INSPRA- eplerenone tablet, film coated
Inspra by
Drug Labeling and Warnings
Inspra by is a Prescription medication manufactured, distributed, or labeled by Viatris Specialty LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use INSPRA safely and effectively. See full prescribing information for INSPRA.
INSPRA® (eplerenone) tablets, for oral use
Initial U.S. Approval: 2002INDICATIONS AND USAGE
INSPRA is an aldosterone antagonist indicated for:
- Improving survival of stable adult patients with symptomatic heart failure with reduced ejection fraction (HFrEF) after an acute myocardial infarction. (1.1)
- The treatment of hypertension in adults, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. (1.2)
DOSAGE AND ADMINISTRATION
HFrEF Post-MI: Initiate treatment with 25 mg once daily. Titrate to maximum of 50 mg once daily within 4 weeks, as tolerated. Dose adjustments may be required based on potassium levels. (2.1)
Hypertension: 50 mg once daily, alone or combined with other antihypertensive agents. For inadequate response, increase to 50 mg twice daily. Higher dosages are not recommended. (2.2)For all patients:
Measure serum potassium before starting INSPRA and periodically thereafter. (2.3)DOSAGE FORMS AND STRENGTHS
Tablets: 25 mg, 50 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Hyperkalemia: Patients with decreased renal function, diabetes, proteinuria or patients who are taking ACEs and ARBs, NSAIDs or moderate CYP3A inhibitors are at increased risk. Monitor serum potassium levels and adjust dose as needed. (5.1)
ADVERSE REACTIONS
HFrEF Post-MI: Most common adverse reactions (>2% and more frequent than with placebo): hyperkalemia and increased creatinine. (6.1)
Hypertension: In clinical studies, adverse reactions with INSPRA were uncommon. (6.1)To report SUSPECTED ADVERSE REACTIONS, contact Viatris at 1-877-446-3679 (1-877-4-INFO-RX) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- CYP3A Inhibitors: In post-MI HFrEF patients, do not exceed 25 mg once daily when used with moderate CYP3A inhibitors (e.g., verapamil, erythromycin, saquinavir, fluconazole). In patients with hypertension, initiate at 25 mg once daily. For inadequate blood pressure response, dosing may be increased to a maximum of 25 mg twice daily. (2.4, 7.1, 12.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Heart Failure Post-Myocardial Infarction
1.2 Hypertension
2 DOSAGE AND ADMINISTRATION
2.1 Heart Failure Post-Myocardial Infarction
2.2 Hypertension
2.3 Recommended Monitoring
2.4 Dose Modification for Use with Moderate CYP3A Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hyperkalemia
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
6.3 Clinical Laboratory Test Findings
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors
7.2 ACE Inhibitors and Angiotensin II Receptor Antagonists
7.3 Lithium
7.4 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Heart Failure Post-Myocardial Infarction
14.2 Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Heart Failure Post-Myocardial Infarction
INSPRA is indicated to improve survival of stable adult patients with symptomatic heart failure with reduced ejection fraction (≤40%) (HFrEF) after an acute myocardial infarction (MI).
1.2 Hypertension
INSPRA is indicated for the treatment of hypertension in adults, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular (CV) events, primarily strokes and MI. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes.
Control of high blood pressure should be part of comprehensive CV risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce CV morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent CV outcome benefit has been a reduction in the risk of stroke, but reductions in MI and CV mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased CV risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
INSPRA may be used alone or in combination with other antihypertensive agents.
-
2 DOSAGE AND ADMINISTRATION
2.1 Heart Failure Post-Myocardial Infarction
Initiate treatment at 25 mg once daily and titrate to the recommended dose of 50 mg once daily, preferably within 4 weeks as tolerated by the patient.
Once treatment with INSPRA has begun, adjust the dose based on the serum potassium level as shown in Table 1.
Table 1. Dose Adjustment in Heart Failure Post-MI Serum Potassium (mEq/L)
Dose Adjustment
<5.0
25 mg every other day to 25 mg once daily
25 mg once daily to 50 mg once daily5.0-5.4
No adjustment
5.5-5.9
50 mg once daily to 25 mg once daily
25 mg once daily to 25 mg every other day
25 mg every other day to withhold≥6.0
Withhold and restart at 25 mg every other day when potassium levels fall to <5.5 mEq/L
2.2 Hypertension
The recommended starting dose of INSPRA is 50 mg administered once daily. The full therapeutic effect of INSPRA is apparent within 4 weeks. For patients with an inadequate blood pressure response to 50 mg once daily increase the dosage of INSPRA to 50 mg twice daily. Higher dosages of INSPRA are not recommended because they have no greater effect on blood pressure than 100 mg and are associated with an increased risk of hyperkalemia [see Clinical Studies (14.2)].
2.3 Recommended Monitoring
Measure serum potassium before initiating INSPRA therapy, within the first week, and at one month after the start of treatment or dose adjustment. Assess serum potassium periodically thereafter.
Check serum potassium and serum creatinine within 3-7 days of a patient initiating a moderate CYP3A inhibitor ACE inhibitors, angiotensin II blockers or nonsteroidal anti-inflammatories.
2.4 Dose Modification for Use with Moderate CYP3A Inhibitors
In post-MI HFrEF patients receiving a moderate CYP3A inhibitor (e.g., erythromycin, saquinavir, verapamil, and fluconazole), do not exceed 25 mg once daily. In patients with hypertension receiving a moderate CYP3A inhibitor, initiate at 25 mg once daily. For inadequate blood pressure response, dosing may be increased to a maximum of 25 mg twice daily [see Drug Interactions (7.1)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
For All Patients
INSPRA is contraindicated in all patients with:
- serum potassium >5.5 mEq/L at initiation,
- creatinine clearance ≤30 mL/min, or
- concomitant administration of strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, nefazodone, troleandomycin, clarithromycin, ritonavir, and nelfinavir) [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
For Patients Treated for Hypertension
INSPRA is contraindicated for the treatment of hypertension in patients with:
- type 2 diabetes with microalbuminuria,
- serum creatinine >2.0 mg/dL in males or >1.8 mg/dL in females,
- creatinine clearance <50 mL/min, or
- concomitant administration of potassium supplements or potassium-sparing diuretics (e.g., amiloride, spironolactone, or triamterene) [see Warnings and Precautions (5.1), Adverse Reactions (6.2), Drug Interactions (7), and Clinical Pharmacology (12.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hyperkalemia
The risk of hyperkalemia is higher in patients with impaired renal function, proteinuria, diabetes and those concomitantly treated with ACEs, ARBs, NSAIDs and moderate CYP3A inhibitors. Minimize the risk of hyperkalemia with proper patient selection and monitoring [see Dosage and Administration (2.1), Contraindications (4), Adverse Reactions (6.2), and Drug Interactions (7)]. Monitor patients for the development of hyperkalemia until the effect of INSPRA is established. Patients who develop hyperkalemia (5.5-5.9 mEq/L) may continue INSPRA therapy with proper dose adjustment. Dose reduction decreases potassium levels. Patients on moderate CYP3A inhibitors that cannot be avoided should have their dose of eplerenone reduced [see Drug Interactions (7.2)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Hyperkalemia [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
Heart Failure Post-Myocardial Infarction
In EPHESUS, safety was evaluated in 3307 patients treated with INSPRA and 3301 placebo-treated patients. The overall incidence of adverse events reported with INSPRA (78.9%) was similar to placebo (79.5%). Adverse events occurred at a similar rate regardless of age, gender, or race. Patients discontinued treatment due to an adverse event at similar rates in either treatment group (4.4% INSPRA vs. 4.3% placebo), with the most common reasons for discontinuation being hyperkalemia, MI, and abnormal renal function.
Adverse reactions that occurred more frequently in patients treated with INSPRA than placebo were hyperkalemia (3.4% vs. 2.0%) and increased creatinine (2.4% vs. 1.5%). Discontinuations due to hyperkalemia or abnormal renal function were less than 1.0% in both groups.
Hypertension
INSPRA has been evaluated for safety in 3091 patients treated for hypertension. A total of 690 patients were treated for over 6 months and 106 patients were treated for over 1 year.
In placebo-controlled studies, the overall rates of adverse events were 47% with INSPRA and 45% with placebo. Adverse events occurred at a similar rate regardless of age, gender, or race. Therapy was discontinued due to an adverse event in 3% of patients treated with INSPRA and 3% of patients given placebo. The most common reasons for discontinuation of INSPRA were headache, dizziness, angina pectoris/MI, and increased GGT.
Gynecomastia and abnormal vaginal bleeding were reported with INSPRA but not with placebo. The rates increased with increasing duration of therapy.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of INSPRA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin: angioedema, rash
6.3 Clinical Laboratory Test Findings
Heart Failure Post-Myocardial Infarction
Creatinine: Increases of more than 0.5 mg/dL were reported for 6.5% of patients administered INSPRA and for 4.9% of placebo-treated patients.
Potassium: In EPHESUS [see Clinical Studies (14.1)], the frequencies of patients with changes in potassium (<3.5 mEq/L or >5.5 mEq/L or ≥6.0 mEq/L) receiving INSPRA compared with placebo are displayed in Table 2.
Table 2. Hypokalemia (<3.5 mEq/L) or Hyperkalemia (>5.5 or ≥6.0 mEq/L) in EPHESUS Potassium (mEq/L)
INSPRA
(N=3251)
n (%)Placebo
(N=3237)
n (%)<3.5
273 (8.4)
424 (13.1)
>5.5
508 (15.6)
363 (11.2)
≥6.0
180 (5.5)
126 (3.9)
Rates of hyperkalemia increased with decreasing renal function.
Table 3. Rates of Hyperkalemia (>5.5 mEq/L) in EPHESUS by Baseline Creatinine Clearance* * Estimated using the Cockroft-Gault formula. Baseline Creatinine Clearance
INSPRA
(N=508)
n (%)Placebo
(N=363)
n (%)≤30 mL/min
160 (32)
82 (23)
31-50 mL/min
122 (24)
46 (13)
51-70 mL/min
86 (17)
48 (13)
>70 mL/min
56 (11)
32 (9)
The rates of hyperkalemia in EPHESUS in the INSPRA-treated group vs. placebo were increased in patients with proteinuria (16% vs 11%), diabetes (18% vs. 13%) or both (26% vs. 16%).
Hypertension
Potassium: In placebo-controlled fixed-dose studies, the mean increases in serum potassium were dose-related and are shown in Table 4 along with the frequencies of values >5.5 mEq/L.
Table 4. Increases in Serum Potassium in the Placebo-Controlled, Fixed-Dose Hypertension Studies of INSPRA Mean Increase mEq/L
% >5.5 mEq/L
Daily Dosage
n
Placebo
194
0
1
25
97
0.08
0
50
245
0.14
0
100
193
0.09
1
-
7 DRUG INTERACTIONS
7.1 CYP3A Inhibitors
Eplerenone metabolism is predominantly mediated via CYP3A. Do not use INSPRA with drugs that are strong inhibitors of CYP3A [see Contraindications (4) and Clinical Pharmacology (12.3)].
In post-MI HFrEF patients taking a moderate CYP3A inhibitor, do not exceed 25 mg once daily. In patients with hypertension taking a moderate CYP3A inhibitor, initiate at 25 mg once daily. For inadequate blood pressure response, dosing may be increased to a maximum of 25 mg twice daily [see Dosage and Administration (2.3, 2.4) and Clinical Pharmacology (12.3)].
7.2 ACE Inhibitors and Angiotensin II Receptor Antagonists
The risk of hyperkalemia increases when eplerenone is used in combination with an ACE inhibitor and/or an ARB. A close monitoring of serum potassium and renal function is recommended, especially in patients at risk for impaired renal function, e.g., the elderly [see Warnings and Precautions (5.1)].
7.3 Lithium
A drug interaction study of eplerenone with lithium has not been conducted. Lithium toxicity has been reported in patients receiving lithium concomitantly with diuretics and ACE inhibitors. Serum lithium levels should be monitored frequently if INSPRA is administered concomitantly with lithium.
7.4 Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
A drug interaction study of eplerenone with an NSAID has not been conducted. The administration of other potassium-sparing antihypertensives with NSAIDs has been shown to reduce the antihypertensive effect in some patients and result in severe hyperkalemia in patients with impaired renal function. Therefore, when INSPRA and NSAIDs are used concomitantly, monitor blood pressure and serum potassium levels.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The available data from published case reports on eplerenone use during pregnancy are insufficient to establish a drug-associated risk of major birth defects, miscarriage, adverse maternal or fetal outcomes (see Clinical Considerations). In animal studies, no adverse developmental effects were observed when eplerenone was administered to pregnant rats and rabbits during organogenesis at exposures 32 and 31 times, respectively the human exposure at the 100 mg/day therapeutic dose.
The estimated background risk of major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Pregnant women with heart failure are at increased risk for preterm birth. Stroke volume and heart rate increase during pregnancy, increasing cardiac output, especially during the first trimester. Clinical classification of heart disease may worsen with pregnancy and lead to maternal death. Closely monitor pregnant patients for destabilization of their heart failure.
Data
Animal Data
Embryo-fetal development studies were conducted with doses up to 1000 mg/kg/day in rats and 300 mg/kg/day in rabbits (exposures up to 32 and 31 times the human AUC for the 100 mg/day therapeutic dose, respectively) administered during organogenesis. No teratogenic effects were seen in rats or rabbits, although decreased rat fetal weights were observed, and decreased body weight in maternal rabbits and increased rabbit fetal resorptions and post-implantation loss were observed at the highest administered dosages.
In a pre- and postnatal development study, pregnant rats were administered eplerenone at doses up to 1000 mg/kg/day from Gestation Day 6 through Lactation Day 20. Decreased pup weights were observed beginning at birth at 1000 mg/kg/day.
8.2 Lactation
Risk Summary
There are no human data available on whether eplerenone is present in human milk, or has effects on breastfed infants or on milk production. Eplerenone was present in the milk of lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
8.3 Females and Males of Reproductive Potential
Infertility
Based on animal data, use of INSPRA may compromise male fertility. In mature rats, male fertility was decreased with eplerenone exposure at 17 times the 100 mg/day human therapeutic dose. Reversibility of effects was not evaluated [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of INSPRA for treatment of hypertension have not been established in pediatric patients. In a 10-week study of 304 hypertensive pediatric patients ages 4 to 16 years treated with INSPRA up to 100 mg per day, doses that produced exposure similar to that in adults, INSPRA did not lower blood pressure effectively. In this study and in a 1-year pediatric safety study in 149 patients (age range 5 to 17 years), the incidence of reported adverse events was similar to that of adults. INSPRA was not studied for treatment of hypertension in pediatric patients younger than 4 years of age because the study in older pediatric patients did not demonstrate effectiveness.
The safety and effectiveness of INSPRA have not been established in pediatric patients with heart failure.
8.5 Geriatric Use
Heart Failure Post-Myocardial Infarction
Of the total number of patients in EPHESUS, 3340 (50%) were 65 and over, while 1326 (20%) were 75 and over. Patients greater than 75 years did not appear to benefit from the use of INSPRA [see Clinical Studies (14.1)].
No differences in overall incidence of adverse events were observed between elderly and younger patients. However, due to age-related decreases in creatinine clearance, the incidence of laboratory-documented hyperkalemia was increased in patients 65 and older [see Warnings and Precautions (5.1)].
Hypertension
Of the total number of subjects in clinical hypertension studies of INSPRA, 1123 (23%) were 65 and over, while 212 (4%) were 75 and over. No overall differences in safety or effectiveness were observed between elderly subjects and younger subjects, however due to age-related decreases in creatine clearance, the risk of hyperkalemia may be increased [see Warnings and Precautions (5.1)].
-
10 OVERDOSAGE
No cases of human overdosage with eplerenone have been reported. Lethality was not observed in mice, rats, or dogs after single oral doses that provided Cmax exposures at least 25 times higher than in humans receiving eplerenone 100 mg/day. Dogs showed emesis, salivation, and tremors at a Cmax 41 times the human therapeutic Cmax, progressing to sedation and convulsions at higher exposures.
The most likely manifestation of human overdosage would be anticipated to be hypotension or hyperkalemia. Eplerenone cannot be removed by hemodialysis. Eplerenone has been shown to bind extensively to charcoal. If symptomatic hypotension should occur, supportive treatment should be instituted. If hyperkalemia develops, standard treatment should be initiated.
-
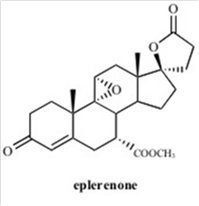
11 DESCRIPTION
INSPRA contains eplerenone, a blocker of aldosterone binding at the mineralocorticoid receptor.
Eplerenone is chemically described as Pregn-4-ene-7,21-dicarboxylic acid, 9,11-epoxy-17-hydroxy-3-oxo-, γ-lactone, methyl ester, (7α,11α,17α)-. Its empirical formula is C24H30O6 and it has a molecular weight of 414.50. The structural formula of eplerenone is represented below:
Eplerenone is an odorless, white to off-white crystalline powder. It is very slightly soluble in water, with its solubility essentially pH-independent. The octanol/water partition coefficient of eplerenone is approximately 7.1 at pH 7.0.
INSPRA tablets for oral administration contain 25 mg or 50 mg of eplerenone and the following inactive ingredients: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, red iron oxide, sodium lauryl sulfate, talc, titanium dioxide, and yellow iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Eplerenone binds to the mineralocorticoid receptor and blocks the binding of aldosterone, a component of the renin-angiotensin-aldosterone-system (RAAS). Aldosterone synthesis, which occurs primarily in the adrenal gland, is modulated by multiple factors, including angiotensin II and non-RAAS mediators such as adrenocorticotropic hormone (ACTH) and potassium. Aldosterone binds to mineralocorticoid receptors in both epithelial (e.g., kidney) and nonepithelial (e.g., heart, blood vessels, and brain) tissues and increases blood pressure through induction of sodium reabsorption and possibly other mechanisms.
Eplerenone has been shown to produce sustained increases in plasma renin and serum aldosterone, consistent with inhibition of the negative regulatory feedback of aldosterone on renin secretion. The resulting increased plasma renin activity and aldosterone circulating levels do not overcome the effects of eplerenone.
Eplerenone selectively binds to human mineralocorticoid receptors relative to its binding to recombinant human glucocorticoid, progesterone, and androgen receptors.
12.2 Pharmacodynamics
There was no significant change in average heart rate among patients treated with INSPRA in the combined clinical studies. No consistent effects of INSPRA on heart rate, QRS duration, or PR or QT interval were observed in 147 normal subjects evaluated for electrocardiographic changes during pharmacokinetic studies.
12.3 Pharmacokinetics
Eplerenone is cleared predominantly by cytochrome P450 (CYP) 3A4 metabolism, with an elimination half-life of 3 to 6 hours. Steady state is reached within 2 days. Absorption is not affected by food. Inhibitors of CYP3A (e.g., ketoconazole, saquinavir) increase blood levels of eplerenone.
Absorption and Distribution
Mean peak plasma concentrations of eplerenone are reached approximately 1.5 to 2 hours following oral administration. Absorption is not affected by food. The absolute bioavailability of eplerenone is 69% following administration of a 100 mg oral tablet. Both peak plasma levels (Cmax) and area under the curve (AUC) are dose proportional for doses of 25 mg to 100 mg and less than proportional at doses above 100 mg. Upon repeat dosing, steady state levels are reached within 2 days.
The plasma protein binding of eplerenone is about 50% and it is primarily bound to alpha 1-acid glycoproteins. The apparent volume of distribution at steady state ranged from 42 to 90 L. Eplerenone does not preferentially bind to red blood cells.
Metabolism and Excretion
Eplerenone metabolism is primarily mediated via CYP3A4. No active metabolites of eplerenone have been identified in human plasma.
Less than 5% of an eplerenone dose is recovered as unchanged drug in the urine and feces. Following a single oral dose of radiolabeled drug, approximately 32% of the dose was excreted in the feces and approximately 67% was excreted in the urine. The elimination half-life of eplerenone is approximately 3 to 6 hours. The apparent plasma clearance is approximately 10 L/hr.
Age, Gender, and Race
The pharmacokinetics of eplerenone at a dose of 100 mg once daily has been investigated in the elderly (≥65 years), in males and females, and in Blacks. At steady state, elderly subjects had increases in Cmax (22%) and AUC (45%) compared with younger subjects (18 to 45 years). The pharmacokinetics of eplerenone did not differ significantly between males and females. At steady state, Cmax was 19% lower and AUC was 26% lower in Blacks [see Dosage and Administration (2.4) and Use in Specific Populations (8.5)].
Renal Impairment
The pharmacokinetics of eplerenone was evaluated in patients with varying degrees of renal impairment and in patients undergoing hemodialysis. Compared with control subjects, steady state AUC and Cmax were increased by 38% and 24%, respectively, in patients with severe renal impairment and were decreased by 26% and 3%, respectively, in patients undergoing hemodialysis. No correlation was observed between plasma clearance of eplerenone and creatinine clearance. Eplerenone is not removed by hemodialysis [see Warnings and Precautions (5.1)].
Hepatic Impairment
The pharmacokinetics of eplerenone 400 mg has been investigated in patients with moderate (Child-Pugh Class B) hepatic impairment and compared with normal subjects. Steady state Cmax and AUC of eplerenone were increased by 3.6% and 42%, respectively.
Heart Failure
The pharmacokinetics of eplerenone 50 mg was evaluated in 8 patients with heart failure (NYHA classification II–IV) and 8 matched (gender, age, weight) healthy controls. Compared with the controls, steady state AUC and Cmax in patients with stable heart failure were 38% and 30% higher, respectively.
Drug-Drug Interactions
Eplerenone is metabolized primarily by CYP3A4. Inhibitors of CYP3A cause increased exposure [see Drug Interactions (7.1)].
Drug-drug interaction studies were conducted with a 100 mg dose of eplerenone.
Following a single dose of INSPRA 100 mg and CYP3A inhibitor ketoconazole 200 mg twice a day, eplerenone’s Cmax was 1.7-fold and AUC was 5.4-fold compared with eplerenone alone.
Administration of eplerenone with moderate CYP3A inhibitors (e.g., erythromycin 500 mg BID, verapamil 240 mg once daily, saquinavir 1200 mg three times a day, fluconazole 200 mg once daily) resulted in increases in Cmax of eplerenone ranging from 40% to 60% and AUC from 100% to 190%.
Grapefruit juice caused a 25% increase in exposure.
Eplerenone is not an inhibitor of CYP1A2, CYP3A4, CYP2C19, CYP2C9, or CYP2D6. Eplerenone did not inhibit the metabolism of amiodarone, amlodipine, astemizole, chlorzoxazone, cisapride, dexamethasone, dextromethorphan, diclofenac, 17α-ethinyl estradiol, fluoxetine, losartan, lovastatin, mephobarbital, methylphenidate, methylprednisolone, metoprolol, midazolam, nifedipine, phenacetin, phenytoin, simvastatin, tolbutamide, triazolam, verapamil, or warfarin in vitro. Eplerenone is not a substrate or an inhibitor of P-Glycoprotein at clinically relevant doses.
No clinically significant drug-drug pharmacokinetic interactions were observed when eplerenone was administered with cisapride, cyclosporine, digoxin, glyburide, midazolam, oral contraceptives (norethindrone/ethinyl estradiol), simvastatin, or warfarin. St. John’s wort (a CYP3A inducer) caused a small (about 30%) decrease in eplerenone AUC.
No significant changes in eplerenone pharmacokinetics were observed when eplerenone was administered with aluminum- and magnesium-containing antacids.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Eplerenone was non-genotoxic in a battery of assays including in vitro bacterial mutagenesis (Ames test in Salmonella spp. and E. coli), in vitro mammalian cell mutagenesis (mouse lymphoma cells), in vitro chromosomal aberration (Chinese hamster ovary cells), in vivo rat bone marrow micronucleus formation, and in vivo/ex vivo unscheduled DNA synthesis in rat liver.
There was no drug-related tumor response in heterozygous P53 deficient mice when tested for 6 months at dosages up to 1000 mg/kg/day (systemic AUC exposures up to 9 times the exposure in humans receiving the 100 mg/day therapeutic dose). Statistically significant increases in benign thyroid tumors were observed after 2 years in both male and female rats when administered eplerenone 250 mg/kg/day (highest dose tested) and in male rats only at 75 mg/kg/day. These dosages provided systemic AUC exposures approximately 2 to 12 times higher than the average human therapeutic exposure at 100 mg/day. Repeat dose administration of eplerenone to rats increases the hepatic conjugation and clearance of thyroxin, which results in increased levels of TSH by a compensatory mechanism. Drugs that have produced thyroid tumors by this rodent-specific mechanism have not shown a similar effect in humans.
Male rats treated with eplerenone at 1000 mg/kg/day for 10 weeks (AUC 17 times that at the 100 mg/day human therapeutic dose) had decreased weights of seminal vesicles and epididymides and slightly decreased fertility. Dogs administered eplerenone at dosages of 15 mg/kg/day and higher (AUC 5 times that at the 100 mg/day human therapeutic dose) had dose-related prostate atrophy. The prostate atrophy was reversible after daily treatment for 1 year at 100 mg/kg/day. Dogs with prostate atrophy showed no decline in libido, sexual performance, or semen quality. Testicular weight and histology were not affected by eplerenone in any test animal species at any dosage.
-
14 CLINICAL STUDIES
14.1 Heart Failure Post-Myocardial Infarction
The eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS) was a multinational, multicenter, double-blind, randomized, placebo-controlled study in patients clinically stable 3 to 14 days after an acute MI with LV dysfunction (as measured by left ventricular ejection fraction [LVEF] ≤40%) and either diabetes or clinical evidence of HF (pulmonary congestion by exam or chest x-ray or S3). Patients with HF of valvular or congenital etiology, patients with unstable post-infarct angina, and patients with serum potassium >5.0 mEq/L or serum creatinine >2.5 mg/dL were to be excluded. Patients were allowed to receive standard post-MI drug therapy and to undergo revascularization by angioplasty or coronary artery bypass graft surgery.
Patients randomized to INSPRA were given an initial dose of 25 mg once daily and titrated to the target dose of 50 mg once daily after 4 weeks if serum potassium was <5.0 mEq/L. Dosage was reduced or suspended anytime during the study if serum potassium levels were ≥5.5 mEq/L [see Dosage and Administration (2.1)].
EPHESUS randomized 6,632 patients (9.3% U.S.) at 671 centers in 27 countries. The study population was primarily white (90%, with 1% Black, 1% Asian, 6% Hispanic, 2% other) and male (71%). The mean age was 64 years (range, 22 to 94 years). The majority of patients had pulmonary congestion (75%) by exam or x-ray and were Killip Class II (64%). The mean ejection fraction was 33%. The average time to enrollment was 7 days post-MI. Medical histories prior to the index MI included hypertension (60%), coronary artery disease (62%), dyslipidemia (48%), angina (41%), type 2 diabetes (30%), previous MI (27%), and HF (15%).
The mean dose of INSPRA was 43 mg/day. Patients also received standard care including aspirin (92%), ACE inhibitors (90%), beta-blockers (83%), nitrates (72%), loop diuretics (66%), or HMG-CoA reductase inhibitors (60%).
Patients were followed for an average of 16 months (range, 0 to 33 months). The ascertainment rate for vital status was 99.7%.
The co-primary endpoints for EPHESUS were (1) the time to death from any cause, and (2) the time to first occurrence of either cardiovascular mortality [defined as sudden cardiac death or death due to progression of HF, stroke, or other CV causes] or CV hospitalization (defined as hospitalization for progression of HF, ventricular arrhythmias, acute MI, or stroke).
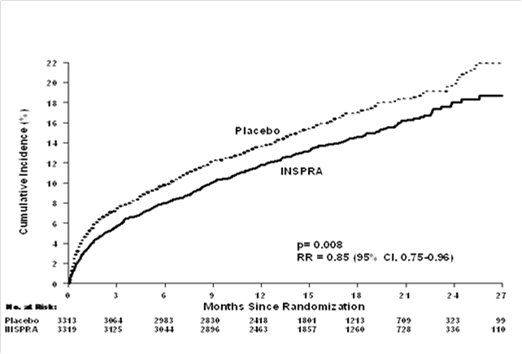
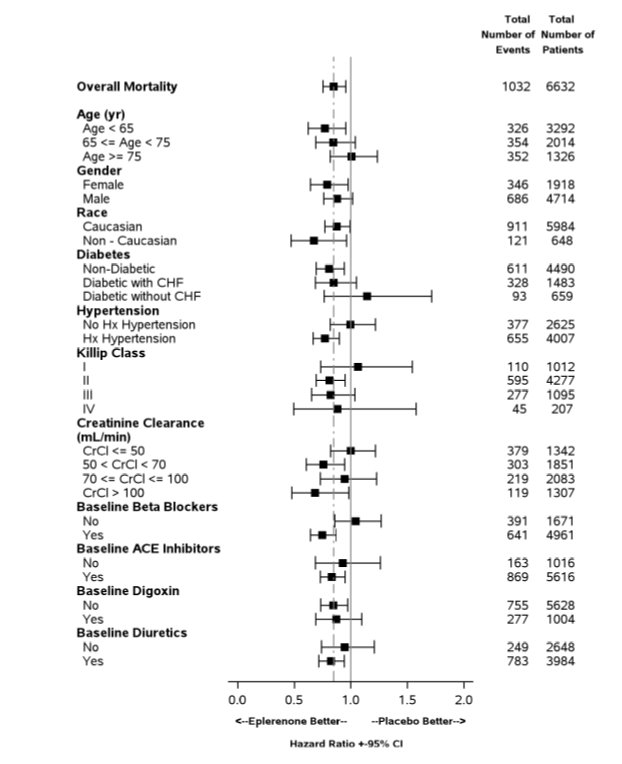
For the co-primary endpoint for death from any cause, there were 478 deaths in the INSPRA group (14.4%) and 554 deaths in the placebo group (16.7%). The risk of death with INSPRA was reduced by 15% [hazard ratio equal to 0.85 (95% confidence interval 0.75 to 0.96; p = 0.008 by log rank test)]. Kaplan-Meier estimates of all-cause mortality are shown in Figure 1 and the components of mortality are provided in Table 5.
Table 5. Components of All-Cause Mortality in EPHESUS INSPRA
(N=3319)
n (%)Placebo
(N=3313)
n (%)Hazard
Ratiop-value
Death from any cause
478 (14.4)
554 (16.7)
0.85
0.008
CV Death
407 (12.3)
483 (14.6)
0.83
0.005
Non-CV Death
60 (1.8)
54 (1.6)
Unknown or unwitnessed death
11 (0.3)
17 (0.5)
Most CV deaths were attributed to sudden death, acute MI, and HF.
The time to first event for the co-primary endpoint of CV death or hospitalization, as defined above, was longer in the INSPRA group (hazard ratio 0.87, 95% confidence interval 0.79 to 0.95, p = 0.002). An analysis that included the time to first occurrence of CV mortality and all CV hospitalizations (atrial arrhythmia, angina, CV procedures, progression of HF, MI, stroke, ventricular arrhythmia, or other CV causes) showed a smaller effect with a hazard ratio of 0.92 (95% confidence interval 0.86 to 0.99; p = 0.028). The combined endpoints, including combined all-cause hospitalization and mortality were driven primarily by CV mortality. The combined endpoints in EPHESUS, including all-cause hospitalization and all-cause mortality, are presented in Table 6.
Table 6. Rates of Death or Hospitalization in EPHESUS - * Co-Primary Endpoint.
Event
INSPRA
n (%)Placebo
n (%)CV death or hospitalization for progression of HF, stroke, MI or ventricular arrhythmia*
885 (26.7)
993 (30.0)
Death
Hospitalization
407 (12.3)
606 (18.3)
483 (14.6)
649 (19.6)
CV death or hospitalization for progression of HF, stroke, MI, ventricular arrhythmia, atrial arrhythmia, angina, CV procedures, or other CV causes (PVD; Hypotension)
1516 (45.7)
1610 (48.6)
Death
Hospitalization
407 (12.3)
1281 (38.6)
483 (14.6)
1307 (39.5)
All-cause death or hospitalization
Death*
Hospitalization
1734 (52.2)
478 (14.4)
1497 (45.1)
1833 (55.3)
554 (16.7)
1530 (46.2)
Mortality hazard ratios varied for some subgroups as shown in Figure 2. Mortality hazard ratios appeared favorable for INSPRA for both genders and for all races or ethnic groups, although the numbers of non-Caucasians were low (648, 10%). Patients with diabetes without clinical evidence of HF and patients greater than 75 years did not appear to benefit from the use of INSPRA. Such subgroup analyses must be interpreted cautiously.
Analyses conducted for a variety of CV biomarkers did not confirm a mechanism of action by which mortality was reduced.
14.2 Hypertension
The safety and efficacy of INSPRA have been evaluated alone and in combination with other antihypertensive agents in clinical studies of 3091 hypertensive patients. The studies included 46% women, 14% Blacks, and 22% elderly (age ≥65). The studies excluded patients with elevated baseline serum potassium (>5.0 mEq/L) and elevated baseline serum creatinine (generally >1.5 mg/dL in males and >1.3 mg/dL in females).
Two fixed-dose, placebo-controlled, 8- to 12-week monotherapy studies in patients with baseline diastolic blood pressures of 95 to 114 mmHg were conducted to assess the antihypertensive effect of INSPRA. In these two studies, 611 patients were randomized to INSPRA and 140 patients to placebo. Patients received INSPRA in doses of 25 mg to 400 mg daily as either a single daily dose or divided into two daily doses. The mean placebo-subtracted reductions in trough cuff blood pressure achieved by INSPRA in these studies at doses up to 200 mg are shown in Figures 3 and 4.
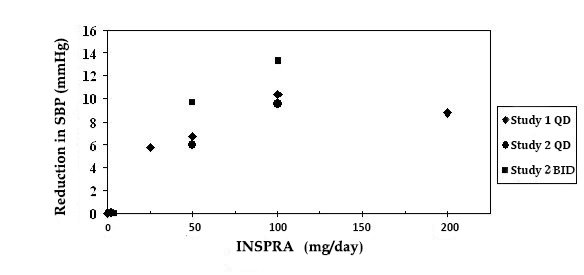
Figure 3. INSPRA Dose Response – Trough Cuff SBP Placebo-Subtracted Adjusted Mean Change from Baseline In Hypertension Studies
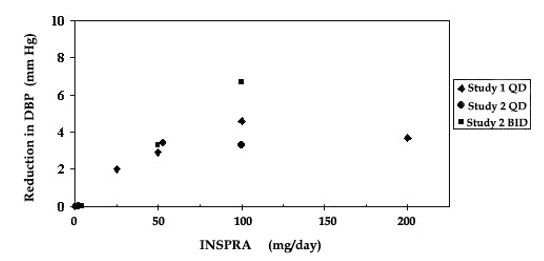
Figure 4. INSPRA Dose Response – Trough Cuff DBP Placebo-Subtracted Adjusted Mean Change from Baseline in Hypertension Studies
Patients treated with INSPRA 50 mg to 200 mg daily experienced significant decreases in sitting systolic and diastolic blood pressure at trough with differences from placebo of 6-13 mmHg (systolic) and 3-7 mmHg (diastolic). These effects were confirmed by assessments with 24-hour ambulatory blood pressure monitoring (ABPM). In these studies, assessments of 24-hour ABPM data demonstrated that INSPRA, administered once or twice daily, maintained antihypertensive efficacy over the entire dosing interval. However, at a total daily dose of 100 mg, INSPRA administered as 50 mg twice per day produced greater trough cuff (4/3 mmHg) and ABPM (2/1 mmHg) blood pressure reductions than 100 mg given once daily.
Blood pressure lowering was apparent within 2 weeks from the start of therapy with INSPRA, with maximal antihypertensive effects achieved within 4 weeks. Stopping INSPRA following treatment for 8 to 24 weeks in six studies did not lead to adverse event rates in the week following withdrawal of INSPRA greater than following placebo or active control withdrawal. Blood pressures in patients not taking other antihypertensives rose 1 week after withdrawal of INSPRA by about 6/3 mmHg, suggesting that the antihypertensive effect of INSPRA was maintained through 8 to 24 weeks.
Blood pressure reductions with INSPRA in the two fixed-dose monotherapy studies and other studies using titrated doses, as well as concomitant treatments, were not significantly different when analyzed by age, gender, or race with one exception. In a study in patients with low renin hypertension, blood pressure reductions in Blacks were smaller than those in whites during the initial titration period with INSPRA.
INSPRA has been studied concomitantly with treatment with ACE inhibitors, ARB, calcium channel blockers, beta-blockers, and hydrochlorothiazide. When administered concomitantly with one of these drugs, INSPRA usually produced its expected antihypertensive effects.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
INSPRA Tablets are yellow, diamond biconvex, and film-coated. They are debossed with “VLE” on one side. They are supplied as follows:
Dose
Deboss
Side 2NDC 58151-xxx-xx
Bottle/30
25 mg
NSR
25142-93
50 mg
NSR
50143-93
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F) [See USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise patients receiving INSPRA:
- Not to use potassium supplements or salt substitutes containing potassium without consulting the prescribing physician [see Warnings and Precautions (5.1)].
- To call their physician if they experience dizziness, diarrhea, vomiting, rapid or irregular heartbeat, lower extremity edema, or difficulty breathing [see Warnings and Precautions (5.1)].
Distributed by:
Viatris Specialty LLC
Morgantown, WV 26505 U.S.A.© 2025 Viatris Inc.
INSPRA is a registered trademark of UPJOHN US 2 LLC, a Viatris Company.
UPJ:INSPRA:R2
-
PRINCIPAL DISPLAY PANEL – 25 mg
NDC: 58151-142-93
INSPRA®
(eplerenone) tablets25 mg
30 Tablets
Rx only
Store at 25°C (77°F);
excursions permitted to
15-30°C (59-86°F)
[see USP Controlled
Room Temperature].Dispense in tight (USP),
child-resistant containers.DOSAGE AND USE
See accompanying
prescribing information.
Each tablet contains
25 mg eplerenone.Distributed by:
Viatris Specialty LLC
Morgantown, WV 26505 U.S.A.© 2023 Viatris Inc.
RUPJ142H
-
PRINCIPAL DISPLAY PANEL – 50 mg
NDC: 58151-143-93
INSPRA®
(eplerenone) tablets50 mg
30 Tablets
Rx only
Store at 25°C (77°F);
excursions permitted to
15-30°C (59-86°F)
[see USP Controlled
Room Temperature].Dispense in tight (USP),
child-resistant containers.DOSAGE AND USE
See accompanying
prescribing information.
Each tablet contains
50 mg eplerenone.Distributed by:
Viatris Specialty LLC
Morgantown, WV 26505 U.S.A.© 2023 Viatris Inc.
RUPJ143H
-
INGREDIENTS AND APPEARANCE
INSPRA
eplerenone tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 58151-142 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPLERENONE (UNII: 6995V82D0B) (EPLERENONE - UNII:6995V82D0B) EPLERENONE 25 mg Inactive Ingredients Ingredient Name Strength LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYSORBATE 80 (UNII: 6OZP39ZG8H) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color YELLOW Score no score Shape DIAMOND Size 7mm Flavor Imprint Code VLE;NSR;25 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58151-142-93 30 in 1 BOTTLE; Type 0: Not a Combination Product 04/19/2024 01/31/2027 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021437 04/19/2024 01/31/2027 INSPRA
eplerenone tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 58151-143 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPLERENONE (UNII: 6995V82D0B) (EPLERENONE - UNII:6995V82D0B) EPLERENONE 50 mg Inactive Ingredients Ingredient Name Strength LACTOSE, UNSPECIFIED FORM (UNII: J2B2A4N98G) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) POLYSORBATE 80 (UNII: 6OZP39ZG8H) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color YELLOW Score no score Shape DIAMOND Size 9mm Flavor Imprint Code VLE;NSR;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58151-143-93 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/17/2024 01/31/2027 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021437 09/17/2024 01/31/2027 Labeler - Viatris Specialty LLC (117455616)

Trademark Results [Inspra]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 INSPRA 90517309 not registered Live/Pending |
Shenzhen Hippo Trading Co.,Ltd. 2021-02-08 |
 INSPRA 78305153 2925085 Dead/Cancelled |
Pharmacia & Upjohn Company 2003-09-25 |
 INSPRA 78188264 not registered Dead/Abandoned |
X.J. Group USA 2002-11-23 |
 INSPRA 76607097 3000360 Dead/Cancelled |
PHARMACIA & UPJOHN COMPANY LLC 2004-08-16 |
 INSPRA 75861567 2865357 Live/Registered |
PHARMACIA & UPJOHN COMPANY LLC 1999-12-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.