VARIZIG (varicella zoster immune globulin- human solution
VARIZIG by
Drug Labeling and Warnings
VARIZIG by is a Prescription medication manufactured, distributed, or labeled by Saol Therapeutics. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VARIZIG® safely and effectively. See full prescribing information for VARIZIG.
VARIZIG® [Varicella Zoster Immune Globulin (Human)]
for intramuscular administration only.
Sterile Solution for Injection
Initial U.S. Approval: 2012
INDICATIONS AND USAGE
VARIZIG is a Varicella Zoster Immune Globulin (Human) indicated for post-exposure prophylaxis in high risk individuals (1). High risk groups include:
- immunocompromised children and adults,
- newborns of mothers with varicella shortly before or after delivery,
- premature infants,
- infants less than one year of age,
- adults without evidence of immunity,
- pregnant women.
VARIZIG administration is intended to reduce the severity of varicella.
DOSAGE AND ADMINISTRATION
Intramuscular use only.
Dosing of VARIZIG is based on body weight. Administer a single dose of VARIZIG intramuscularly as recommended in the following table (2.1):
Weight of Patient (kg) Dose (IU) Number of Vials ≤2.0 62.5 0.5 2.1–10.0 125 1 10.1–20.0 250 2 20.1–30.0 375 3 30.1–40.0 500 4 >40.1 625 5 Discard any partial vials.
The intramuscular dose should be divided and administered in two sites, dependent on patient size. Do not exceed 3 mL per injection site (2.2).
DOSAGE FORMS AND STRENGTHS
VARIZIG is supplied as a sterile solution for intramuscular injection and is available in a single-use vial of 125 IU in 1.2 mL (3).
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions from clinical trials are pain at the injection site (3%) and headache (2%) (6).
To report SUSPECTED ADVERSE REACTIONS, contact Saol Therapeutics at 1-833-644-4216 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Efficacy of live attenuated virus vaccines may be impaired by immune globulin administration; revaccination may be necessary (7).
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Preparation and Handling
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Events
5.2 Coagulation Disorders
5.3 Hypersensitivity
5.4 Transmissible Infectious Agents
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Immunocompromised Patients
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
14 CLINICAL STUDIES
14.1 Pregnant Women Exposed to Varicella Zoster Virus
14.2 High Risk Patients Exposed to Varicella Zoster Virus
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VARIZIG® [Varicella Zoster Immune Globulin (Human)] is indicated for post-exposure prophylaxis of varicella in high risk individuals. High risk groups include:
- immunocompromised children and adults,
- newborns of mothers with varicella shortly before or after delivery,
- premature infants,
- neonates and infants less than one year of age,
- adults without evidence of immunity,
- pregnant women.
VARIZIG administration is intended to reduce the severity of varicella. Administer VARIZIG as soon as possible following varicella zoster virus (VZV) exposure, ideally within 96 hours for greatest effectiveness.
- There is no convincing evidence that VARIZIG reduces the incidence of chickenpox infection after exposure to VZV.
- There is no convincing evidence that established infections with VZV can be modified by VARIZIG administration.
- There is no indication for the prophylactic use of VARIZIG in immunodeficient children or adults when there is a past history of varicella, unless the patient is undergoing bone marrow transplantation.
-
2 DOSAGE AND ADMINISTRATION
For intramuscular use only.
2.1 Preparation and Handling
Each vial of VARIZIG contains a minimum potency of 125 IU in 1.2 mL.
Bring VARIZIG to room temperature prior to use.
Inspect VARIZIG for particulate matter and discoloration prior to administration. Do not use if the solution is cloudy or contains particulates.
VARIZIG is for single use only. Discard any unused portion.
Dosing of VARIZIG is based on body weight. Administer a single dose of VARIZIG intramuscularly as recommended in Table 1.
The minimum dose is 62.5 International Units (IU) for small infants under two kilograms body weight; the maximum dose of 625 IU should be administered for all patients greater than 40 kilograms in weight.
Table 1 VARIZIG Dose and Volume of Administration *Extractable volumes are confirmed using a 21 gauge needle as per USP General Chapters <1> Injections.
Weight of Patient VARIZIG Dose Volume to Administer* (milliliters) Kilograms Pounds IU Number of Vials ≤2.0 ≤4.4 62.5 0.5 0.6 2.1–10.0 4.5–22.0 125 1 1.2 10.1–20.0 22.1–44.0 250 2 2.4 20.1–30.0 44.1–66.0 375 3 3.6 30.1–40.0 66.1–88.0 500 4 4.8 ≥40.1 ≥88.1 625 5 6.0 Consider a second full dose of VARIZIG for high risk patients who have additional exposures to varicella greater than three weeks after initial VARIZIG administration.
2.2 Administration
For intramuscular use only.
Divide the intramuscular dose and administer in two or more injection sites, depending on patient size. Do not exceed 3 milliliters per injection site.
Inject into the deltoid muscle or the anterolateral aspects of the upper thigh. Due to the risk of sciatic nerve injury, do not use the gluteal region as a routine injection site. If the gluteal region is used, only use the upper, outer quadrant.
To prevent the transmission of infectious agents from one person to another, use a new disposable sterile syringe and needle for each individual patient.
-
3 DOSAGE FORMS AND STRENGTHS
VARIZIG is supplied as a sterile solution for intramuscular injection and is available in a single-use vial of 125 IU. Each 125 IU vial of VARIZIG contains less than 156 milligrams of total protein, mostly human immune globulin G (IgG). VARIZIG contains no preservative and is intended for single use only. VARIZIG does not contain mercury.
-
4 CONTRAINDICATIONS
- Individuals known to have anaphylactic or severe systemic (hypersensitivity) reactions to human immune globulin preparations should not receive VARIZIG.
- IgA-deficient patients with antibodies against IgA and a history of hypersensitivity may have an anaphylactoid reaction.
- VARIZIG contains less than 40 micrograms per milliliter of IgA.
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Events
Thrombotic events may occur during or following treatment with immune globulin products (1, 2, 3). Patients at risk include those with a history of atherosclerosis, multiple cardiovascular risk factors, advanced age, impaired cardiac output, coagulation disorders, prolonged periods of immobilization, and/or known/suspected hyperviscosity. Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies.
5.2 Coagulation Disorders
Administer VARIZIG intramuscularly only. In patients who have severe thrombocytopenia or any coagulation disorder that would contraindicate intramuscular injections, only administer VARIZIG if the expected benefits outweigh the potential risks.
5.3 Hypersensitivity
Severe hypersensitivity reactions may occur following VARIZIG administration. Administer VARIZIG in a setting with appropriate equipment, medication and personnel trained in the management of hypersensitivity, anaphylaxis and shock. In the case of hypersensitivity, discontinue administration of VARIZIG immediately and provide appropriate treatment.
VARIZIG contains trace amounts of IgA (less than 40 micrograms per milliliter). Patients with known antibodies to IgA have a greater risk of severe hypersensitivity and anaphylactic reactions. VARIZIG is contraindicated in IgA deficient patients with antibodies against IgA and history of hypersensitivity reactions [see 4 CONTRAINDICATIONS].
5.4 Transmissible Infectious Agents
Because VARIZIG is made from human plasma, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. The plasma donors are screened for the presence of certain infectious agents and the manufacturing process for VARIZIG includes measures to inactivate and remove certain viruses [see 11 DESCRIPTION]. Despite these measures, products derived from human plasma can still potentially transmit diseases. No cases of transmission of viral diseases, vCJD or CJD have been associated with the use of VARIZIG.
Report all infections thought by a physician to have been transmitted by VARIZIG to Saol Therapeutics at 1-833-644-4216. Discuss the risks and benefits of this product with the patient before administering it to the patient [see 17 PATIENT COUNSELING INFORMATION].
-
6 ADVERSE REACTIONS
The most serious adverse drug reactions observed in clinical trials for all subjects and patients (n=601) include pyrexia, nausea, and vomiting.
The most common adverse drug reactions (reported by ≥ 1% of subjects) observed in clinical trials for all subjects and patients (n=601) are the following:
- injection site pain (3%),
- headache (2%),
- rash (including terms pruritus, rash, rash erythematous, rash vesicular and urticaria) (1%),
- fatigue (1%),
- chills (1%),
- nausea (1%).
All other adverse drug reactions occurred in less than 1%.
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Six hundred and one (n=601) high risk individuals received VARIZIG intramuscularly in two clinical trials which included pregnant women, infants and immunocompromised pediatric and adult patients. The highest incidence of adverse reactions occurred in pregnant women (n=166), including injection site pain (7%), rash (including terms pruritus, rash, rash erythematous, and rash vesicular) (4%), headache (3%), and fatigue (2%). All other adverse reactions occurred in 1% or less of clinical trial subjects within each high risk group. A single incidence of serum sickness (approximately one in 600 patients treated with VARIZIG) was observed in an immunocompromised adolescent patient.
There were eight reported adverse events associated with the coagulation system including, deep vein thrombosis (n=1), disseminated intravascular coagulation (n=1) , intracranial hemorrhage (n=2), coagulopathy (n=2), intraventricular hemorrhage (n=1), and pulmonary hemorrhage (n=1) in 621 subjects in the open-label, Expanded Access Protocol (EAP); the study was not designed to differentiate between adverse events attributed to the underlying medical condition and adverse reactions to VARIZIG.
-
7 DRUG INTERACTIONS
The passive transfer of antibodies with immune globulin administration may impair the efficacy of live attenuated virus vaccines such as measles, rubella, mumps and varicella. Defer vaccination with live virus vaccines until approximately three months after VARIZIG administration. Inform the immunizing physician of recent therapy with VARIZIG so that appropriate measures can be taken [see 17 PATIENT COUNSELING INFORMATION].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy category C. Animal reproduction studies have not been conducted with VARIZIG. It also is not known whether VARIZIG can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. VARIZIG should be given to a pregnant woman only if clearly needed.
The safety and effectiveness of VARIZIG have been evaluated for post-exposure prophylaxis in clinical trials in 166 pregnant women [see 6 ADVERSE REACTIONS and 14 CLINICAL STUDIES].
8.3 Nursing Mothers
It is not known whether VARIZIG is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when VARIZIG is administered to a nursing mother.
8.4 Pediatric Use
The dosing recommendations in the treatment of pediatric patients are by body weight [see 2 DOSAGE AND ADMINISTRATION].
The safety and effectiveness of VARIZIG have been evaluated for post-exposure prophylaxis in the VARIZIG expanded access clinical trial in 374 pediatric patients, including immunocompromised pediatric patients:
- 94 preterm newborns and infants,
- 53 term newborns,
- 45 infants and toddlers,
- 176 children and,
- 43 adolescents.
In the EAP, follow up data were available for 110 VARIZIG treatments in infants (including newborns, pre-term infants, and infants <1 year old). Three severe infections were reported, all three with pox count >100, one of which also had pneumonia and another one also developed probable varicella encephalitis.
8.5 Geriatric Use
Clinical studies of VARIZIG administered intramuscularly for post-exposure prophylaxis did not include sufficient numbers of geriatric subjects (aged 65 and over) to determine whether they respond differently from younger subjects.
Use caution when administering VARIZIG to patients age 65 and over who are judged to be at increased risk of thrombotic events [see 5 WARNINGS AND PRECAUTIONS]. Do not exceed recommended doses and administer VARIZIG intramuscularly only.
8.6 Immunocompromised Patients
In the EAP, both adult (n=37) and pediatric immunocompromised subjects (n=235) were treated. Twelve immunocompromised subjects developed clinical varicella and none developed varicella pneumonitis; however at least five are reported to have received concomitant acyclovir and due to incomplete reporting, it is not known if others also received acyclovir.
- 10 OVERDOSAGE
-
11 DESCRIPTION
VARIZIG [Varicella Zoster Immune Globulin (Human)] is a solvent/detergent-treated sterile liquid preparation of purified human immune globulin G (IgG) containing antibodies to varicella zoster virus (anti-VZV). VZV is the causative agent of chickenpox. VARIZIG is prepared from plasma donated by healthy, screened donors with high titers of antibodies to VZV, which is purified by an anion-exchange column chromatography manufacturing method. This donor selection process includes donors with high anti-VZV titers due to recent natural infection by VZV, or due to recurrent zoster infection (shingles).
VARIZIG is intended for single use and should be administered intramuscularly [see 2 DOSAGE AND ADMINISTRATION].
The product potency is expressed in IU by comparison to the World Health Organization (WHO) international reference preparation for anti-VZV immune globulin. Each vial contains 125 IU of anti-VZV. VARIZIG is formulated with 10% maltose and 0.03% polysorbate 80. VARIZIG has a pH of 5.0 – 6.5 and contains no preservative.
The presence of anti-Protein S antibodies has been reported to arise transiently in patients after VZV infection (4). Low levels of anti-Protein S antibodies have been reported in VARIZIG.
The source plasma used in the manufacture of this product was tested by FDA licensed nucleic acid testing (NAT) for human immunodeficiency virus-1 (HIV-1), hepatitis B virus (HBV) and hepatitis C virus (HCV) and found to be negative. Plasma also was tested by in-process NAT for hepatitis A virus (HAV) and parvovirus B19 (B19) via minipool testing; the limit for B19 in the manufacturing pool is set not to exceed 104 IU of B19 DNA per milliliter.
The manufacturing process contains two steps implemented specifically for virus clearance. The solvent/detergent step (using tri-n-butyl phosphate and Triton® X-100) is effective in the inactivation of enveloped viruses, such as HBV, HCV and HIV-1. Virus filtration, using a Planova® 20N virus filter, is effective for the removal of viruses based on their size, including some non-enveloped viruses. These two viral clearance steps are designed to increase product safety by reducing the risk of transmission of enveloped and non-enveloped viruses. In addition to these two specific steps, the process step of anion-exchange chromatography was identified as contributing to the overall viral clearance capacity for small non-enveloped viruses.
The inactivation and reduction of known enveloped and non-enveloped model viruses were validated in laboratory studies as summarized in Table 2. The viruses employed for spiking studies were selected to represent those viruses that are potential contaminants in the product, and to represent a wide range of physiochemical properties in order to challenge the manufacturing process’s ability for viral clearance in general.
Table 2 Virus Reduction Values (Log10) Obtained through Validation Studies *The PRV was retained by the 0.1 µm pre-filter during the virus validation. Since manufacturing employs a 0.1 µm pre-filter before the 20N filter, the claim of ≥5.6 reduction is considered applicable.
Abbreviations:
HIV-1: human immunodeficiency virus-1; relevant virus for human immunodeficiency virus-1 and model for HIV-2
BVDV: bovine viral diarrhea virus; model virus for hepatitis C virus (HCV) and West Nile virus (WNV)
PRV: pseudorabies virus; model for large enveloped DNA viruses, including herpes
HAV: human hepatitis A virus; relevant virus for HAV and model for small non-enveloped viruses in general
EMC: encephalomyocarditis virus; model for HAV and for small non-enveloped viruses in general
MMV: murine minute virus; model for human parvovirus B19 and for small non-enveloped viruses in general
n.e.: not evaluated
Enveloped Non-Enveloped Genome RNA DNA RNA DNA Virus HIV-1 BVDV PRV HAV EMC MMV PPV Family retro flavi herpes picorna parvo Size (nm) 80–100 50–70 120–200 25–30 30 20–25 18–24 Anion Exchange Chromatography (partitioning) Not evaluated 2.3 n.e. 3.4 n.e. 20N Filtration (size exclusion) ≥4.7 ≥3.5 ≥5.6* n.e. 4.8 n.e. 4.1 Solvent/Detergent (inactivation) ≥4.7 ≥7.3 ≥5.5 Not evaluated Total Reduction (log10) ≥9.4 ≥10.8 ≥11.1 2.3 4.8 3.4 4.1 -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
VARIZIG provides passive immunization for non-immune individuals exposed to VZV, reducing the severity of varicella infections (5).
12.3 Pharmacokinetics
In a comparative pharmacokinetic clinical trial, 35 volunteers were administered an intramuscular dose of 12.5 IU/kg of VARIZIG (n=18) or the comparator product VZIG (n=17). The dose of 12.5 IU/kg of VZIG or VARIZIG given to the subjects was based on the assumption that the potency was similar for both products. For the bioequivalence analysis, a potency correction factor was applied (concentrations of VARIZIG were multiplied by 2.3) to account for higher measured potency of the comparator product. The mean peak concentration (Cmax) of varicella antibodies occurred within five days of administration for both products (Table 3). In the trial, baseline levels of anti-VZV antibodies ranged from 0 to 720 mIU/mL, therefore baseline levels were taken into account for pharmacokinetic calculations, to better represent the indicated population. After potency correction, baseline correction, and exclusion of subjects with baseline values of anti-VZV antibody levels of >200 mIU/mL, the two products were pharmacokinetically comparable.
Table 3 Pharmacokinetic Comparison of VARIZIG and VZIG * Potency and subgroup analysis were implemented for pharmacokinetic calculations. Study subjects with elevated baseline anti-VZV levels (>200 mIU/mL) from both treatment groups were excluded from pharmacokinetic calculations.
** The half-life is expected to vary from patient to patient.
PK Parameters* VARIZIG VZIG Ratio
90% Confidence IntervalAUC0-28
(mIUxDay/mL)2472 ± 970 2347 ± 535 84.1–124.6 AUC0-84
(mIUxDay/mL)4087 ± 1620 3916 ± 964 82.0–125.6 Cmax
(mIU/mL)136 ± 66 138 ± 22 76.5–112.8 Tmax
(Days)4.5 ± 2.8 3.3 ± 1.5 Not applicable t1/2**
(Days)26.2 ± 4.6 23.1 ± 8.6 Not applicable CL/F
(mL/Day)0.204 ± 0.045 0.199 ± 0.087 Not applicable -
14 CLINICAL STUDIES
14.1 Pregnant Women Exposed to Varicella Zoster Virus
A randomized, open-label, multicenter, active controlled clinical trial was conducted in 60 pregnant women without immunity to VZV as confirmed by a latex agglutination test. Patients were stratified on the basis of time from first exposure to varicella as follows:
- one to four days post-exposure and,
- five to 14 days post-exposure.
The women were randomized into one of three study arms as follows:
- a single intravenous dose of 125 IU/10 kg body weight to a maximum dose of 625 IU of VARIZIG,
- a single intramuscular dose of 125 IU/10 kg body weight to a maximum dose of 625 IU of VARIZIG or,
- a single intramuscular dose of 125 IU/10 kg body weight to a maximum dose of 625 IU of VZIG (licensed comparator product).
Patients were followed for 42 days.
Incidence of clinical varicella was similar across all treatment groups with an overall incidence of 33%; however, in the subset of 28 subjects with more than 24 hours exposure to varicella, the incidence of clinical varicella in the combined treatment groups was 64%.
Mean weighted constitutional illness scores (CIS) (6) were similar across all groups and none of the subjects had serious complications of varicella. The small number of subjects in each treatment stratum and the lack of agreed upon pre-specified hypothesis testing precluded formal statistical comparisons between groups.
14.2 High Risk Patients Exposed to Varicella Zoster Virus
An open-label, Expanded Access Protocol (EAP) conducted in the United States of America was designed to provide VARIZIG to high risk individuals who were exposed to varicella zoster virus (VZV). Although the study was not designed to evaluate efficacy, the objective of the study was to further assess and confirm the safety and efficacy of intramuscular injection of VARIZIG in the prevention or reduction of severity of complications from varicella infections in the indicated high risk populations. Initially, enrollment was limited to allow treatment with VARIZIG only within 96 hours of exposure, but the protocol was amended to expand the treatment window to 10 days post-exposure.
The incidence of clinical varicella (chickenpox lesions), was compared to predefined historical reference rates. The incidence of severe varicella complications, including pneumonia, encephalitis, severe varicella with pox counts >100 pox, mortality and all complications was also evaluated. The overall incidence of clinical varicella was evaluated in an interim analysis, where 10% (31/311) of high risk individuals exposed to VZV and treated with VARIZIG for all combined populations, for whom complete or partial efficacy data was available. Clinical varicella was observed in 8.4% (13/154) of immunocompromised pediatric and adult patients, in 6.8 % (5/74) of pregnant women, in 14.8% (12/81) of infants and one healthy adult (Table 4). Clinical varicella was more common after prolonged VZV exposure. The final report confirmed the efficacy results in the interim analysis (Table 5 ). In addition, a comparison of the incidence of varicella based on treatment window revealed that treatment between 5 and 10 days post-exposure was no different from treatment within 96 hours.
Table 4 Comparison of Varicella Incidence in Subjects Treated with VARIZIG to Historical Incidence of Varicella in Untreated Individuals – Interim analysis 1 1 n = number of VARIZIG doses for post-exposure prophylaxis of varicella in efficacy population.
2 One sample two-sided exact binomial test.
* Statistically significant (α=0.05).
High Risk Population Historical Incidence of Varicella in Untreated Individuals n1 Incidence of Varicella in VARIZIG-treated Subjects 95% Confidence Interval p-value2 Pregnant Women 70% 74 6.8% (n=5) (2.2–15.1%) <0.0001* Immunocompromised patients 88% 154 8.4% (n=13) (4.6–14.0%) <0.0001* Infants 50% 81 14.8% (n=12) (7.9–24.5%) <0.0001* Table 5 Updated Summary of Incidence of Varicella in Subjects Treated with VARIZIG – Final Report 1 n = number of VARIZIG doses for post-exposure prophylaxis of varicella in efficacy population
High Risk Population All VARIZIG Treated Subjects n1 Incidence of Varicella in VARIZIG-treated Subjects 95% Confidence Interval Pregnant Women 137 7.3% (n=10) (3.6% - 13.0%) Immunocompromised patients 269 4.5% (n=12) (2.3% - 7.7%) Infants including newborns, pre-term infants and infants <1 year 105 11.4% (n=12) (6.1% - 19.1%) -
15 REFERENCES
- Dalakas MC. High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology. 1994; 44:223-6.
- Woodruff RK, Grigg AP, Firkin FC, Smith IL. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet. 1986; 2:217-8.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M. Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol. 2000; 65:30-4.
- Josephson C, Nuss R, Jacobson L, Hacker MR, Murphy J, Weinberg A et al. The Varicella-Autoantibody Syndrome. Pediatr Res. 2001; 50:345-52.
- CDC. Prevention of varicella. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR. 2007; 56(No. RR-4):1-30.
- Koren G, Money D, Boucher M, Aoki F, Petric M, Innocencion G et al. Serum concentrations, efficacy, and safety of a new, intravenously administered varicella zoster immune globulin in pregnant women. J Clin Pharmacol. 2002; 42(3):267-74.
-
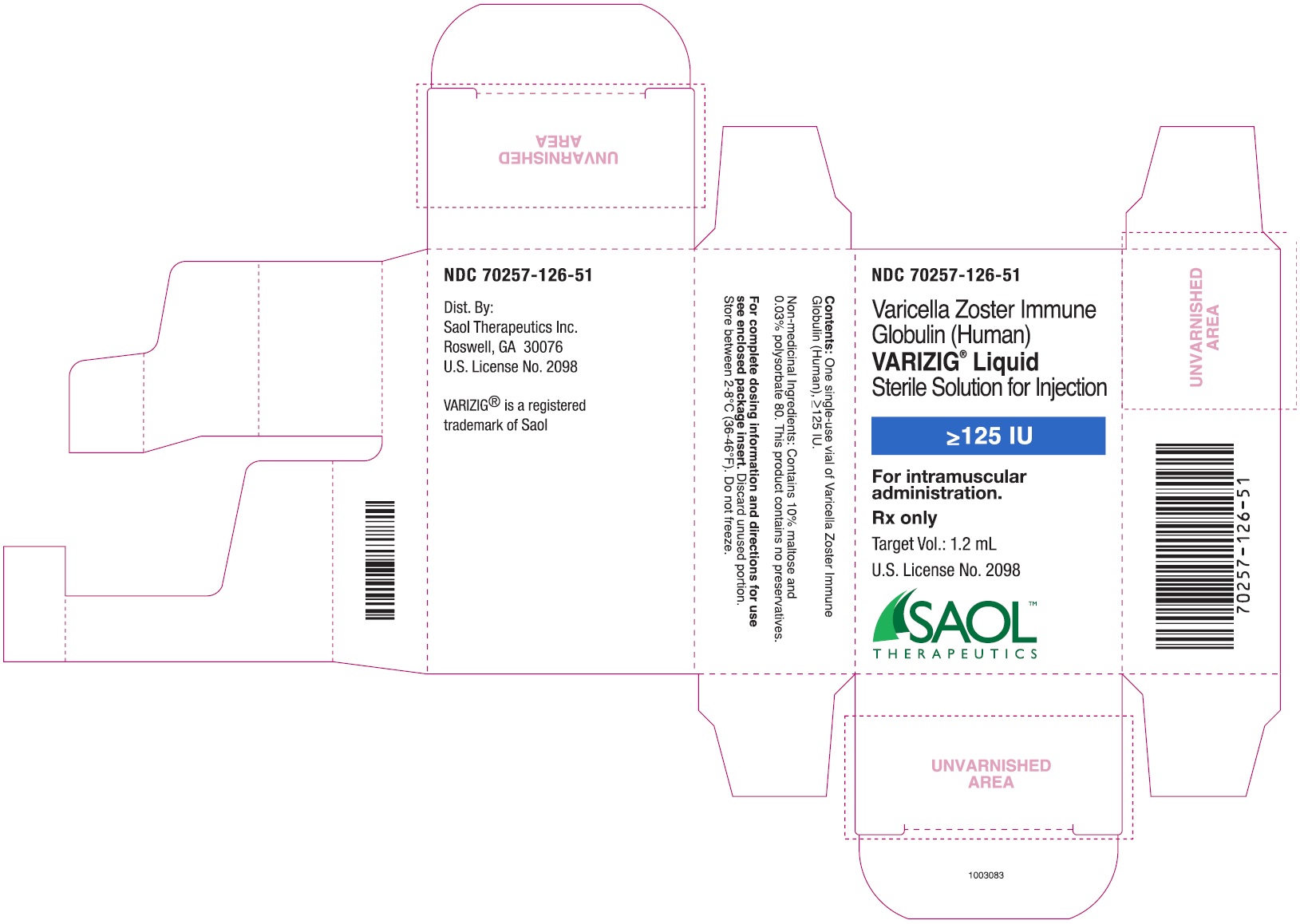
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
NDC: 70257-126-51: VARIZIG [Varicella Zoster Immune Globulin (Human)] is supplied as a sterile liquid approximately 125 IU of anti-VZV in a 3 mL type 1 glass tubing vial fitted with a 13 mm rubber stopper and a 13 mm flip-off seal, and a package insert.
-
17 PATIENT COUNSELING INFORMATION
Inform patients of the following:
- VARIZIG is intended to reduce the severity of chickenpox infections. Please see your doctor if you develop the signs and symptoms of varicella.
- VARIZIG is prepared from human plasma and therefore, may contain infectious agents such as viruses that can cause disease.
- The risk that products derived from human plasma will transmit an infectious agent has been reduced by screening plasma donors for prior exposure to certain viruses, by testing for the presence of certain current virus infections, and by inactivating and/or removing certain viruses during manufacturing.
- Despite these measures, products derived from human plasma can still potentially transmit disease.
- There is also the possibility that unknown infectious agents may be present in such products.
Tell patients that persons known to have severe, potentially life-threatening reactions to human immune globulin products should not receive VARIZIG or any other immune globulin products unless the risk has been justified.
Tell patients that persons who are deficient in IgA may have the potential for developing anti-IgA antibodies and have severe potentially life threatening allergic reactions.
- In the case of allergic or anaphylactic reaction, administration should be stopped immediately.
- In the case of shock, the current medical standards for treatment of shock should be administered.
Inform patients that administration of immune globulin may interfere with the response to live virus vaccines (e.g. measles, mumps, rubella and varicella), and instruct them to notify their immunizing physician of recent therapy with VARIZIG.
VARIZIG® [Varicella Zoster Immune Globulin (Human)] Sterile Solution for Injection and any and all Saol brand, product, service and feature names, logos, slogans are trademarks or registered trademarks of Saol. All rights reserved.
PLANOVA® is a registered trademark of Asahi Kasei Medical Co., Ltd, TRITON® is a registered trademark of Union Carbide Corporation.
Distributed by:
Saol Therapeutics Inc.
Roswell, GA 30076
U.S. License No. 2098
- PRINCIPAL DISPLAY PANEL - NDC: 70257-126-11 - 1.25 mL Vial Label
- PRINCIPAL DISPLAY PANEL - NDC: 70257-126-51 - 1.25 mL Carton Label
-
INGREDIENTS AND APPEARANCE
VARIZIG
varicella zoster immune globulin (human) solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70257-126 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN VARICELLA-ZOSTER IMMUNE GLOBULIN (UNII: 33T61IWL27) (HUMAN VARICELLA-ZOSTER IMMUNE GLOBULIN - UNII:33T61IWL27) HUMAN VARICELLA-ZOSTER IMMUNE GLOBULIN 125 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength MALTOSE (UNII: XJ6S9RV06F) POLYSORBATE 80 (UNII: 6OZP39ZG8H) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70257-126-51 1 in 1 CARTON 03/01/2019 1 NDC: 70257-126-11 1.25 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125430 03/01/2019 Labeler - Saol Therapeutics Inc. (080040201)

Trademark Results [VARIZIG]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VARIZIG 78679351 3240650 Live/Registered |
SAOL INTERNATIONAL LIMITED 2005-07-27 |
 VARIZIG 75934913 not registered Dead/Abandoned |
Cangene Corporation 2000-03-03 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.