INTUNIV- guanfacine tablet, extended release
Intuniv by
Drug Labeling and Warnings
Intuniv by is a Prescription medication manufactured, distributed, or labeled by TYA Pharmaceuticals. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use INTUNIV safely and effectively. See full prescribing information for INTUNIV . INTUNIV (guanfacine) extended-release tablets, for oral use Initial U.S. Approval: 1986 ®®
®
RECENT MAJOR CHANGES
Dosage and Administration, Dose Selection ( ), 02/2013 2.2
INDICATIONS AND USAGE
INTUNIV is a central alpha -adrenergic receptor agonist indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) as monotherapy and as adjunctive therapy to stimulant medications ( ). ®2A1
DOSAGE AND ADMINISTRATION
- Recommended dose: 1 to 4 mg once daily in the morning or evening ( ). 2.2
- Begin at a dose of 1 mg once daily and adjust in increments of no more than 1 mg/week ( ). 2.2
- Do not crush, chew or break tablets before swallowing ( ). 2.1
- Do not administer with high-fat meals, because of increased exposure ( ). 2.1
- Do not substitute for immediate-release guanfacine tablets on a mg-per-mg basis, because of differing pharmacokinetic profiles ( ). 2.3
- If switching from immediate-release guanfacine, discontinue that treatment and titrate with INTUNIV as directed ( ). ®2.3
- Consider dosing on a mg/kg basis. Improvements observed starting at doses of 0.05-0.08 mg/kg once daily. Doses up to 0.12 mg/kg once daily may provide additional benefit ( ). 2.2
- When discontinuing, taper the dose in decrements of no more than 1 mg every 3 to 7 days ( ). 2.5
DOSAGE FORMS AND STRENGTHS
Extended-release tablets: 1 mg, 2 mg, 3 mg and 4 mg ( ) 3
CONTRAINDICATIONS
History of hypersensitivity to INTUNIV , its inactive ingredients, or other products containing guanfacine ( ). ®4
WARNINGS AND PRECAUTIONS
- Hypotension, bradycardia, and syncope: Use INTUNIV with caution in patients at risk for hypotension, bradycardia, heart block, or syncope (e.g., those taking antihypertensives). Measure heart rate and blood pressure prior to initiation of therapy, following dose increases, and periodically while on therapy. Advise patients to avoid becoming dehydrated or overheated ( ). ®5.1
- Sedation and somnolence: Occur commonly with INTUNIV . Consider the potential for additive sedative effects with CNS depressant drugs. Caution patients against operating heavy equipment or driving until they know how they respond to INTUNIV ( ). ®®5.2
ADVERSE REACTIONS
Most common adverse reactions (≥5% and at least twice placebo rate) in the monotherapy trials: somnolence, fatigue, nausea, lethargy, and hypotension ( ). Most common adverse reactions (≥5% and at least twice placebo rate) in the adjunctive trial: somnolence, fatigue, insomnia, dizziness, and abdominal pain ( ). 66
To report SUSPECTED ADVERSE REACTIONS, contact Shire US Inc. at 1-800-828-2088 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.DRUG INTERACTIONS
- Strong CYP3A4 inhibitors (e.g., ketoconazole): Coadministration increases guanfacine exposure. Guanfacine dose should be limited to no more than 2 mg/day. When discontinuing CYP3A4 inhibitors, guanfacine dose should be doubled based on patient tolerability. The maximum dose should not exceed 4 mg/day ( and ). 2.77
- Strong CYP3A4 inducers (e.g., rifampin): Coadministration decreases guanfacine exposure. Guanfacine dose may be titrated up to 8 mg/day. When discontinuing CYP3A4 inducers, guanfacine dose should be decreased by half in 1-2 weeks based on patient tolerability. The maximum dose should not exceed 4 mg/day ( and ). 2.77
USE IN SPECIFIC POPULATIONS
Hepatic or Renal Impairment: dose reduction may be required in patients with clinically significant impairment of hepatic or renal function ( ). 8.6
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2014
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Instruction for Use
2.2 Dose Selection
2.3 Switching from Immediate-Release Guanfacine to INTUNIV
2.4 Maintenance Treatment
2.5 Discontinuation
2.6 Missed Doses
2.7 Dose Adjustment with Concomitant Use of Strong CYP3A4 Inhibitors or Inducers
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension, Bradycardia, and Syncope
5.2 Sedation and Somnolence
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Patients with Renal or Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Safety and Efficacy Studies
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
INTUNIV is indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) as monotherapy and as adjunctive therapy to stimulant medications. The efficacy of INTUNIV was studied for the treatment of ADHD in three controlled monotherapy clinical trials (up to 8 weeks in duration) and one controlled adjunctive trial with psychostimulants (8 weeks in duration) in children and adolescents ages 6-17 who met DSM-IV criteria for ADHD [ ]. The effectiveness of INTUNIV for longer-term use (more than 8 weeks) has not been systematically evaluated in controlled trials. ®®®see Clinical Studies (14)®
-
2 DOSAGE AND ADMINISTRATION
2.1 General Instruction for Use
Do not administer with high fat meals, due to increased exposure. Swallow tablets whole. Do not crush, chew, or break tablets because this will increase the rate of guanfacine release.
2.2 Dose Selection
INTUNIV should be taken once daily, either in the morning or evening, at approximately same time each day. Begin at a dose of 1 mg/day, and adjust in increments of no more than 1 mg/week. Maintain the dose within the range of 1 mg to 4 mg once daily, depending on clinical response and tolerability, for both monotherapy and adjunctive therapy to a psychostimulant. Doses above 4 mg/day have not been systematically studied in controlled clinical studies . [ ] see Clinical Studies (14.1)
Clinically relevant improvements were observed beginning at doses in the range 0.05-0.08 mg/kg once daily in both mono- and adjunctive therapy. Efficacy increased with increasing weight-adjusted dose (mg/kg). If well tolerated, doses up to 0.12 mg/kg once daily may provide additional benefit.
In clinical trials, there were dose-related and exposure-related risks for several clinically significant adverse reactions (hypotension, bradycardia, sedative events). Thus, consideration should be given to dosing INTUNIV on a mg/kg basis, in order to balance the exposure-related potential benefits and risks of treatment. ®
2.3 Switching from Immediate-Release Guanfacine to INTUNIV
If switching from immediate-release guanfacine, discontinue that treatment, and titrate with INTUNIV following above recommended schedule. ®
Do not substitute for immediate-release guanfacine tablets on a milligram-per-milligram basis, because of differing pharmacokinetic profiles. INTUNIV has a delayed T , reduced C and lower bioavailability compared to those of the same dose of immediate-release guanfacine . ®maxmax[ ] see Clinical Pharmacology (12.3)
2.4 Maintenance Treatment
It is generally agreed that pharmacological treatment of ADHD may be needed for an extended period. The effectiveness of INTUNIV for longer-term use (more than 9 weeks) has not been systematically evaluated in controlled trials. Therefore the physician electing to use INTUNIV for extended periods should periodically re-evaluate the long-term usefulness of the drug for the individual patient. ®®
2.5 Discontinuation
Infrequent, transient elevations in blood pressure above original baseline (i.e., rebound) have been reported to occur upon abrupt discontinuation of guanfacine. To minimize these effects, the dose should generally be tapered in decrements of no more than 1 mg every 3 to 7 days.
2.6 Missed Doses
When reinitiating patients to the previous maintenance dose after two or more missed consecutive doses, physicians should consider titration based on patient tolerability.
2.7 Dose Adjustment with Concomitant Use of Strong CYP3A4 Inhibitors or Inducers
Dosage adjustments for INTUNIV are recommended with concomitant use of strong CYP3A4 inhibitors (e.g., boceprevir, clarithromycin, conivaptan, grapefruit juice, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, mibefradil, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, and voriconazole), or CYP3A4 inducers (e.g., avasimibe, carbamazepine, phenytoin, rifampin, and St.John’s wort) (Table 1) . ®[] see Drug Interactions (7)
Table 1: Dose Adjustments in Patients Taking Concomitant CYP3A4 Inhibitors or Inducers
Comedications Scenarios Initiate INTUNIV when taking comedications ®
Continue INTUNIV when adding a comedication ®
Stop a comedication when continuing INTUNIV
®Strong CYP3A4 Inhibitors
INTUNIV dose should be limited to 2 mg/day ®
INTUNIV dose should be decreased by half. ®
INTUNIV dose should be doubled based on patient tolerability. The maximum dose should not exceed 4 mg/day ®
Strong CYP3A4 Inducers
INTUNIV dose may be titrated up to 8 mg/day. Consider faster titration (e.g. in increments of 2 mg/week) ®
Consider increase INTUNIV dose gradually in 1-2 weeks to 2 fold of the original dose based on patient tolerability.
®
INTUNIV dose should be decreased by half in 1-2 weeks based on patient tolerability. The maximum dose should not exceed 4 mg/day ®
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Patients with a history of hypersensitivity to INTUNIV , its inactive ingredients or other products containing guanfacine should not take INTUNIV . ®[ ] see Description (11)®
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypotension, Bradycardia, and Syncope
Treatment with INTUNIV can cause dose-dependent decreases in blood pressure and heart rate. Decreases were less pronounced over time of treatment. Orthostatic hypotension and syncope have been reported . ®[] see Adverse Reactions(6.1)
Measure heart rate and blood pressure prior to initiation of therapy, following dose increases, and periodically while on therapy. Use INTUNIV with caution in patients with a history of hypotension, heart block, bradycardia, cardiovascular disease, or who have a history of syncope or may have a condition that predisposes them to syncope, such as hypotension, orthostatic hypotension, bradycardia, or dehydration. Use INTUNIV with caution in patients treated concomitantly with antihypertensives or other drugs that can reduce blood pressure or heart rate or increase the risk of syncope. Advise patients to avoid becoming dehydrated or overheated. ®®
5.2 Sedation and Somnolence
Somnolence and sedation were commonly reported adverse reactions in clinical studies [ . Before using INTUNIV with other centrally active depressants (such as phenothiazines, barbiturates, or benzodiazepines), consider the potential for additive sedative effects. Caution patients against operating heavy equipment or driving until they know how they respond to treatment with INTUNIV Advise patients to avoid use with alcohol. ] see Adverse Reactions (6.1)®®.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labelling:
- Hypotension, bradycardia, and syncope [ ] see Warnings and Precautions (5.1)
- Sedation and somnolence [ ] see Warnings and Precautions (5.2)
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
A total of 2,028 subjects have been exposed to INTUNIV while participating in clinical trials. This includes 1,533 patients from completed studies in children and adolescents, and 495 subjects in completed studies in adult healthy volunteers. ®
The mean duration of exposure of 446 patients that previously participated in two 2-year, open-label long-term studies was approximately 10 months.
Monotherapy Trials
The most commonly observed adverse reactions (incidence ≥ 5% and at least twice the rate for placebo) in the monotherapy trials (Studies 1 and 2) with INTUNIV were: somnolence, fatigue, nausea, lethargy, and hypotension. Most Common Adverse Reactions -®
Twelve percent (12%) of patients receiving INTUNIV discontinued from the monotherapy clinical studies (Studies 1 and 2) due to adverse reactions, compared to 4% in the placebo group. The most common adverse reactions leading to discontinuation of INTUNIV -treated patients from the studies were somnolence/sedation (6%) and fatigue (2%). Less common adverse reactions leading to discontinuation (occurring in approximately 1% of patients) included: hypotension, headache, and dizziness. Adverse Reactions Leading to Discontinuation -®®
Adjunctive Trial
The most commonly observed adverse reactions (incidence ≥ 5% and at least twice the rate for placebo) in the adjunctive trial with INTUNIV were: somnolence, fatigue, insomnia, dizziness, and abdominal pain. Most Common Adverse Reactions -®
In the adjunctive clinical study, 3% of patients receiving INTUNIV discontinued due to adverse reactions, compared to 1% in the placebo group. Each adverse reaction leading to discontinuation occurred in less than 1% of INTUNIV -treated patients. Adverse Reactions Leading to Discontinuation –®®
Short Term Monotherapy Clinical Studies
- Two short-term, placebo-controlled, double-blind pivotal studies (Studies 1 and 2) were conducted in children and adolescents with ADHD, using fixed doses of INTUNIV (1 mg, 2 mg, 3 mg, and 4 mg/day). The most commonly reported adverse reactions (occurring in ≥ 2% of patients) that were considered drug-related and reported in a greater percentage of patients taking INTUNIV compared to patients taking placebo are shown in Table 2. Adverse reactions that were dose-related include: somnolence/sedation, abdominal pain, dizziness, hypotension, dry mouth and constipation. Common Adverse Reactions®®
Table 2: Percentage of Patients Experiencing Common (≥ 2%) Adverse Reactions in Short-Term
Monotherapy Studies 1 and 2Adverse Reaction Term All Doses of INTUNIV (N=513)
®
Placebo
(N=149)Somnolence a 38% 12% Headache 24% 19% Fatigue 14% 3% Abdominal pain b 11% 9% Hypotension c 7% 3% Nausea 6% 2% Lethargy 6% 3% Dizziness 6% 4% Irritability 6% 4% Decreased appetite 5% 3% Dry mouth 4% 1% Constipation 3% 1% a: The somnolence term includes somnolence, sedation, and hypersomnia. b: The abdominal pain term includes abdominal pain, abdominal pain upper, and abdominal pain lower. c: The hypotension term includes hypotension, orthostatic hypotension, and decreased blood pressure.
In an 8-week, placebo-controlled study in children 6-12 years of age with ADHD in which INTUNIV was dosed once (1-4 mg/day) in the morning or evening (Study 4), the safety profile was consistent with the once daily morning dosing of INTUNIV . ®®
Short Term Adjunctive Clinical Study
- A 8-week, placebo-controlled, double-blind, dose-optimized pivotal study (Study 3) was conducted in children and adolescents aged 6-17 years with a diagnosis of ADHD who were identified as having a sub-optimal response to psychostimulants. Patients received INTUNIV (1 mg, 2 mg, 3 mg, and 4 mg/day) or placebo, dosed in the morning or in the evening, in combination with their morning dose of psychostimulant. The most commonly reported adverse reactions (occurring in ≥ 2% of patients in the overall INTUNIV group) that were reported in a greater percentage of patients taking INTUNIV compared to patients taking placebo are shown in Table 3. Common Adverse Reactions®®®
Table 3: Percentage of Patients Experiencing Common (≥ 2%) Adverse Reactions in Short-Term Adjunctive Study 3
Adverse Reaction Term All Doses of INTUNIV ®
(N=302) aPlacebo (N=153)
Headache 21% 13% Somnolence b 18% 7% Insomnia c 12% 6% Fatigue 10% 3% Abdominal pain d 10% 3% Dizziness 8% 4% Decreased appetite 7% 4% Nausea 5% 3% Diarrhea 4% 1% Hypotension e 3% 0% Affect lability 2% 1% Bradycardia 2% 0% Constipation 2% 0% Dry mouth 2% 0% a: The morning and evening dose groups of INTUNIV are combined. b: The somnolence term includes somnolence, sedation, and hypersomnia. c: The insomnia term includes insomnia, initial insomnia, and middle insomnia. d: The abdominal pain term includes abdominal pain, abdominal pain upper, and abdominal pain lower. e: The hypotension term includes hypotension, orthostatic hypotension, and decreased blood pressure. ®
Effects on Blood Pressure and Heart Rate
In the monotherapy pediatric, short-term, controlled trials (Studies 1 and 2), the maximum mean changes from baseline in systolic blood pressure, diastolic blood pressure, and pulse were −5 mmHg, −3 mmHg, and −6 bpm, respectively, for all dose groups combined (generally one week after reaching target doses of 1 mg/day, 2 mg/day, 3 mg/day or 4 mg/day). These changes were dose dependent. Decreases in blood pressure and heart rate were usually modest and asymptomatic; however, hypotension and bradycardia can occur. Hypotension was reported as an adverse reaction for 7% of the INTUNIV group and 3% of the placebo group. This includes orthostatic hypotension, which was reported for 1% of the INTUNIV group and none in the placebo group. In the adjunctive trial, hypotension (3%) and bradycardia (2%) were observed in patients treated with INTUNIV as compared to none in the placebo group. In long-term, open label studies, (mean exposure of approximately 10 months), maximum decreases in systolic and diastolic blood pressure occurred in the first month of therapy. Decreases were less pronounced over time. Syncope occurred in 1% of pediatric subjects in the clinical program. The majority of these cases occurred in the long-term, open-label studies. ®®®
Other Adverse Reactions Observed in Clinical Studies
Table 4 includes additional adverse reactions observed in short-term, placebo-controlled and long-term, open-label clinical studies not included elsewhere in section 6.1, listed by organ system.
Table 4: Other adverse reactions observed in clinical studies
Body System
Adverse Reaction Cardiac
Atrioventricular block, sinus arrhythmia Gastrointestinal
Dyspepsia, stomach discomfort, vomiting General
Asthenia, chest pain Immune System Disorders
Hypersensitivity Investigations
Increased alanine amino transferase, increased weight Nervous system
Convulsion Psychiatric
Agitation, anxiety, depression, nightmare Renal
Increased urinary frequency, enuresis Respiratory
Asthma Vascular
Hypertension, pallor 6.2 Post-marketing Experience
The following adverse reactions have been identified during post-approval use of guanfacine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
An open-label post-marketing study involving 21,718 patients was conducted to assess the safety of guanfacine (as the hydrochloride) 1 mg/day given at bedtime for 28 days. Guanfacine was administered with or without other antihypertensive agents. Adverse events reported in the post-marketing study at an incidence greater than 1% included dry mouth, dizziness, somnolence, fatigue, headache and nausea. The most commonly reported adverse events in this study were the same as those observed in controlled clinical trials.
Less frequent, possibly guanfacine-related events observed in the post-marketing study and/or reported spontaneously, not included in , include: section 6.1
edema, malaise, tremor General:
palpitations, tachycardia Cardiovascular:
paresthesias, vertigo Central Nervous System:
blurred vision Eye Disorders:
arthralgia, leg cramps, leg pain, myalgia Musculo-Skeletal System:
confusion, hallucinations Psychiatric:
impotence Reproductive System, Male:
dyspnea Respiratory System:
alopecia, dermatitis, exfoliative dermatitis, pruritus, rash Skin and Appendages:
alterations in taste Special Senses:
-
7 DRUG INTERACTIONS
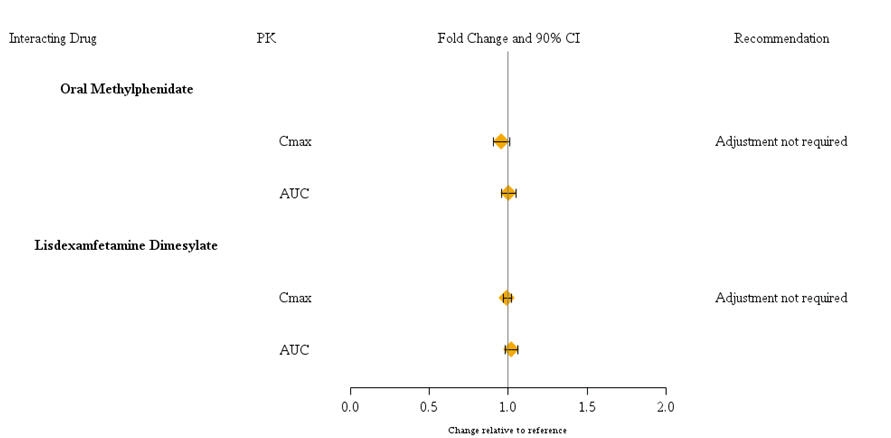
Guanfacine is primarily metabolized by CYP3A4 and its plasma concentrations can be affected significantly by CYP3A4 inhibitors or inducers (Figure 1). Dose adjustments are recommended . Guanfacine does not significantly affect exposures of methylphenidate and lisdexamfetamine when coadministered (Figure 2). Therefore, no dose adjustments in methylphenidate or lisdexamfetamine are necessary. [] see Dosage and Administration (2.7)
Figure 1: Impact of Other Drugs on the Pharmacokinetics (PK) of Intuniv
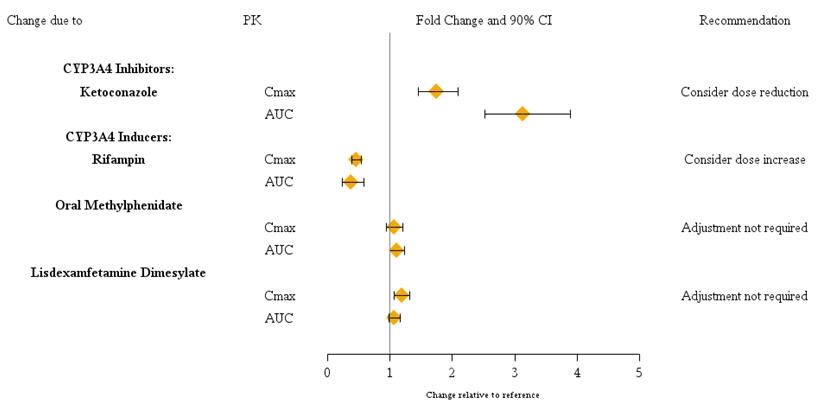
Figure 2: Impact of Intuniv on the Pharmacokinetics (PK) of Other Drugs
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category B
Risk Summary
There are no adequate and well-controlled studies of INTUNIV in pregnant women. No fetal harm was observed in rats and rabbits with administration of guanfacine at 6 and 4 times, respectively, the maximum recommended human dose. Because animal studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Animal data
Reproduction studies conducted in rats have shown that guanfacine crosses the placenta. However, administration of guanfacine to rats and rabbits at 6 and 4 times, respectively, the maximum recommended human dose of 4 mg/day on a mg/m basis resulted in no evidence of harm to the fetus. Higher doses (20 times the maximum recommended human dose in both rabbits and rats) were associated with reduced fetal survival and maternal toxicity. 2
8.3 Nursing Mothers
It is not known whether guanfacine is excreted in human milk; however, guanfacine is excreted in rat milk. Because many drugs are excreted in human milk, caution should be exercised when INTUNIV is administered to a nursing woman. Observe human milk-fed infants for sedation and somnolence. ®
8.4 Pediatric Use
Safety and efficacy of INTUNIV in pediatric patients less than 6 years of age have not been established. ®
Animal Data
In studies in juvenile rats, guanfacine alone produced a slight delay in sexual maturation in males and females at 2-3 times the maximum recommended human dose (MRHD). Guanfacine in combination with methylphenidate produced a slight delay in sexual maturation and decreased growth as measured by a decrease in bone length in males at a dose of guanfacine comparable to the MRHD and a dose of methylphenidate approximately 4 times the MRHD.
In a study where juvenile rats were treated with guanfacine alone from 7 to 59 days of age, development was delayed as indicated by a slight delay in sexual maturation and decreased body weight gain in males at 2 mg/kg/day and in females at 3 mg/kg/day. The No Adverse Effect Level (NOAEL) for delayed sexual maturation was 1 mg/kg/day, which is equivalent to the MRHD of 4 mg/day, on a mg/m basis. The effects on fertility were not evaluated in this study. 2
In a study where juvenile rats were treated with guanfacine in combination with methylphenidate from 7 to 59 days of age, a decrease in ulna bone length and a slight delay in sexual maturation were observed in males given 1 mg/kg/day of guanfacine in combination with 50 mg/kg/day of methylphenidate. The NOAELs for these findings were 0.3 mg/kg of guanfacine in combination with 16 mg/kg/day of methylphenidate, which are equivalent to 0.3 and 1.4 times the MRHD of 4 mg/day and 54 mg/day for guanfacine and methylphenidate, respectively, on a mg/m basis. These findings were not observed with guanfacine alone at 1 mg/kg/day or methylphenidate alone at 50 mg/kg/day. 2
8.5 Geriatric Use
The safety and efficacy of INTUNIV in geriatric patients have not been established. ®
8.6 Use in Patients with Renal or Hepatic Impairment
Renal Impairment
The impact of renal impairment on the pharmacokinetics of guanfacine in children was not assessed. In adult patients with impaired renal function, the cumulative urinary excretion of guanfacine and the renal clearance diminished as renal function decreased. In patients on hemodialysis, the dialysis clearance was about 15% of the total clearance. The low dialysis clearance suggests that the hepatic elimination (metabolism) increases as renal function decreases. It may be necessary to adjust the dose in patients with significant impairment of renal function.
Hepatic Impairment
The impact of hepatic impairment on PK of guanfacine in children was not assessed. Guanfacine in adults is cleared both by the liver and the kidney, and approximately 50% of the clearance of guanfacine is hepatic. It may be necessary to adjust the dose in patients with significant impairment of hepatic function.
- 9 DRUG ABUSE AND DEPENDENCE
-
10 OVERDOSAGE
Symptoms
Post-marketing reports of guanfacine overdosage indicate that hypotension, drowsiness, lethargy, and bradycardia have been observed following overdose. Initial hypertension may develop early and may be followed by hypotension. Similar symptoms have been described in voluntary reports to the American Association of Poison Control Center’s National Poison Data System. Miosis of the pupils may be noted on examination No fatal overdoses of guanfacine have been reported in published literature. .
Treatment
Consult a Certified Poison Control Center by calling 1-800-222-1222 for up to date guidance and advice.
Management of INTUNIV overdose should include monitoring for and the treatment of initial hypertension, if that occurs, as well as hypotension, bradycardia, lethargy and respiratory depression. Children and adolescents who develop lethargy should be observed for the development of more serious toxicity including coma, bradycardia and hypotension for up to 24 hours, due to the possibility of delayed onset hypotension. ®
-
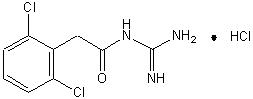
11 DESCRIPTION
INTUNIV is a once-daily, extended-release formulation of guanfacine hydrochloride (HCl) in a matrix tablet formulation for oral administration only. The chemical designation is N-amidino-2-(2,6-dichlorophenyl) acetamide monohydrochloride. The molecular formula is C H Cl N O·HCl corresponding to a molecular weight of 282.55. The chemical structure is: ®9923
Guanfacine HCl is a white to off-white crystalline powder, sparingly soluble in water (approximately 1 mg/mL) and alcohol and slightly soluble in acetone. The only organic solvent in which it has relatively high solubility is methanol (>30 mg/mL). Each tablet contains guanfacine HCl equivalent to 1 mg, 2 mg, 3 mg, or 4 mg of guanfacine base. The tablets also contain hypromellose, methacrylic acid copolymer, lactose, povidone, crospovidone, microcrystalline cellulose, fumaric acid, and glyceryl behenate. In addition, the 3mg and 4mg tablets also contain green pigment blend PB-1763.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Guanfacine is a central alpha -adrenergic receptor agonist. Guanfacine is not a central nervous system (CNS) stimulant. The mechanism of action of guanfacine in ADHD is not known. 2A
12.2 Pharmacodynamics
Guanfacine is a selective central alpha -adrenergic receptor agonist in that it has a 15-20 times higher affinity for this receptor subtype than for the alpha or alpha subtypes. 2A2B2C
Guanfacine is a known antihypertensive agent. By stimulating central alpha -adrenergic receptors, guanfacine reduces sympathetic nerve impulses from the vasomotor center to the heart and blood vessels. This results in a decrease in peripheral vascular resistance and a reduction in heart rate. 2A
Effects on Height, Weight, and Body Mass Index (BMI)
Patients taking INTUNIV demonstrated similar growth compared to normative data. Patients taking INTUNIV had a mean increase in weight of 0.5 kg (1 lb) compared to those receiving placebo over a comparative treatment period. Patients receiving INTUNIV for at least 12 months in open-label studies gained an average of 8 kg (17 lbs) in weight and 8 cm (3 in) in height. The height, weight, and BMI percentile remained stable in patients at 12 months in the long-term studies compared to when they began receiving INTUNIV . ®®®®
Effect on ECG
The effect of two dose levels of immediate-release guanfacine (4 mg and 8 mg) on the QT interval was evaluated in a double-blind, randomized, placebo- and active-controlled, cross-over study in healthy adults. A dose-dependent decrease in heart rate was observed during the first 12 hours, at time of maximal concentrations. The mean change in heart rate was -13 bpm at 4 mg and -22 bpm at 8 mg. An apparent increase in mean QTc was observed for both doses. However, guanfacine does not appear to interfere with cardiac repolarization of the form associated with pro-arrhythmic drugs. This finding has no known clinical relevance.
12.3 Pharmacokinetics
Absorption and Distribution
Guanfacine is readily absorbed and approximately 70% bound to plasma proteins independent of drug concentration. After oral administration of INTUNIV the time to peak plasma concentration is approximately 5 hours in children and adolescents with ADHD. ®
Immediate-release guanfacine and INTUNIV have different pharmacokinetic characteristics; dose substitution on a milligram for milligram basis will result in differences in exposure. ®
A comparison across studies suggests that the C is 60% lower and AUC 43% lower, respectively, for INTUNIV compared to immediate-release guanfacine. Therefore, the relative bioavailability of INTUNIV to immediate-release guanfacine is 58%. The mean pharmacokinetic parameters in adults following the administration of INTUNIV 1 mg once daily and immediate-release guanfacine 1mg once daily are summarized in Table 5. max0-∞®®®
Table 5: Pharmacokinetic Parameters in Adults Parameter INTUNIV ®
1 mg once daily
(n=52)Immediate-release
guanfacine
1 mg once daily
(n=12)C (ng/mL) max
1.0 ± 0.3 2.5 ± 0.6 AUC (ng.h/mL) 0-∞
32 ± 9 56 ± 15 t (h) max
6.0 (4.0 - 8.0) 3.0 (1.5-4.0) t (h) 1/2
18 ± 4 16 ± 3 Note: Values are mean +/- SD, except for t which is median (range) max
Exposure to guanfacine was higher in children (ages 6-12) compared to adolescents (ages 13-17) and adults. After oral administration of multiple doses of INTUNIV 4 mg, the C was 10 ng/mL compared to 7 ng/mL and the AUC was 162 ng h/mL compared to 116 ng h/mL in children (ages 6-12) and adolescents (ages 13-17), respectively. These differences are probably attributable to the lower body weight of children compared to adolescents and adults. ®max
The pharmacokinetics were affected by intake of food when a single dose of INTUNIV 4 mg was administered with a high-fat breakfast. The mean exposure increased (C ~75% and AUC ~40%) compared to dosing in a fasted state. ®max
Dose Proportionality
Following administration of INTUNIV in single doses of 1 mg, 2 mg, 3 mg, and 4 mg to adults, C and AUC of guanfacine were proportional to dose. ®max0-∞
Metabolism and Elimination
In vitro studies with human liver microsomes and recombinant CYP’s demonstrated that guanfacine was primarily metabolized by CYP3A4. In pooled human hepatic microsomes, guanfacine did not inhibit the activities of the major cytochrome P450 isoenzymes (CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 or CYP3A4/5). Guanfacine is a substrate of CYP3A4/5 and exposure is affected by CYP3A4/5 inducers/inhibitors.
Renal and Hepatic Impairment
The impact of renal impairment on PK of guanfacine in children was not assessed . [] see Use in Specific Populations (8.6)
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis No carcinogenic effect of guanfacine was observed in studies of 78 weeks in mice or 102 weeks in rats at doses up to 6-7 times the maximum recommended human dose of 4 mg/day on a mg/ m basis.
2Mutagenesis Guanfacine was not genotoxic in a variety of test models, including the Ames test and an chromosomal aberration test; however, a marginal increase in numerical aberrations (polyploidy) was observed in the latter study.
in vitroImpairment of Fertility No adverse effects were observed in fertility studies in male and female rats at doses up to 30 times the maximum recommended human dose on a mg/ m basis.
2 -
14 CLINICAL STUDIES
14.1 Safety and Efficacy Studies
The efficacy of INTUNIV in the treatment of ADHD was established in 3 placebo-controlled monotherapy trials (Studies 1, 2, and 4) and in 1 placebo-controlled adjunctive trial with psychostimulants (Study 3) in pediatric population. Studies 1, 2, and 3 were conducted in children and adolescents ages 6-17 and Study 4 was conducted in children ages 6-12 years. ®
Studies 1 and 2: Fixed-dose INTUNIV Monotherapy ®
Study 1 was a double-blind, placebo-controlled, parallel-group, fixed dose study, in which efficacy of once daily dosing with INTUNIV (2 mg, 3 mg and 4 mg) was evaluated for 5 weeks (n=345). Study 2 was a double-blind, placebo-controlled, parallel-group, fixed-dose study, in which efficacy of once daily dosing with INTUNIV (1 mg, 2 mg, 3 mg and 4 mg) was evaluated for 6 weeks (n=324). In both studies, randomized subjects in 2 mg, 3 mg and 4 mg dose groups were titrated to their target fixed dose, and continued on the same dose until a dose tapering phase started The lowest dose of 1 mg used in Study 2 was assigned only to patients less than 50 kg (110 lbs). Patients who weighed less than 25 kg (55 lbs) were not included in either study. ®®
Signs and symptoms of ADHD were evaluated on a once weekly basis using the clinician administered and scored ADHD Rating Scale (ADHD-RS-IV), which includes both hyperactive/impulsive and inattentive subscales. The primary efficacy outcome was the change from baseline to endpoint in ADHD-RS-IV total scores. Endpoint was defined as the last post-randomization treatment week for which a valid score was obtained prior to dose tapering (up to Week 5 in Study 1 and up to Week 6 in Study 2).
The mean reductions in ADHD-RS-IV total scores at endpoint were statistically significantly greater for INTUNIV compared to placebo for Studies 1 and 2. Placebo-adjusted changes from baseline were statistically significant for each of the 2 mg, 3 mg, and 4 mg INTUNIV randomized treatment groups in both studies, as well as the 1 mg INTUNIV treatment group (for patients 55-110 lbs) that was included only in Study 2 (see Table 6). ®®®
Dose-responsive efficacy was evident, particularly when data were examined on a weight-adjusted (mg/kg) basis. When evaluated over the dose range of 0.01-0.17 mg/kg/day, clinically relevant improvements were observed beginning at doses in the range 0.05-0.08 mg/kg/day. Doses up to 0.12 mg/kg/day were shown to provide additional benefit.
Controlled, monotherapy long-term efficacy studies (>9 weeks) have not been conducted.
In the monotherapy trials (Studies 1 and 2), subgroup analyses were performed to identify any differences in response based on gender or age (6-12 vs. 13-17). Analyses of the primary outcome did not suggest any differential responsiveness on the basis of gender. Analyses by age revealed a statistically significant treatment effect only in the 6-12 age subgroup. Due to the relatively small proportion of adolescent patients (ages 13-17) enrolled into these studies (approximately 25%), these data may not be sufficient to demonstrate efficacy in the adolescent patients. In these studies, patients were randomized to a fixed dose of INTUNIV rather than optimized by body weight. Therefore, some adolescent patients were randomized to a dose that might have resulted in relatively lower plasma guanfacine concentrations compared to the younger patients. Over half (55%) of the adolescent patients received doses of 0.01-0.04mg/kg. In studies in which systematic pharmacokinetic data were obtained, there was a strong inverse correlation between body weight and plasma guanfacine concentrations. ®
Table 6: Fixed dose Studies
Study
(Age Range)
Primary Efficacy Measure
Treatment Group Placebo Intuniv® 1mg
Intuniv® 2mg
Intuniv® 3mg
Intuniv® 4mg
(6 – 17 years) 1
Mean Baseline (SD)
38.1 (9.34)
-- 36.1 (9.99) 36.8 (8.72) 38.4 (9.21) LS Mean Change from Baseline (SE)
-8.5 (1.42)
-- -15.9 (1.37) -16.0 (1.38) -18.5 (1.39) LS Mean Difference from Placebo (95% CI)
-- -- -7.4 (-11.3, -3.5) a
-7.5 (-11.4, -3.6) a
-10.0 (-13.9, -6.1) a
(6 – 17 years) 2
Mean Baseline (SD)
39.3 (8.85)
41.7 (7.81)
39.9 (8.74)
39.1 (9.22)
40.6 (8.57)
LS Mean Change from Baseline (SE)
-12.7 (1.60)
-19.4 (1.69) -18.1 (1.60)
-20.0 (1.64) -20.6 (1.60) LS Mean Difference from Placebo (95% CI)
-- -6.8 (-11.3, -2.2) a
-5.4 (-9.9, -0.9) a
-7.3 (-11.8, -2.8) a
-7.9 (-12.3, -3.4) a
LS Mean: least-square mean; SD: standard deviation; SE: standard error; 95% CI (unadjusted) Doses were shown to be statistically significantly superior to placebo.
a
Study 3: Flexible-dose INTUNIV as Adjunctive Therapy to Psychostimulants ®Study 3 was a double-blind, randomized, placebo-controlled, dose-optimization study, in which efficacy of once daily optimized dosing (morning or evening) with INTUNIV (1mg, 2mg, 3mg and 4mg), when co-administered with psychostimulants, was evaluated for 8 weeks, in children and adolescents aged 6-17 years with a diagnosis of ADHD, with a sub-optimal response to stimulants (n=455). Subjects were started at the 1 mg INTUNIV dose level and were titrated weekly over a 5-week dose-optimization period to an optimal INTUNIV dose not to exceed 4 mg/day based on tolerability and clinical response. The dose was then maintained for a 3-week dose maintenance period before entry to 1 week of dose tapering. Subjects took INTUNIV either in the morning or the evening while maintaining their current dose of psychostimulant treatment given each morning. Allowable psychostimulants in the study were ADDERALL XR , VYVANSE , CONCERTA , FOCALIN XR , RITALIN LA , METADATE CD or FDA-approved generic equivalents. ®®®®®®®®®®
Symptoms of ADHD were evaluated on a weekly basis by clinicians using the ADHD Rating Scale (ADHD-RS-IV), which includes both hyperactive/impulsive and inattentive subscales. The primary efficacy outcome was the change from baseline to endpoint in ADHD-RS-IV total scores. Endpoint was defined as the last post-randomization treatment week prior to dose tapering for which a valid score was obtained (up to Week 8).
Mean reductions in ADHD-RS-IV total scores at endpoint were statistically significantly greater for INTUNIV given in combination with a psychostimulant compared to placebo given with a psychostimulant for Study 3, for both morning and evening INTUNIV dosing (see Table 7). Nearly two-thirds (64.2%) of subjects reached optimal doses in the 0.05-0.12 mg/kg/day range. ®®
Study 4: Flexible-dose INTUNIV Monotherapy ®
Study 4 was a double-blind, randomized, placebo-controlled, dose-optimization study, in which efficacy of once daily dosing (morning or evening) with INTUNIV (1mg, 2mg, 3mg, and 4mg) was evaluated for 8 weeks in children aged 6-12 years (n=340). ®
Signs and symptoms of ADHD were evaluated on a once weekly basis using the clinician administered and scored ADHD Rating Scale (ADHD-RS-IV), which includes both hyperactive/impulsive and inattentive subscales. The primary efficacy outcome was the change from baseline score at endpoint on the ADHD-RS-IV total scores. Endpoint was defined as the last post-randomization treatment week for which a valid score was obtained prior to dose tapering (up to Week 8).
Mean reductions in ADHD-RS-IV total scores at endpoint were statistically significantly greater for INTUNIV compared to placebo in both AM and PM dosing groups of INTUNIV (see Table 7). ®®
Table 7: Flexible-Dose studiesStudy (Age Range)
Treatment Group Placebo Intuniv®1mg – 4mg AM
PM
(6 – 17 years) 3 a
Mean Baseline (SD)
37.7 (7.75)
37.6 (8.13)
37.0 (7.65)
LS Mean Change from Baseline (SE)
-15.9 (0.96)
-20.3 (0.97)
-21.2 (0.97)
LS Mean Difference from Placebo (95% CI)
-- -4.5 (-7.5, -1.4) b
-5.3 (-8.3, -2.3) b
(6 – 12 years) 4
Mean Baseline (SD)
42.9 (6.21)
41.7 (6.39)
41.6 (6.66)
LS Mean Change from Baseline (SE)
-10.6 (1.20)
-20.0 (1.23)
-20.4 (1.19)
LS Mean Difference from Placebo (95% CI)
-- -9.4 (-12.8, -6.0) b
-9.8 (-13.1, -6.4) b
LS Mean: least-square mean; SD: standard deviation; SE: standard error; 95% CI (unadjusted) Treatment was given in combination with a psychostimulant. Doses were shown to be statistically significantly superior to placebo.
a
b - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information)
Dosing and Administration
Instruct patients to swallow INTUNIV whole with water, milk or other liquid. Patients should not take INTUNIV together with a high-fat meal, since this can raise blood levels of INTUNIV . Instruct the parent or caregiver to supervise the child or adolescent taking INTUNIV and to keep the bottle of tablets out of reach of children. ®Tablets should not be crushed, chewed or broken prior to administration because this may increase the rate of release of the active drug .®®®
Instruct patients on how to properly taper the medication, if the physician decides to discontinue treatment. [ ]. see Dosage and Administration (2.5)
Adverse Reactions
Advise patients that sedation can occur, particularly early in treatment or with dose increases. Caution against operating heavy equipment or driving until they know how they respond to treatment with INTUNIV Headache and abdominal pain can also occur. If any of these symptoms persist, or other symptoms occur, the patient should be advised to discuss the symptoms with the physician. ®[ ]. see Warnings and Precautions (5.2)
Advise patients to avoid becoming dehydrated or overheated, which may potentially increase the risks of hypotension and syncope Advise patients to avoid use with alcohol. [ ]. see Warnings and Precautions (5.1)
-
PATIENT PACKAGE INSERT
Patient Information INTUNIV (
®in-TOO-niv)
(guanfacine)
Extended-Release Tablets
Read the Patient Information that comes with INTUNIV before you start taking it and each time you get a refill. There may be new information. ®This leaflet does not take the place of talking with your doctor about your medical condition or your treatment.
What is INTUNIV ? ®
INTUNIV is a prescription medicine used to treat the symptoms of attention deficit/hyperactivity disorder (ADHD). ®
INTUNIV is not a central nervous system (CNS) stimulant. ®
What should I tell my doctor before taking INTUNIV ? ®
Before you take INTUNIV , tell your doctor if you: ®
- have heart problems or a low heart rate
- have fainted
- have low blood pressure
- have liver or kidney problems
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if INTUNIV will harm your unborn baby. Talk to your doctor if you are pregnant or plan to become pregnant.
®
- are breast-feeding or plan to breast-feed. It is not known if INTUNIV passes into your breast milk. Talk to your doctor about the best way to feed your baby while taking INTUNIV . ®®
Tell your doctor about all of the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
INTUNIV may affect the way other medicines work, and other medicines may affect how INTUNIV works. ®®
Especially tell your doctor if you take:
- ketoconazole
- medicines that can affect enzyme metabolism
- high blood pressure medicine
- sedatives
- benzodiazepines
- barbiturates
- antipsychotics
Ask your doctor or pharmacist for a list of these medicines, if you are not sure.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take INTUNIV ? ®
- Take INTUNIV exactly as your doctor tells you.
®
- Your doctor may change your dose. Do not change your dose of INTUNIV without talking to your doctor.
®
- Do not stop taking INTUNIV without talking to your doctor.
®
- INTUNIV should be taken 1 time a day in the morning or in the evening, either alone or in combination with an ADHD stimulant medication that your doctor may prescribe. Your doctor will tell you when to take INTUNIV and when to take your ADHD stimulant medication.
®®
- INTUNIV should be swallowed whole with a small amount of water, milk, or other liquid.
®
- Do not crush, chew, or break INTUNIV . Tell your doctor if you can not swallow INTUNIV whole.
®®
- Do not take INTUNIV with a high-fat meal.
®
- Your doctor will check your blood pressure and heart rate while you take INTUNIV .
®
- If you take too much INTUNIV , call your local Poison Control Center or go to the nearest emergency room right away. ®
What should I avoid while taking INTUNIV ? ®
- Do not drive, operate heavy machinery, or do other dangerous activities until you know how INTUNIV affects you. INTUNIV can slow your thinking and motor skills.
®®
- Do not drink alcohol or take other medicines that make you sleepy or dizzy while taking INTUNIV until you talk with your doctor. INTUNIV taken with alcohol or medicines that cause sleepiness or dizziness may make your sleepiness or dizziness worse. ®®
What are the possible side effects of INTUNIV ? ®
INTUNIV may cause serious side effects including: ®
- low blood pressure
- low heart rate
- fainting
- sleepiness
Get medical help right away, if you have any of the symptoms listed above.
The most common side effects of INTUNIV include: ®- sleepiness
- tiredness
- trouble sleeping
- low blood pressure
- nausea
- stomach pain
- dizziness
Tell the doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of INTUNIV . For more information, ask your doctor or pharmacist. ®
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store INTUNIV ? ®
- Store INTUNIV between 59 F to 86 F (15 C to 30 C) ®oooo
Keep INTUNIV and all medicines out of the reach of children. ®
General Information about INTUNIV ®
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information Leaflet. Do not use INTUNIV for a condition for which it was not prescribed. Do not give INTUNIV to other people, even if they have the same symptoms that you have. It may harm them. ®®
This leaflet summarizes the most important information about INTUNIV . If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about INTUNIV that is written for health professionals. For more information, go to ®®
www.INTUNIV.comor call 1-800-828-2088.What are the ingredients in INTUNIV ? ®
guanfacine hydrochloride Active ingredient:
hypromellose, methacrylic acid copolymer, lactose, povidone, crospovidone, microcrystalline cellulose, fumaric acid, and glycerol behenate. In addition, the 3mg and 4mg tablets also contain green pigment blend PB-1763. Inactive ingredients:
Manufactured for Shire US Inc., Wayne, PA 19087. INTUNIV is a registered trademark of Shire LLC. ©2013 Shire US Inc. This product is covered by US patents including 6,287,599; 6,811,794. Version: 08 2013
®
- have heart problems or a low heart rate
- INTUNIV (GUANFACINE) TABLET, EXTENDED RELEASE INTUNIV (GUANFACINE) KIT
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
INTUNIV
guanfacine tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64725-0515(NDC:54092-515) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GUANFACINE HYDROCHLORIDE (UNII: PML56A160O) (GUANFACINE - UNII:30OMY4G3MK) GUANFACINE 2 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSE 2208 (15000 MPA.S) (UNII: Z78RG6M2N2) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) POVIDONES (UNII: FZ989GH94E) CROSPOVIDONE (UNII: 68401960MK) METHACRYLIC ACID - METHYL METHACRYLATE COPOLYMER (1:1) (UNII: 74G4R6TH13) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) FUMARIC ACID (UNII: 88XHZ13131) Product Characteristics Color WHITE (white) Score no score Shape OVAL Size 12mm Flavor Imprint Code 503;2mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64725-0515-1 30 in 1 CONTAINER Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022037 09/02/2009 INTUNIV
guanfacine tablet, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64725-0513(NDC:54092-513) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GUANFACINE HYDROCHLORIDE (UNII: PML56A160O) (GUANFACINE - UNII:30OMY4G3MK) GUANFACINE 1 mg Inactive Ingredients Ingredient Name Strength HYPROMELLOSE 2208 (15000 MPA.S) (UNII: Z78RG6M2N2) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) POVIDONES (UNII: FZ989GH94E) CROSPOVIDONE (UNII: 68401960MK) METHACRYLIC ACID - METHYL METHACRYLATE COPOLYMER (1:1) (UNII: 74G4R6TH13) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) FUMARIC ACID (UNII: 88XHZ13131) Product Characteristics Color WHITE (white) Score no score Shape ROUND Size 7mm Flavor Imprint Code 503;1mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64725-0513-1 100 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022037 09/02/2009 Labeler - TYA Pharmaceuticals (938389038) Registrant - TYA Pharmaceuticals (938389038) Establishment Name Address ID/FEI Business Operations TYA Pharmaceuticals 938389038 RELABEL(64725-0515, 64725-0513) , REPACK(64725-0515)


Trademark Results [Intuniv]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 INTUNIV 78649257 not registered Dead/Abandoned |
Shire LLC 2005-06-13 |
 INTUNIV 77754292 3877676 Dead/Cancelled |
Shire LLC 2009-06-08 |
 INTUNIV 77754273 3877675 Dead/Cancelled |
Shire LLC 2009-06-08 |
 INTUNIV 77674806 3871096 Live/Registered |
Shire LLC 2009-02-20 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.