland and inflammation of duodenum; and epithelial hyperplasia of colon and cecum), bone marrow (hypocellularity and lymphoid depletion), and liver. Tooth abnormalities and whitening as well as effects on bones including reduced bone mineral content and density, physeal hypertrophy, and decreased cortical bone also occurred at all dose levels. Recovery was not assessed at the 2 mg/kg dose level (approximately 0.14 times the clinical dose of 140 mg based on body surface area) due to high levels of mortality. At the low dose level, effects on bone parameters were partially resolved but effects on the kidney and epididymis/testis persisted after treatment ceased.
8.5 Geriatric Use
Clinical studies of COMETRIQ did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.
8.6 Hepatic Impairment
Increased exposure to cabozantinib has been observed in patients with mild to moderate hepatic impairment. Reduce the starting dose of COMETRIQ in patients with mild (Child-Pugh score (C-P) A) or moderate (C-P B) hepatic impairment. COMETRIQ is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Dosage adjustment is not required in patients with mild or moderate renal impairment. There is no experience with COMETRIQ in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
One case of overdosage was reported in a patient who inadvertently took twice the intended dose (200 mg daily) for nine days. The patient suffered Grade 3 memory impairment, Grade 3 mental status changes, Grade 3 cognitive disturbance, Grade 2 weight loss, and Grade 1 increase in BUN. The extent of recovery was not documented.
-
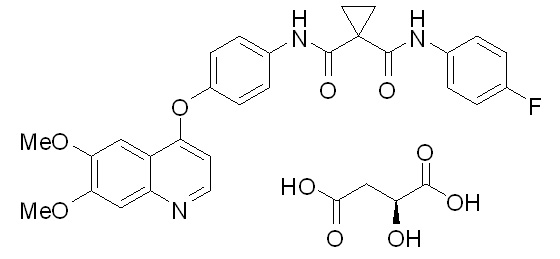
11 DESCRIPTION
COMETRIQ is the (S)-malate salt of cabozantinib, a kinase inhibitor. Cabozantinib (S)-malate is described chemically as N-(4-(6,7-dimethoxyquinolin-4-yloxy)phenyl)-N'-(4-fluorophenyl)cyclopropane- 1,1-dicarboxamide, (2S)-hydroxybutanedioate. The molecular formula is C28H24FN3O5C4H6O5 and the molecular weight is 635.6 Daltons as malate salt. The chemical structure of cabozantinib (S)-malate salt is:
Cabozantinib (S)-malate salt is a white to off-white solid that is practically insoluble in aqueous media.
COMETRIQ (cabozantinib) capsules for oral use are supplied as printed hard gelatin capsules containing cabozantinib (S)-malate equivalent to 20 mg or 80 mg cabozantinib and the following inactive ingredients: silicified microcrystalline cellulose, croscarmellose sodium, sodium starch glycolate, fumed silica, and stearic acid.
The grey gelatin capsule shells contain black iron oxide and titanium dioxide and the Swedish orange gelatin capsule shells contain red iron oxide, and titanium dioxide. The printing ink contains shellac glaze, black iron oxide, N-butyl alcohol, isopropyl alcohol, propylene glycol, and ammonium hydroxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
In vitro biochemical and/or cellular assays have shown that cabozantinib inhibits the tyrosine kinase activity of RET, MET, VEGFR-1, -2 and -3, KIT, TRKB, FLT-3, AXL, ROS1, TYRO3, MER, and TIE-2. These receptor tyrosine kinases are involved in both normal cellular function and pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, drug resistance, and maintenance of the tumor microenvironment.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of orally administered COMETRIQ 140 mg on QTc interval was evaluated in a randomized, double-blinded, placebo-controlled study in patients with MTC. A mean increase in QTcF of 10 - 15 ms was observed at 4 weeks after initiating COMETRIQ. A concentration-QTc relationship could not be definitively established. Changes in cardiac wave form morphology or new rhythms were not observed. No COMETRIQ-treated patients had a QTcF > 500 ms [see Clinical Studies (14)].
12.3 Pharmacokinetics
A population pharmacokinetic analysis of cabozantinib was performed using data collected from 289 patients with solid tumors including MTC following oral administration of 140 mg daily doses. Repeat daily dosing of COMETRIQ at 140 mg for 19 days resulted in 4- to 5-fold mean cabozantinib accumulation (based on AUC) compared to a single dose administration; steady state was achieved by Day 15.
Absorption
Following oral administration of COMETRIQ, median time to peak cabozantinib plasma concentrations (Tmax) ranged from 2 to 5 hours post-dose.
A 19% increase in the Cmax of the tablet formulation (CABOMETYX™) compared to the capsule formulation (COMETRIQ) was observed following a single 140 mg dose. A less than 10% difference in the AUC was observed between cabozantinib tablet (CABOMETYX) and capsule (COMETRIQ) formulations [see Dosage and Administration (2.1)].
Cabozantinib Cmax and AUC values increased by 41% and 57%, respectively, following a high-fat meal relative to fasted conditions in healthy subjects administered a single 140 mg oral COMETRIQ dose.
Distribution
The oral volume of distribution (V/F) of cabozantinib is approximately 349 L. Cabozantinib is highly protein bound in human plasma (≥ 99.7%).
Elimination
The predicted effective half-life is approximately 55 hours and the clearance (CL/F) at steady-state is estimated to be 4.4 L/hr.
Metabolism
Cabozantinib is a substrate of CYP3A4 in vitro.
Excretion
Approximately 81% of the total administered radioactivity was recovered within a 48-day collection period following a single 140 mg dose of an investigational 14C-cabozantinib formulation in healthy subjects. Approximately 54% was recovered in feces and 27% in urine. Unchanged cabozantinib accounted for 43% of the total radioactivity in feces and was not detectable in urine following a 72 hour collection.
Specific Populations
The following patient characteristics did not result in a clinically relevant difference in the pharmacokinetics of cabozantinib: age (20-86 years), sex, race (Whites and non-Whites), or mild to moderate renal impairment (eGFR greater than or equal to 30 mL/min/1.73 m2 as estimated by MDRD (modification of diet in renal disease equation)). The pharmacokinetics of cabozantinib is unknown in patients with worse than moderate renal impairment (eGFR less than 29 mL/min/1.73m2) as estimated by MDRD equation or renal impairment requiring dialysis.
Patients with Hepatic Impairment
Following a single oral 60 mg COMETRIQ, mean AUC0-inf for cabozantinib increased by 81% in subjects with mild (Child-Pugh A) hepatic impairment and 63% in subjects with moderate (Child-Pugh B) hepatic impairment compared to subjects with normal hepatic function [see Dosage and Administration (2.5), Use in Specific Populations (8.6)].
The pharmacokinetics of cabozantinib has not been studied in patients with severe (Child-Pugh C) hepatic impairment [see Use in Specific Populations (8.6)].
Drug Interaction Studies
CYP3A4 Inhibition on Cabozantinib
Administration of a strong CYP3A4 inhibitor, ketoconazole (400 mg daily for 27 days) to healthy subjects increased single-dose plasma cabozantinib exposure (AUC0-inf) by 38%.
CYP3A4 Induction on Cabozantinib
Administration of a strong CYP3A4 inducer, rifampin (600 mg daily for 31 days) to healthy subjects decreased single-dose plasma cabozantinib exposure (AUC0-inf) by 77%.
Cabozantinib on CYP2C8 substrates
No clinically-significant effect on single-dose rosiglitazone (a CYP2C8 substrate) plasma exposure (Cmax and AUC) was observed when co-administered with cabozantinib at steady-state plasma concentrations (≥ 100 mg/day daily for a minimum of 21 days) in patients with solid tumors.
Gastric pH modifying agents on Cabozantinib
No clinically-significant effect on plasma cabozantinib exposure (AUC) was observed following co-administration of the proton pump inhibitor (PPI) esomeprazole (40 mg daily for 6 days) with a single dose of 100 mg cabozantinib to healthy volunteers.
In vitro Studies
Metabolic Pathways:
Inhibition of CYP3A4 reduced the formation of the XL184 N-oxide metabolite by >80%. Inhibition of CYP2C9 had a minimal effect on cabozantinib metabolite formation (i.e., a <20% reduction). Inhibition of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 and CYP2E1 had no effect on cabozantinib metabolite formation.
Although cabozantinib is an inhibitor of CYP2C8 in vitro, a clinical study of this potential interaction concluded that concurrent use did not result in a clinically relevant effect on CYP2C8 substrate exposure. Given this finding, other less sensitive substrates of pathways affected by cabozantinib in vitro (i.e., CYP2C9, CYP2C19, and CYP3A4) were not evaluated in a clinical study because, although a clinically relevant exposure effect cannot be ruled out, it is unlikely. Cabozantinib does not inhibit CYP1A2 and CYP2D6 isozymes in vitro.
Cabozantinib is an inducer of CYP1A1 mRNA; however, the clinical relevance of this finding is unknown. Cabozantinib does not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP3A4.
Drug Transporter Systems:
Cabozantinib is an inhibitor, but not a substrate, of P-gp transport activities and has the potential to increase plasma concentrations of co-administered substrates of P-gp. The clinical relevance of this finding is unknown.
Cabozantinib is a substrate of MRP2 in vitro and MRP2 inhibitors have the potential to increase plasma concentrations of cabozantinib. The clinical relevance of this finding is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of cabozantinib has been evaluated in two species: rasH2 transgenic mice and Sprague-Dawley rats. In the 2-year rat carcinogenicity study, once daily oral administration of cabozantinib resulted in a statistically significant increase in the incidence of malignant/complex malignant pheochromocytoma in combination with benign pheochromocytoma or in benign pheochromocytoma alone in male rats at a dose of 1 mg/kg (approximately 0.6 times the human exposure by AUC at the recommended 140 mg dose). Cabozantinib was not carcinogenic in a 26-week carcinogenicity study in rasH2 transgenic mice at a slightly higher exposure than the intended human therapeutic exposure.
Cabozantinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay and was not clastogenic in both the in vitro cytogenetic assay using human lymphocytes or in the in vivo mouse micronucleus assay.
Based on nonclinical findings, male and female fertility may be impaired by treatment with COMETRIQ. In a fertility study in which cabozantinib was administered to male and female rats at doses of 1, 2.5, and 5 mg/kg/day, male fertility was significantly compromised at doses equal to or greater than 2.5 mg/kg/day (approximately equal to the human exposure by AUC at the recommended dose), with a decrease in sperm counts and reproductive organ weights. In females, fertility was significantly reduced at doses equal to or greater than 1 mg/kg/day (approximately 50% of the human exposure by AUC at the 140 mg recommended dose) with a significant decrease in the number of live embryos and a significant increase in pre- and post-implantation losses.
Observations of effects on reproductive tract tissues in general toxicology studies were supportive of effects noted in the dedicated fertility study and included hypospermia and absence of corpora lutea in male and female dogs in a 6-month repeat dose study at exposures equal to 6% and 3%, respectively, the human exposure by AUC at the recommended dose. In addition, female rats administered 5 mg/kg/day for 14 days (approximately equal to the human exposure by AUC at the 140 mg recommended dose) exhibited ovarian necrosis.
-
14 CLINICAL STUDIES
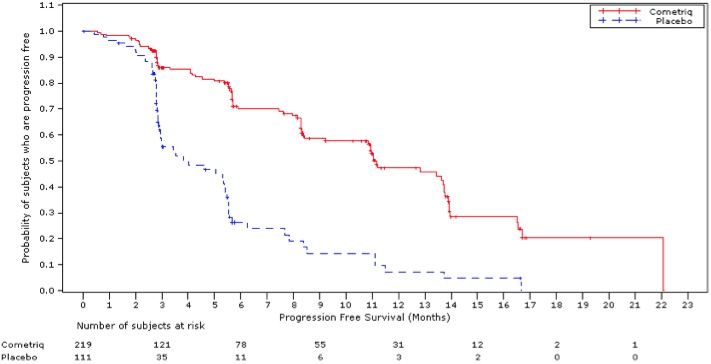
The safety and efficacy of COMETRIQ was assessed in an international, multi-center, randomized, double-blind, controlled trial (Study 1) of 330 patients with metastatic medullary thyroid carcinoma (MTC). Patients were required to have evidence of actively progressive disease within 14 months prior to study entry confirmed by an Independent Radiology Review Committee (IRRC) masked to treatment assignment (89%) or the treating physician (11%). Patients were randomized (2:1) to receive COMETRIQ 140 mg (n = 219) or placebo (n = 111) orally once daily, without food, until disease progression determined by the treating physician or until intolerable toxicity. Randomization was stratified by age (≤ 65 years vs. > 65 years) and prior use of a tyrosine kinase inhibitor (TKI) (yes vs. no). No cross-over was allowed at the time of progression. The main efficacy outcome measures of progression-free survival (PFS), objective response (OR), and response duration were based on IRRC-confirmed events using modified RECIST criteria.
Of 330 patients randomized, 67% were male, the median age was 55 years, 23% were 65 years or older, 89% were white, 54% had a baseline ECOG performance status of 0, and 92% had undergone a thyroidectomy. The RET mutation status determined by a research-use assay was positive in 51%, negative in 14%, and was unknown in 35%. Twenty-five percent (25%) had two or more prior systemic therapies and 21% had been previously treated with a TKI.
A statistically significant prolongation in PFS was demonstrated among COMETRIQ-treated patients compared to those receiving placebo [HR 0.28 (95% CI: 0.19, 0.40); p<0.0001], with median PFS times of 11.2 months and 4.0 months in the COMETRIQ and placebo arms, respectively.
Partial responses were observed only among patients in the COMETRIQ arm (27% vs. 0; p<0.0001). The median duration of objective responses was 14.7 months (95% CI: 11.1, 19.3) for patients treated with COMETRIQ. There was no statistically significant difference in overall survival (median OS: 26.6 months in the COMETRIQ arm vs. 21.1 months in the placebo arm [HR = 0.85 (95% CI: 0.64, 1.12), p = 0.2409]).
Figure 1: Progression-Free Survival
-
16 HOW SUPPLIED/STORAGE AND HANDLING
COMETRIQ 20 mg capsules are supplied as hard gelatin capsules with grey cap and grey body, printed with "XL184 20mg" in black ink and containing cabozantinib (S)-malate salt equivalent to 20 mg cabozantinib.
COMETRIQ 80 mg capsules are supplied as hard gelatin capsules with Swedish orange cap and Swedish orange body, printed with "XL184 80mg" in black ink and containing cabozantinib (S)- malate salt equivalent to 80 mg cabozantinib.
COMETRIQ capsules are supplied as follows:
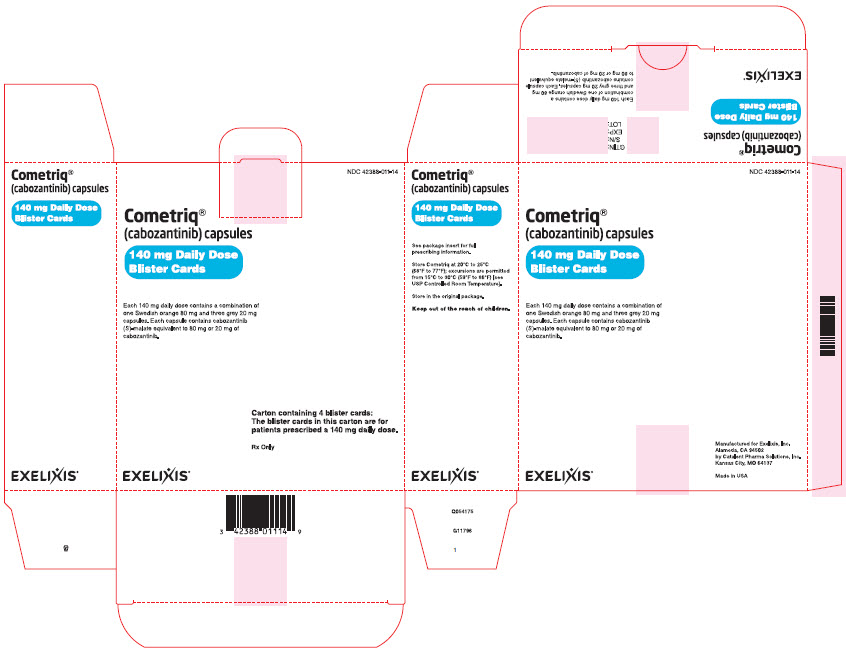
Cartons- 140 mg daily-dose carton NDC#42388-011-14
Containing four 140 mg daily-dose blister cards
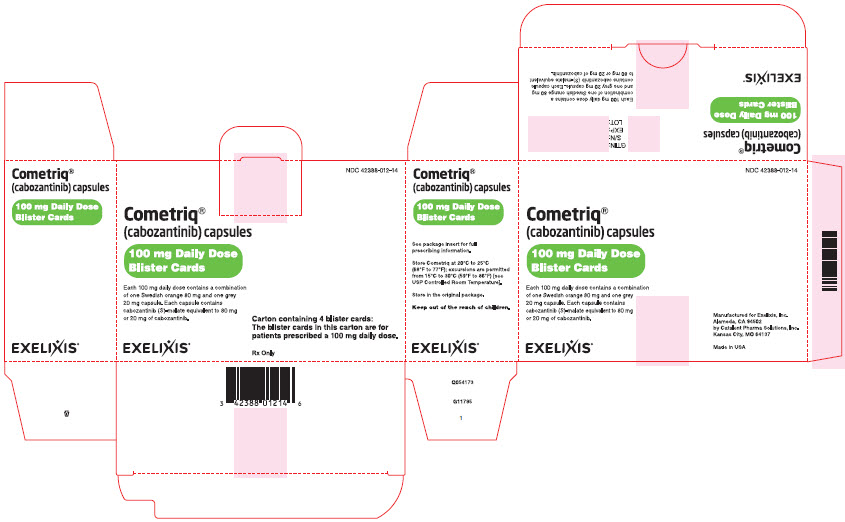
(each blister card contains seven 80-mg and twenty-one 20-mg capsules) - 100 mg daily-dose carton NDC#42388-012-14
Containing four 100 mg daily-dose blister cards
(each blister card contains seven 80-mg and seven 20-mg capsules) - 60 mg daily-dose carton NDC#42388-013-14
Containing four 60 mg daily-dose blister cards
(each blister card contains twenty-one 20-mg capsules)
Store COMETRIQ at 20°C to 25°C (68°F to 77°F); excursions are permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
- 140 mg daily-dose carton NDC#42388-011-14
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
- Perforations and fistulas: Advise patients that gastrointestinal disorders such as diarrhea, nausea, vomiting, and constipation may develop during COMETRIQ treatment and to seek immediate medical attention if they experience persistent or severe abdominal pain because cases of gastrointestinal perforation and fistula have been reported in patients taking COMETRIQ [see Warnings and Precautions (5.1)].
- Hemorrhage: Instruct patients to contact their healthcare provider to seek immediate medical attention for signs or symptoms of unusual severe bleeding or hemorrhage [see Warnings and Precautions (5.2)].
- Thrombotic events: Venous and arterial thrombotic events have been reported. Advise patients to report signs or symptoms of an arterial thrombosis. Venous thromboembolic events including pulmonary embolus have been reported. Advise patients to contact their health care provider if new onset of dyspnea, chest pain, or localized limb edema occurs [see Warnings and Precautions (5.3)].
- Impaired wound healing: Advise patients that COMETRIQ may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.4)].
- Hypertension and hypertensive crisis: Inform patients of the signs and symptoms of hypertension. Advise patients to undergo routine blood pressure monitoring and to contact their health care provider if blood pressure is elevated or if they experience signs or symptoms of hypertension [see Warnings and Precautions (5.5)].
- Osteonecrosis of the jaw: Advise patients regarding good oral hygiene practices. Advise patients to immediately contact their healthcare provider for signs or symptoms associated with osteonecrosis of the jaw [see Warnings and Precautions (5.6)].
- Diarrhea: Advise patients to notify their healthcare provider at the first signs of poorly formed or loose stool or an increased frequency of bowel movements [see Warnings and Precautions (5.7)].
- Palmar-plantar erythrodysesthesia: Advise patients to contact their healthcare provider for progressive or intolerable rash [see Warnings and Precautions (5.8)].
- Reversible posterior leukoencephalopathy syndrome: Advise patients to immediately contact their health care provider for new onset or worsening neurological function [see Warnings and Precautions (5.10)].
-
Embryo-fetal toxicity:
- Advise females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.11), Use in Specific Populations (8.1)].
- Advise patients of reproductive potential to use effective contraception during treatment with COMETRIQ and for at least 4 months after the final dose [see Use in Specific Populations (8.3)].
- Lactation: Advise women not to breastfeed during treatment with COMETRIQ and for 4 months following the last dose [see Use in Specific Populations (8.2)].
- Drug interactions: Advise patients to inform their healthcare provider of all prescription or nonprescription medications, vitamins or herbal products. Inform patients to avoid grapefruit, grapefruit juice, and St. John’s wort [see Drug Interactions (7.1), (7.2)].
Important administration information
- Instruct patients not to eat for at least 2 hours before and at least 1 hour after taking COMETRIQ. Instruct patients that COMETRIQ capsules should not be opened or crushed and to take COMETRIQ capsules with a full glass (at least 8 ounces) of water.
Manufactured for Exelixis, Inc. Alameda, CA 94502
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
COMETRIQ® (Ko-me-trik)
cabozantinib
capsulesWhat is COMETRIQ?
COMETRIQ is a prescription medicine used to treat people with medullary thyroid cancer that has spread to other parts of the body.
It is not known if COMETRIQ is safe and effective in children.
Before you take COMETRIQ, tell your healthcare provider about all of your medical conditions including if you:
- have a recent history of coughing up blood or bleeding or any unusual bleeding
- have an open wound
- have high blood pressure
- plan to have any surgery, a dental procedure, or have had a recent surgery. You should stop taking COMETRIQ at least 3 weeks before planned surgery. See "What are the possible side effects of COMETRIQ?"
- have liver problems
- are pregnant or plan to become pregnant. COMETRIQ can harm your unborn baby. If you are able to become pregnant, you should use effective birth control during treatment and for 4 months after the final dose of COMETRIQ. Talk to your healthcare provider about birth control methods that may be right for you. If you become pregnant or think you are pregnant, tell your healthcare provider right away.
- are breastfeeding or plan to breastfeed. It is not known if COMETRIQ passes into your breast milk. Do not breastfeed during treatment and for 4 months after the final dose of COMETRIQ.
Tell your healthcare provider about all the medicines you take, including prescription or over-the-counter medicines, vitamins, and herbal supplements. COMETRIQ and certain other medicines may affect each other causing side effects.
How should I take COMETRIQ?
- Take COMETRIQ exactly as your healthcare provider tells you to take it.
- Do not take COMETRIQ with food. Do not eat for at least 2 hours before and at least 1 hour after taking COMETRIQ.
- Swallow COMETRIQ capsules whole with a full glass (at least 8 ounces) of water.
- Do not crush or open COMETRIQ capsules.
- If you miss a dose and your next dose is in
- less than 12 hours, take your next dose at the normal time. Do not make up the missed dose.
- hours or more, take the missed dose as soon as you remember. Take your next dose at the normal time.
What should I avoid while taking COMETRIQ?
Do not drink grapefruit juice, eat grapefruit or supplements that contain grapefruit during treatment with COMETRIQ.
What are the possible side effects of COMETRIQ?
COMETRIQ may cause serious side effects, including:
- a tear in your stomach or intestinal wall (perforation) or an abnormal connection between 2 parts of your body (fistula) that may lead to death. Tell your healthcare provider right away if you get tenderness or pain in your stomach-area (abdomen).
-
bleeding (hemorrhage). COMETRIQ can cause severe bleeding that may lead to death. Tell your healthcare provider right away if you get any signs of bleeding during treatment with COMETRIQ, including:
- coughing up blood or blood clots
- vomiting blood or if your vomit looks like coffee-grounds
- red or black (looks like tar) stools
- menstrual bleeding that is heavier than normal
- any unusual or heavy bleeding
-
blood clots, stroke, heart attack, and chest pain. Get emergency help right away if you get:
- swelling or pain in your arms or legs
- shortness of breath
- feel lightheaded or faint
- sweating more than usual
- numbness or weakness of your face, arm or leg, especially on one side of your body
- sudden confusion, trouble speaking or understanding
- sudden trouble seeing in one or both eyes
- sudden trouble walking
- dizziness, loss of balance or coordination
- a sudden severe headache
-
wound healing problems. Wound healing problems have happened in some people who take COMETRIQ. Tell your healthcare provider if you plan to have any surgery before or during treatment with COMETRIQ.
- You should stop taking COMETRIQ at least 3 weeks before planned surgery.
- Your healthcare provider should tell you when you may start taking COMETRIQ again after surgery.
- high blood pressure (hypertension). Hypertension is common with COMETRIQ and can be severe. Your healthcare provider will check your blood pressure before starting COMETRIQ and during treatment with COMETRIQ. If needed, your healthcare provider may prescribe medicine to treat your high blood pressure.
- severe jaw bone problems (osteonecrosis). Symptoms may include jaw pain, toothache, or sores on your gums. Your healthcare provider should examine your mouth before you start and during treatment with COMETRIQ. Tell your dentist that you are taking COMETRIQ. It is important for you to practice good mouth care during treatment with COMETRIQ.
- diarrhea. Diarrhea is common with COMETRIQ and can be severe. If needed, your healthcare provider may prescribe medicine to treat your diarrhea. Tell your healthcare provider right away, if you have frequent loose, watery bowel movements.
- a skin problem called hand-foot skin reaction. Hand-foot skin reactions are common with COMETRIQ and can be severe. Tell your healthcare provider right away if you have rashes, redness, pain, swelling, or blisters on the palms of your hands or soles of your feet.
- protein in your urine and possible kidney problems. Symptoms may include swelling in your hands, arms, legs, or feet.
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS). A condition called reversible posterior leukoencephalopathy syndrome can happen during treatment with COMETRIQ. Tell your healthcare provider right away if you have headaches, seizures, confusion, changes in vision, or problems thinking.
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with COMETRIQ if you have certain side effects.
The most common side effects of COMETRIQ are:
- diarrhea
- redness, swelling or pain in your mouth or throat, or mouth sores. Tell your healthcare provider if these symptoms prevent you from eating or drinking.
- weight loss
- decreased appetite
- nausea
- tiredness
- hair color turning lighter
- change in taste
- pain in your abdomen
- constipation
- increased liver function blood tests
- decreased calcium and phosphate blood levels
- decreased white blood cell counts
- decreased platelet counts
- increased bilirubin blood levels
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of COMETRIQ. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store COMETRIQ?
- Store COMETRIQ at room temperature 68°F to 77°F (20°C to 25°C).
Keep COMETRIQ and all medicines out of the reach of children.
General information about COMETRIQ.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use COMETRIQ for a condition for which it was not prescribed. Do not give COMETRIQ to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about COMETRIQ that is written for health professionals.
What are the ingredients in COMETRIQ?
Active ingredient: cabozantinib
Inactive ingredients: silicified microcrystalline cellulose, croscarmellose sodium, sodium starch glycolate, fumed silica, and stearic acid
Capsule shells: Grey gelatin capsule shells contain black iron oxide and titanium dioxide. Swedish orange gelatin capsule shells contain red iron oxide, and titanium dioxide.
The printing ink contains shellac glaze, black iron oxide, N-butyl alcohol, isopropyl alcohol, propylene glycol, and ammonium hydroxide.
Manufactured for Exelixis, Inc. Alameda, CA 94502
For more information, go to www.cometriq.com or call 1-855-292-3935.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised 01/2020
-
PRINCIPAL DISPLAY PANEL
Package Label - CARTON - 140 mg Daily Dose of COMETRIQPRINCIPAL DISPLAY PANEL
Cometriq®
(cabozantinib) capsules
140 mg Daily Dose
Blister Cards
Each 140 mg daily dose contains a combination of
one Swedish orange 80 mg and three grey 20 mg
capsules. Each capsule contains cabozantinib
(S)-malate equivalent to 80 mg or 20 mg of
cabozantinib.
EXELIXIS®
NDC: 42388-011-14
Carton containing 4 blister cards:
The blister cards in this carton are for
patients prescribed a 140 mg daily dose.
Rx Only
-
PRINCIPAL DISPLAY PANEL
Package Label - CARTON - 100 mg Daily Dose of COMETRIQPRINCIPAL DISPLAY PANEL
Cometriq®
(cabozantinib) capsules
100 mg Daily Dose
Blister Cards
Each 100 mg daily dose contains a combination
of one Swedish orange 80 mg and one grey
20 mg capsule. Each capsule contains
cabozantinib (S)-malate equivalent to 80 mg
or 20 mg of cabozantinib.
EXELIXIS®
NDC: 42388-012-14
Carton containing 4 blister cards:
The blister cards in this carton are for
patients prescribed a 100 mg daily dose.
Rx Only
-
PRINCIPAL DISPLAY PANEL
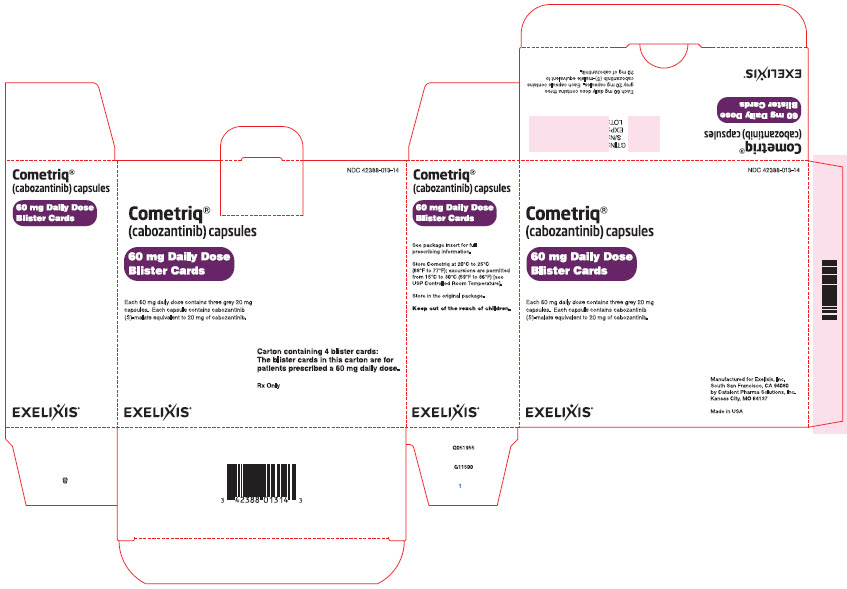
Package Label - CARTON - 60 mg Daily Dose of COMETRIQPRINCIPAL DISPLAY PANEL
Cometriq®
(cabozantinib) capsules
60 mg Daily Dose
Blister Cards
Each 60 mg daily dose contains three grey 20 mg
capsules. Each capsule contains cabozantinib
(S)-malate equivalent to 20 mg of cabozantinib.
EXELIXIS®
NDC: 42388-013-14
Carton containing 4 blister cards:
The blister cards in this carton are for
patients prescribed a 60 mg daily dose.
Rx Only
-
INGREDIENTS AND APPEARANCE
COMETRIQ
cabozantinib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42388-011 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42388-011-14 4 in 1 CARTON 11/29/2012 1 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 7 Part 2 21 Part 1 of 2 COMETRIQ
cabozantinib capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabozantinib s-malate (UNII: DR7ST46X58) (cabozantinib - UNII:1C39JW444G) cabozantinib 80 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) sodium starch glycolate type a potato (UNII: 5856J3G2A2) silicon dioxide (UNII: ETJ7Z6XBU4) stearic acid (UNII: 4ELV7Z65AP) gelatin (UNII: 2G86QN327L) ferric oxide red (UNII: 1K09F3G675) ferrosoferric oxide (UNII: XM0M87F357) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) butyl alcohol (UNII: 8PJ61P6TS3) isopropyl alcohol (UNII: ND2M416302) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) Product Characteristics Color ORANGE (Swedish Orange) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XL184;80mg Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 Part 2 of 2 COMETRIQ
cabozantinib capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabozantinib s-malate (UNII: DR7ST46X58) (cabozantinib - UNII:1C39JW444G) cabozantinib 20 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) sodium starch glycolate type a potato (UNII: 5856J3G2A2) silicon dioxide (UNII: ETJ7Z6XBU4) stearic acid (UNII: 4ELV7Z65AP) gelatin (UNII: 2G86QN327L) ferrosoferric oxide (UNII: XM0M87F357) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) butyl alcohol (UNII: 8PJ61P6TS3) isopropyl alcohol (UNII: ND2M416302) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) Product Characteristics Color GRAY Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XL184;20mg Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 COMETRIQ
cabozantinib kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42388-012 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42388-012-14 4 in 1 CARTON 11/29/2012 1 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 7 Part 2 7 Part 1 of 2 COMETRIQ
cabozantinib capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabozantinib s-malate (UNII: DR7ST46X58) (cabozantinib - UNII:1C39JW444G) cabozantinib 80 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) sodium starch glycolate type a potato (UNII: 5856J3G2A2) silicon dioxide (UNII: ETJ7Z6XBU4) stearic acid (UNII: 4ELV7Z65AP) gelatin (UNII: 2G86QN327L) ferric oxide red (UNII: 1K09F3G675) ferrosoferric oxide (UNII: XM0M87F357) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) butyl alcohol (UNII: 8PJ61P6TS3) isopropyl alcohol (UNII: ND2M416302) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) Product Characteristics Color ORANGE (Swedish Orange) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XL184;80mg Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 Part 2 of 2 COMETRIQ
cabozantinib capsuleProduct Information Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabozantinib s-malate (UNII: DR7ST46X58) (cabozantinib - UNII:1C39JW444G) cabozantinib 20 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) sodium starch glycolate type a potato (UNII: 5856J3G2A2) silicon dioxide (UNII: ETJ7Z6XBU4) stearic acid (UNII: 4ELV7Z65AP) gelatin (UNII: 2G86QN327L) ferrosoferric oxide (UNII: XM0M87F357) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) butyl alcohol (UNII: 8PJ61P6TS3) isopropyl alcohol (UNII: ND2M416302) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) Product Characteristics Color GRAY Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XL184;20mg Contains Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 COMETRIQ
cabozantinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42388-013 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength cabozantinib s-malate (UNII: DR7ST46X58) (cabozantinib - UNII:1C39JW444G) cabozantinib 20 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) sodium starch glycolate type a potato (UNII: 5856J3G2A2) silicon dioxide (UNII: ETJ7Z6XBU4) stearic acid (UNII: 4ELV7Z65AP) gelatin (UNII: 2G86QN327L) ferrosoferric oxide (UNII: XM0M87F357) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) butyl alcohol (UNII: 8PJ61P6TS3) isopropyl alcohol (UNII: ND2M416302) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) Product Characteristics Color GRAY Score no score Shape CAPSULE Size 19mm Flavor Imprint Code XL184;20mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42388-013-14 4 in 1 CARTON 11/29/2012 1 21 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203756 11/29/2012 Labeler - Exelixis, Inc. (785991949) Establishment Name Address ID/FEI Business Operations Catalent CTS, LLC 962674474 MANUFACTURE(42388-011, 42388-012, 42388-013) , LABEL(42388-011, 42388-012, 42388-013) , PACK(42388-011, 42388-012, 42388-013) , ANALYSIS(42388-011, 42388-012, 42388-013) Establishment Name Address ID/FEI Business Operations Catalent U.K. Packaging Limited 232616826 LABEL(42388-011, 42388-012, 42388-013)
Trademark Results [COMETRIQ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 COMETRIQ 85331878 not registered Dead/Abandoned |
Exelixis, Inc. 2011-05-26 |
 COMETRIQ 85331865 4354494 Live/Registered |
Exelixis, Inc. 2011-05-26 |
 COMETRIQ 77197101 not registered Dead/Abandoned |
PowerOneData, Inc. 2007-06-04 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.