EKTERLY- sebetralstat tablet
EKTERLY by
Drug Labeling and Warnings
EKTERLY by is a Prescription medication manufactured, distributed, or labeled by KalVista Pharmaceuticals Ltd, Catalent Nottingham Limited, Almac Pharma Services Limited, Zach System S.A.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EKTERLY safely and effectively. See full prescribing information for EKTERLY.
EKTERLY (sebetralstat) tablets, for oral use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
EKTERLY® is a plasma kallikrein inhibitor indicated for the treatment of acute attacks of hereditary angioedema (HAE) in adult and pediatric patients aged 12 years and older. (1)
DOSAGE AND ADMINISTRATION
Recommended Dosage: one dose of 600 mg (2 tablets) taken orally at the earliest recognition of an HAE attack. (2.1)
- A second dose of 600 mg (2 tablets) may be taken 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur. (2.1)
- Maximum Recommended Dosage: 1,200 mg in any 24-hour period. (2.1)
- See full prescribing information for dosage modification for concomitant use with CYP3A4 inhibitors and inducers. (2.2)
- See full prescribing information for recommended dosage for patients with hepatic impairment. (2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 300 mg (3)
CONTRAINDICATIONS
None (4)
ADVERSE REACTIONS
The most common adverse reaction (incidence ≥2%) is headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact KalVista Pharmaceuticals, Inc. at 1-855-258-4782 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- Strong and Moderate CYP3A4 Inhibitors: Avoid use with strong CYP3A4 inhibitors. In patients taking moderate CYP3A4 inhibitors, take one dose of 300 mg. A second dose of 300 mg may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur. (2.2, 7.1, 12.3)
- CYP3A4 Inducers: Avoid use with moderate or strong CYP3A4 inducers. (2.2, 7.1, 12.3)
USE IN SPECIFIC POPULATIONS
Hepatic Impairment: Avoid use of EKTERLY in patients with severe hepatic impairment (Child-Pugh Class C). In patients with moderate hepatic impairment (Child-Pugh Class B) take one dose of 300 mg. A second dose of 300 mg may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur. (2.3, 8.6, 12.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modification for CYP3A4 Inhibitors and Inducers
2.3 Recommended Dosage in Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on EKTERLY
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of EKTERLY is one dose of 600 mg (two tablets) orally at the earliest recognition of an acute HAE attack.
- A second dose of 600 mg (two tablets) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur.
- Maximum recommended dosage is 1,200 mg (four tablets) in any 24-hour period.
2.2 Dosage Modification for CYP3A4 Inhibitors and Inducers
Refer to Table 1 for dosage modification of EKTERLY when used concomitantly with CYP3A4 inhibitors and inducers.
Table 1: Dosage Modification of EKTERLY for Concomitant use with CYP3A4 Inhibitors and Inducers Dosage Modification for Concomitant use of EKTERLY with CYP3A4 Inhibitors
Strong CYP3A4 Inhibitors
Avoid concomitant use with EKTERLY.
Moderate CYP3A4 Inhibitors
Reduce EKTERLY dosage to 300 mg (one tablet) orally at earliest recognition of an acute HAE attack.
A second dose of 300 mg (one tablet) may be taken at least 3 hours after first dose if response is inadequate or if symptoms worsen or recur [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Weak CYP3A4 Inhibitors
No dosage modification of EKTERLY [see Dosage and Administration (2.1)].
Dosage Modification for Concomitant use of EKTERLY with CYP3A4 Inducers
Strong and Moderate CYP3A4 Inducers
Avoid concomitant use with EKTERLY.
Weak CYP3A4 Inducers
No dosage modification of EKTERLY [see Dosage and Administration (2.1)].
2.3 Recommended Dosage in Patients with Hepatic Impairment
Refer to Table 2 for recommended dosage of EKTERLY in patients with hepatic impairment.
Table 2: Recommended Dosage of EKTERLY in Patients with Hepatic Impairment Recommended Dosage of EKTERLY in Patients with Hepatic Impairment
Severe Hepatic Impairment (Child-Pugh Class C)
Avoid use of EKTERLY [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Moderate Hepatic Impairment (Child-Pugh Class B)
Recommended dosage of EKTERLY is one dose of 300 mg (one tablet) orally at the earliest recognition of an acute HAE attack. A second dose of 300 mg (one tablet) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Mild Hepatic Impairment (Child-Pugh Class A)
Recommended dosage is the same as the recommended dosage in patients with normal hepatic function [see Dosage and Administration (2.1)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of EKTERLY is based on data from a double-blind, randomized, placebo-controlled, three-way, crossover clinical trial (KONFIDENT) [see Clinical Studies (14)]. In KONFIDENT, a total of 110 patients aged 12 years and older with HAE treated 264 attacks. In the safety population, 93 patients received EKTERLY 600 mg, 86 patients received EKTERLY 300 mg, and 83 patients received placebo. While EKTERLY 300 mg was included in KONFIDENT, the safety data is based on the recommended dosage of EKTERLY 600 mg.
Table 3 displays adverse reaction(s) with an incidence of ≥2% in the EKTERLY 600 mg treated patients and more common than placebo.
Table 3: Adverse Reaction(s) with EKTERLY with Incidence ≥2% and More Common than Placebo in Patients with Hereditary Angioedema (KONFIDENT) aOne (1) patient assigned to administer placebo actually received EKTERLY 600 mg. Safety results are presented by actual treatment received. Adverse Reaction
EKTERLY
600 mg
(N = 93)Placebo
(N = 83)an (%)
n (%)
Headache
3 (3.2)
1 (1.2)
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on EKTERLY
Strong CYP3A4 Inhibitors
- Avoid use of EKTERLY with strong CYP3A4 inhibitors [see Clinical Pharmacology (12.3)].
Sebetralstat is a substrate of CYP3A4. Concomitant use of sebetralstat with a strong CYP3A4 inhibitor increases sebetralstat exposure, which may increase the risk of sebetralstat adverse reactions.
Moderate and Weak CYP3A4 Inhibitors
- Reduce dose of EKTERLY to one dose of 300 mg (one tablet) orally at the earliest recognition of an HAE attack when used concomitantly with moderate CYP3A4 inhibitors. A second dose of 300 mg (one tablet) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
Sebetralstat is a substrate of CYP3A4. Concomitant use of sebetralstat with a moderate CYP3A4 inhibitor increases sebetralstat exposure, which may increase the risk of sebetralstat adverse reactions.
- No dose modification is recommended when EKTERLY is used concomitantly with a weak CYP3A4 inhibitor [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
Strong and Moderate CYP3A4 Inducers
- Use of EKTERLY with strong or moderate CYP3A4 inducers is not recommended.
Sebetralstat is a substrate of CYP3A4. Concomitant use of sebetralstat with a strong or moderate CYP3A4 inducer decreases sebetralstat exposure, which may decrease efficacy.
Weak CYP3A4 Inducers
No dose modification is recommended when EKTERLY is used concomitantly with weak CYP3A4 inducers [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on EKTERLY in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. In animal reproduction studies, oral administration of sebetralstat to pregnant rats and rabbits during organogenesis produced no evidence of fetal harm with dose exposures up to approximately 15 and 11 times, respectively, the human exposure at the maximum recommended human dose (MRHD) of up to 1,200 mg (on an area under the curve [AUC] basis). Sebetralstat produced an increase in embryofetal losses and fetal malformations in rats at an exposure that was 60 times the MRHD (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryofetal development study with pregnant rats dosed by the oral route during the period of organogenesis from gestation days 6 to 17, sebetralstat caused a dose-related increase in the incidence of external and visceral malformations, described as cleft palates and ventricular septal defects and an increase in embryofetal losses (early and late fetal deaths, mean number of live fetuses, and post-implantation loss at an exposure that was 60 times the MRHD (on an AUC basis with a maternal dose of 600 mg/kg/day)). Maternal toxicity, as evidenced by decreased body weight gains, was observed at exposures 60 times the MRHD (on an AUC basis with a maternal oral dose of 600 mg/kg/day). No fetal or maternal toxicities were observed at exposures up to 15 times the MRHD (on an AUC basis with a maternal oral dose of 300 mg/kg/day).
In an embryofetal development study with pregnant rabbits dosed by the oral route during the period of organogenesis from gestation days 6 to 18, maternal toxicity was evidenced by a decrease in body weight gain at an exposure that was 11 times the MRHD (on an AUC basis with a maternal dose of 300 mg/kg/day). No adverse effects on maternal toxicity were observed at an exposure that was 2 times the MRHD (on an AUC basis with a maternal dose of 100 mg/kg/day). No adverse effects on fetal toxicity were observed at an exposure that was 11 times the MRHD (on an AUC basis with a maternal oral dose of 300 mg/kg/day).
In a prenatal and postnatal development study with pregnant rats dosed by the oral route during the periods of gestation and lactation from gestation day 6 to lactation day 20, sebetralstat had no effects on the growth and development of offspring at an exposure that was 40 times the MRHD (on an AUC basis with a maternal oral dose of 450 mg/kg/day).
8.2 Lactation
Risk Summary
There are no data on the presence of sebetralstat or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Radioactivity is present in animal milk after dosing with radiolabeled sebetralstat. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Data in the rat have shown excretion of sebetralstat and/or its metabolites in milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for EKTERLY and any potential adverse effects on the breastfed child from EKTERLY or from the underlying maternal condition.
Data
Animal Data
In a single dose placental transfer and milk secretion study in lactating rats dosed with radiolabeled sebetralstat by the oral route, total radioactivity was detected in fetal plasma at 1 hour post dose. Similar concentrations of total radioactivity were observed in the milk and plasma of lactating females, with the maximum radioactive concentration observed at 1 hour post dose that returned to background levels by 24 hours post dose. The concentration of total radioactivity in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
The safety and effectiveness of EKTERLY for treatment of acute attacks of hereditary angioedema (HAE) have been established in pediatric patients aged 12 years and older. Use of EKTERLY for this indication is supported by evidence from an adequate and well-controlled study (KONFIDENT) in adults and pediatric patients aged 12 years and older with additional population pharmacokinetic (PK) analysis from adults and pediatric patients aged 12 years and older, which showed no clinically significant differences in PK based on body weight or age. Results of the subgroup analysis for pediatric patients aged 12 years and older were consistent with study results for adult patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14)].
The safety and effectiveness of EKTERLY in pediatric patients aged <12 years of age have not been established.
8.5 Geriatric Use
Clinical studies of EKTERLY did not include sufficient numbers of patients 65 years of age and older to determine whether they respond differently from younger adult patients.
8.6 Hepatic Impairment
No dose adjustment of EKTERLY is recommended for patients with mild hepatic impairment (Child-Pugh Class A). For patients with moderate hepatic impairment (Child-Pugh Class B), the recommended dose of EKTERLY is one dose of 300 mg (one tablet) orally at the earliest recognition of an HAE attack. A second dose of 300 mg (one tablet) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur. Avoid use of EKTERLY in patients with severe hepatic impairment (Child-Pugh Class C) [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
The active ingredient of EKTERLY is sebetralstat, a plasma kallikrein inhibitor. The chemical name of sebetralstat is N-[(3-fluoro-4-methoxypyridin-2-yl) methyl]-3-(methoxymethyl)-1-({4-[(2-oxo-1,2-dihydropyridin-1-yl) methyl]phenyl}methyl)-1H-pyrazole-4-carboxamide. The chemical structure of sebetralstat is:
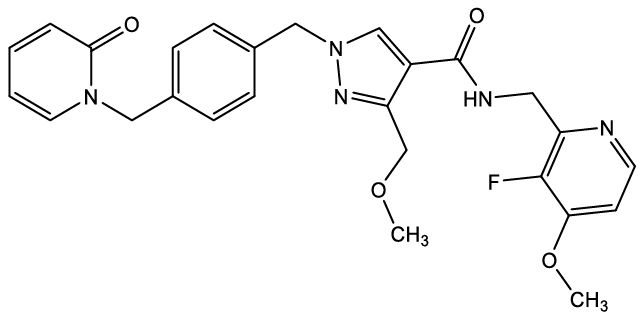
The molecular formula is C26H26FN5O4 and molecular weight is 491.5. Sebetralstat is a white to off-white crystalline powder. It is slightly soluble in ethanol, acetone and isopropanol, and practically insoluble in water.
EKTERLY is supplied as 300 mg film-coated tablets for oral administration. Each tablet contains the active ingredient sebetralstat. The inactive ingredients include croscarmellose sodium, glycerol mono and dicaprylocaprate, guar gum/guar galactomannan, hypromellose, iron oxide black, iron oxide yellow, macrogol polyvinyl alcohol graft copolymer, magnesium stearate, maltodextrin, medium chain triglycerides, microcrystalline cellulose, polyvinyl alcohol, povidone, talc and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sebetralstat is a competitive, reversible inhibitor of plasma kallikrein. Plasma kallikrein is a serine protease that cleaves high molecular weight kininogen (HK) releasing bradykinin which increases vascular permeability through activation of bradykinin receptors causing edema. Sebetralstat inhibits the cleavage of HK and reduces production of bradykinin, thereby treating the clinical symptoms of an acute, episodic attack of HAE. Sebetralstat also inhibits the positive feedback mechanism of the kallikrein kinin system by plasma kallikrein, thereby reducing factor XIIa and additional plasma kallikrein generation.
12.2 Pharmacodynamics
Concentration-dependent inhibition of plasma kallikrein, measured as a reduction from baseline of specific enzyme activity, was observed in exploratory assays. Following administration of a single oral dose of 600 mg sebetralstat to healthy subjects, greater than 90% mean inhibition of plasma kallikrein activity was observed beginning at 30 minutes after dosing and maintained through about 6 hours post-dose.
Cardiac Electrophysiology
The largest mean increase in QTc interval was 10.4 msec (upper confidence interval = 15.3 msec) after administration of sebetralstat (2.5 times the maximum recommended dose) in healthy subjects. The increase in the QTc interval was concentration dependent.
12.3 Pharmacokinetics
Following a single oral dose of 600 mg sebetralstat in subjects with HAE, the geometric mean (CV%) Cmax is 6,080 ng/mL (40%) and AUC0-inf is 17,600 ngh/mL (36%). Following administration of a second oral dose of 600 mg sebetralstat 3 hours after the first dose, Cmax increases 10% and AUC0-inf increases 90% when compared to those observed following a single dose.
No clinically relevant differences in sebetralstat pharmacokinetics were observed between healthy subjects and patients with HAE.
Absorption
After a single oral dose of 600 mg sebetralstat, the median time to peak plasma concentration is approximately 1 hour.
Effect of Food
No clinically relevant differences in sebetralstat pharmacokinetics were observed following administration of a high-fat meal (900-1,000 calories in total – 500-600, 250 and 150 calories from fat, carbohydrate and protein respectively).
Distribution
The estimated typical apparent volume of distribution (Vz/F) for sebetralstat is 70.1 L (95% CI: 64.8, 75.4). Sebetralstat plasma protein binding is 77% in vitro.
Elimination
Following administration of a single oral dose of 600 mg sebetralstat, the mean (SD) elimination half-life of sebetralstat ranged from 5.3 (2.3) to 8.9 (5.1) hours. The estimated typical apparent clearance (CL/F) is 30.7 L/h (95% CI: 29.1, 32.2).
Metabolism
Sebetralstat is primarily metabolized by CYP3A4 and to a lesser extent by CYP2C8.
Excretion
Following administration of a single oral dose of 600 mg radiolabeled sebetralstat to healthy male subjects, approximately 63% of the dose was recovered in feces (12.5% as unchanged sebetralstat) and 32% in urine (8.7% as unchanged sebetralstat).
Specific Populations
No clinically relevant differences in the pharmacokinetics of sebetralstat were observed based on body weight (45-135 kg), age (19-68 years), sex, race (71% White, 17% Black, 11% Asian), and mild renal impairment (eGFR 60-89 mL/min/1.73 m2). The effect of moderate and severe renal impairment (eGFR <60 mL/min/1.73 m2) on sebetralstat pharmacokinetics is unknown.
Patients with Hepatic Impairment
The pharmacokinetics of sebetralstat were evaluated in subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment following administration of a single 600 mg dose. In subjects with mild hepatic impairment Cmax was increased by 7% and AUC increased by 16% compared to subjects with normal hepatic function. In subjects with moderate hepatic impairment Cmax was increased by 63% and AUC was increased by 100% compared to subjects with normal hepatic function. The effect of severe hepatic impairment (Child-Pugh Class C) on sebetralstat pharmacokinetics is unknown [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A4 Inhibitors: Sebetralstat Cmax increased 2.4-fold and AUC0-inf increased 5.2-fold following concomitant administration with itraconazole (a strong CYP3A4 inhibitor) 200 mg once daily for 6 days [see Drug Interactions (7.1)].
Moderate CYP3A4 Inhibitors: Sebetralstat Cmax increased 1.8-fold and AUC0-inf increased 2-fold following concomitant administration with verapamil (a moderate CYP3A4 inhibitor) 240 mg once daily for 6 days [see Drug Interactions (7.1)].
Strong CYP3A4 Inducers: Sebetralstat Cmax decreased by 66% and AUC0-inf decreased by 83% following concomitant administration with phenytoin (a strong CYP3A4 inducer) 100 mg three times daily for 15 days [see Drug Interactions (7.1)].
Moderate CYP3A4 Inducers: Sebetralstat Cmax decreased by 63% and AUC0-inf decreased by 79% following concomitant administration with efavirenz (a moderate CYP3A4 inducer) 600 mg once daily for 14 days [see Drug Interactions (7.1)].
Acid-Reducing Agents: Sebetralstat Cmax is estimated to decrease by up to 56% and AUC0-inf is estimated to decrease by up to 30% at pH 6 relative to pH 0.5.
Other Drugs: No clinically significant differences in sebetralstat pharmacokinetics were observed when administered concomitantly with quinidine (P-gp inhibitor), eltrombopag (BCRP inhibitor), cimetidine (evaluated as a weak CYP3A4 inhibitor), or modafinil (weak CYP3A4 inducer).
In Vitro Studies
CYP450 Enzymes: Sebetralstat is a substrate of CYP3A4 and CYP2C8, and is not a substrate of CYP1A2, CYP2A6, CYP2B6, CYP2C9, CYP2C19, CYP2D6, or CYP2E1. Sebetralstat is an inhibitor of CYP2C9 and CYP3A4. Sebetralstat did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C19, or CYP2D6.
Transporter Systems: Sebetralstat is a substrate of P-gp and BCRP, and is not a substrate of OATP1B1, OATP1B3, OAT1, OAT3, OCT2, or MATE1. Sebetralstat is an inhibitor of BCRP, OATP1B1, OATP1B3, OAT3, OCT2, MATE1, MATE2-K, and BSEP. Sebetralstat did not inhibit P-gp or OAT1.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
A 6-month carcinogenicity study in rasH2-Tg transgenic mice was conducted to assess the carcinogenic potential of sebetralstat. Sebetralstat demonstrated no tumorigenic potential in a study with male and female mice that received oral doses up to 200 mg/kg/day and 300 mg/kg/day, respectively.
Mutagenesis
Sebetralstat was not mutagenic or clastogenic in the following assays: in vitro bacterial reverse mutation (Ames) test, in vitro chromosomal aberration assay in human peripheral blood lymphocytes, and in vivo rat micronucleus assay.
Impairment of Fertility
Fertility and reproductive performance were unaffected in male or female rats that received sebetralstat by the oral route at dose levels up to 600 mg/kg/day (approximately 38 times the MRHD in adults on an AUC basis).
-
14 CLINICAL STUDIES
The efficacy of EKTERLY for the treatment of acute attacks of hereditary angioedema (HAE) in adult and pediatric patients aged 12 years and older was evaluated in a double-blind, randomized, placebo-controlled, multicenter crossover clinical trial (KONFIDENT [NCT05259917]). KONFIDENT employed a 3-way, complete crossover design comparing EKTERLY 600 mg and EKTERLY 300 mg to placebo. If needed (as determined by the patient) a second dose could be administered after 3 hours. Patients were required to treat an eligible HAE attack prior to crossover to the next treatment period. Severe laryngeal attacks were not treated in KONFIDENT. The duration of trial participation was approximately 25 weeks. Although evaluated in KONFIDENT, EKTERLY 300 mg is not a recommended dose for treatment of acute HAE attacks.
The demographics and baseline characteristics of patients in KONFIDENT are provided in Table 4.
Table 4: Demographics and Baseline Characteristics of Patients in KONFIDENT Trial KONFIDENT
(N=110)Female, n (%)
66 (60)
Mean age, years (SD)
38 (15)
12 to <18 years, n (%)
13 (12)
≥18 years, n (%)
97 (88)
Race, n (%)
Asian
10 (9)
Black or African American
1 (1)
White
92 (84)
Other
1 (1)
Not reported
6 (5)
Hispanic or Latino, n (%)
7 (6)
HAE Type I, n (%)
101 (92)
Background HAE prophylaxis, n (%)
24 (22)
A total of 110 patients were randomized and experienced at least one HAE attack. Of the 264 treated HAE attacks, 142 (54%) had peripheral symptoms only, 85 (32%) had abdominal symptoms only, 27 (10%) had abdominal and peripheral symptoms, 8 (3%) had mild to moderate laryngeal symptoms, and 2 (1%) had missing attack location.
The primary endpoint for KONFIDENT was the ‘time to beginning of symptom relief’ defined as at least “a little better” at two consecutive time points within 12 hours of first dose administration, assessed using a seven-point scale Patient Reported Global Impression of Change (PGI-C) ranging from “much worse” to “much better”.
As shown in Figure 1, there was a statistically significant faster time to the beginning of symptom relief for EKTERLY 600 mg compared to placebo. A total of 71 out of 93 (76%) patients administered EKTERLY 600 mg and 41 out of 84 (49%) patients administered placebo achieved the primary endpoint. The median time to beginning of symptom relief within 12 hours of first dose was 2.0 hours (95% CI: 1.5, 2.8) in patients administered EKTERLY 600 mg.
Figure 1: Kaplan-Meier for Time to Beginning of Symptom Relief Within 12 Hours of First Dose Administration with EKTERLY in KONFIDENT
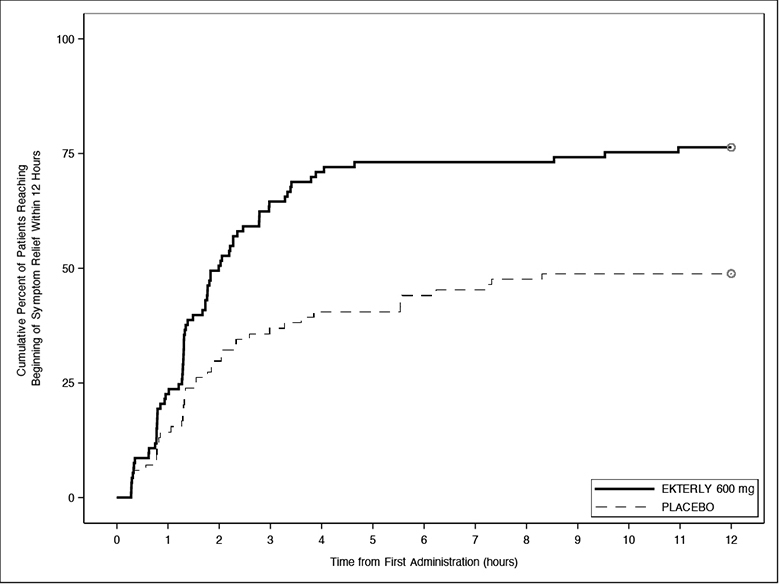
Note: In the EKTERLY 600 mg group, 38% of patients administered a second dose within 12 hours.
Less than 50% of placebo patients reached beginning of symptom relief within 12 hours; therefore, the median time could not be estimated.
Patients who did not achieve the endpoint, received alternate on-demand therapy, or lacked at least 2 consecutive post-baseline assessments were right-censored at 12 hours.
Secondary Endpoints
The first key secondary endpoint was the ‘time to first incidence of reduction in severity’ at two consecutive time points within 12 hours of first dose administration, assessed using a five-point scale Patient Global Impression of Severity (PGI-S) ranging from “none” to “severe”.
The ‘time to first incidence of reduction in severity’ (Figure 2) was statistically significantly faster for EKTERLY 600 mg compared to placebo. A total of 49 out of 93 (53%) patients administered EKTERLY 600 mg and 26 out of 84 (31%) patients administered placebo achieved reduction in severity within 12 hours. The median time to achieve this endpoint was 9.1 hours (95% CI: 3.8, not reached) in patients administered EKTERLY 600 mg.
Figure 2: Kaplan-Meier for Time to First Incidence of Reduction in Severity Within 12 Hours of First Dose Administration with EKTERLY in KONFIDENT
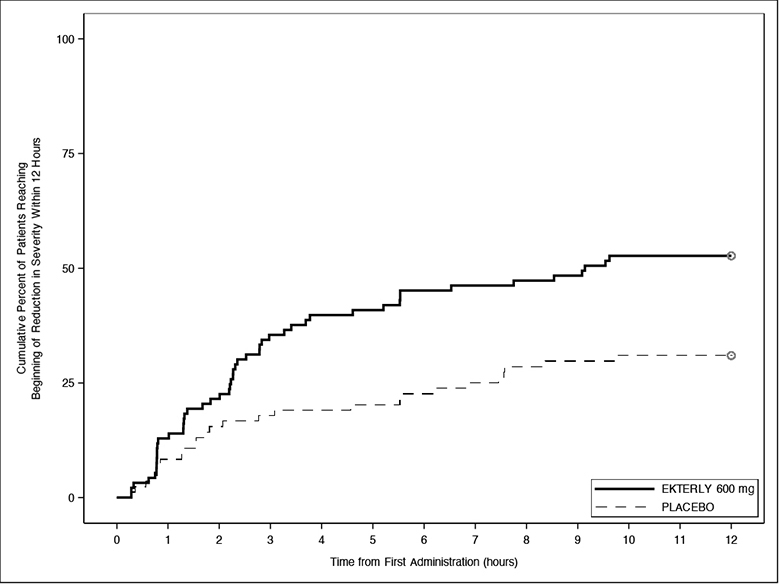
Note: In the EKTERLY 600 mg group, 38% of patients administered a second dose within 12 hours.
Less than 50% of placebo patients reached beginning of reduction in severity within 12 hours; therefore, the median time could not be estimated.
Patients who did not achieve the endpoint, received alternate on-demand therapy, or lacked at least 2 consecutive post-baseline assessments were right-censored at 12 hours.
The second key secondary endpoint was ‘time to attack resolution’ defined as PGI-S of “none” within 24 hours of first dose administration.
The ‘time to attack resolution’ (Figure 3) was statistically significantly faster for EKTERLY 600 mg compared to placebo. A total of 46 out of 93 (49%) patients administered EKTERLY 600 mg and 23 out of 84 (27%) patients administered placebo achieved attack resolution within 24 hours.
Figure 3: Kaplan-Meier for Time to Attack Resolution Within 24 Hours of First Dose Administration with EKTERLY in KONFIDENT
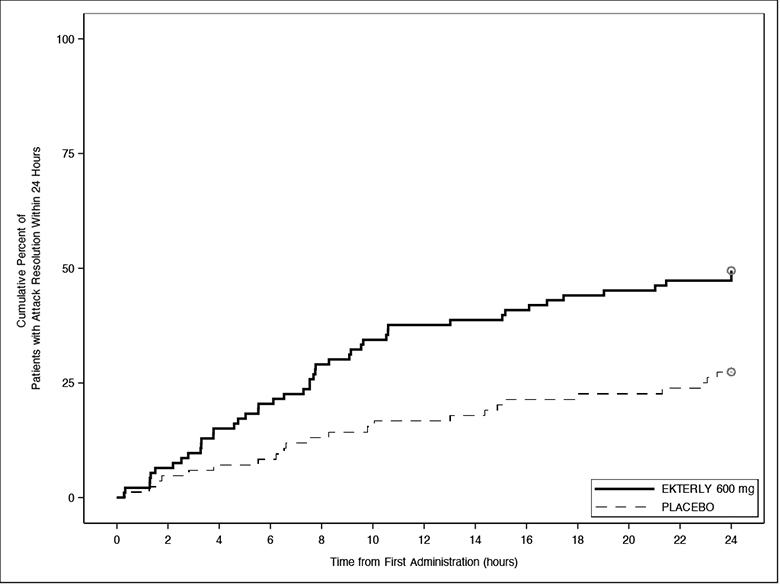
Note: In the EKTERLY 600 mg group, 39% of patients administered a second dose within 24 hours.
Less than 50% of EKTERLY 600 mg and placebo patients had attack resolution within 24 hours; therefore, the median time could not be estimated.
Patients who did not achieve the endpoint, received alternate on-demand therapy, or lacked post-baseline assessments were right-censored at 24 hours.
Other Endpoints
The time to beginning of at least “better” at two consecutive time points on the PGI-C within 12 hours of the first dose administration was assessed. A total of 54 out of 93 (58%) patients administered EKTERLY 600 mg and 21 out of 84 (25%) patients administered placebo achieved this endpoint. The median time was 4.6 hours (95% CI: 3.3, 9.5) in patients administered EKTERLY 600 mg.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
EKTERLY (sebetralstat) tablets: 300 mg, yellow, oval, biconvex, film-coated tablets debossed with
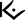 on one side and “300” on the other side.
on one side and “300” on the other side.- EKTERLY is supplied in a carton containing four child-resistant blister cards. Each child-resistant blister card contains 1 x 300 mg tablet (NDC: 82928-300-04).
- Each carton contains a tamper evident seal. Do not use if tamper evident seal is broken or missing.
Store at 20°C to 25°C (68°F to 77°F). Excursions permitted between 15°C and 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Dosage Modification for Strong and Moderate CYP3A4 Inhibitors
Advise patients not to use EKTERLY with strong CYP3A4 inhibitors [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)]. When used concomitantly with moderate CYP3A4 inhibitors, advise patients to reduce dose to one tablet of 300 mg (total dose 300 mg) at the earliest recognition of an HAE attack. A second dose of 300 mg (one tablet) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur [see Dosage and Administration (2.2), Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Recommended Dosage in Patients with Hepatic Impairment
Advise patients with severe hepatic impairment to avoid use of EKTERLY. In patients with moderate hepatic impairment, advise patients to take one tablet of 300 mg (total dose 300 mg) at earliest recognition of an HAE attack. A second dose of 300 mg (one tablet) may be taken at least 3 hours after the first dose if response is inadequate, or if symptoms worsen or recur [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
For more information, visit www.EKTERLY.com
EKTERLY® is a registered trademark of KalVista Pharmaceuticals Limited.
Distributed by:
KalVista Pharmaceuticals, Inc.
200 Crossing Boulevard
Framingham, MA 01702 -
PATIENT PACKAGE INSERT
PATIENT INFORMATION
EKTERLY (ek-TURR-lee)
(sebetralstat)
tablets, for oral useWhat is EKTERLY?
- EKTERLY is a prescription medicine used to treat sudden (acute) attacks of hereditary angioedema (HAE) in adults and children aged 12 years of age and older.
- It is not known if EKTERLY is safe and effective in children under 12 years of age.
Before you take EKTERLY, tell your healthcare provider about all of your medical conditions, including if you:
- are pregnant or planning to become pregnant. It is not known if EKTERLY can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if EKTERLY passes into your breastmilk. Talk to your healthcare provider about the best way to feed your baby while taking EKTERLY.
- have liver problems.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- Taking EKTERLY with certain other medicines can cause side effects or affect how well EKTERLY or the other medicines work.
- itraconazole
- phenytoin
- efavirenz
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take EKTERLY?
- Take EKTERLY exactly as your healthcare provider tells you to take it.
- Take 2 tablets, by mouth, at the earliest sign of an acute attack of hereditary angioedema. If you do not feel better or if your symptoms worsen or come back, take another dose (2 tablets) at least 3 hours after the first dose.
- Your healthcare provider may change how much EKTERLY you receive if you take certain medicines or if you have liver problems:
- Take 1 tablet, by mouth, at the earliest sign of an acute attack of hereditary angioedema. If you do not feel better or if your symptoms worsen or come back take another dose (1 tablet) at least 3 hours after the first dose.
- If you take too much EKTERLY, call your healthcare provider or Poison Help line at 1-800-222-1222 or go to the nearest hospital emergency room right away.
What are the possible side effects of EKTERLY?
The most common side effects of EKTERLY include:- headache
For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store EKTERLY?
- Store EKTERLY in the blister package that it comes in.
- Store EKTERLY at room temperature between 68°F to 77°F (20°C to 25°C).
Keep EKTERLY and all medicines out of the reach of children.
General information about the safe and effective use of EKTERLY
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EKTERLY for a condition for which it was not prescribed. Do not give EKTERLY to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EKTERLY that is written for health professionals.What are the ingredients in EKTERLY?
Active ingredient: sebetralstat
Inactive ingredients: croscarmellose sodium, glycerol mono and dicaprylocaprate, guar gum/guar galactomannan, hypromellose, iron oxide black, iron oxide yellow, macrogol polyvinyl alcohol graft copolymer, magnesium stearate, maltodextrin, medium chain triglycerides, microcrystalline cellulose, polyvinyl alcohol, povidone, talc and titanium dioxide.
Distributed by KalVista Pharmaceuticals, Inc, 200 Crossing Boulevard, Framingham, MA 01702
EKTERLY is a registered trademark of KalVista Pharmaceuticals Limited
©2025 KalVista Pharmaceuticals, Inc. All rights reserved.
For more information, visit www.EKTERLY.com or call 1-855-258-4782.This Patient Information has been approved by the U.S. Food and Drug Administration Issued: 07/2025
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
EKTERLY
sebetralstat tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 82928-300 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Sebetralstat (UNII: O5ZD2TU2B7) (Sebetralstat - UNII:O5ZD2TU2B7) Sebetralstat 300 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE 101 (UNII: 7T9FYH5QMK) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) POVIDONE K30 (UNII: U725QWY32X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL GRAFT POLYETHYLENE GLYCOL COPOLYMER (3:1; 45000 MW) (UNII: 23ZQ42JZZH) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GLYCERYL MONO- AND DICAPRYLOCAPRATE (UNII: U72Q2I8C85) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) FERROSOFERRIC OXIDE (UNII: XM0M87F357) MALTODEXTRIN (UNII: 7CVR7L4A2D) GUAR GUM (UNII: E89I1637KE) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color yellow Score no score Shape OVAL Size 15mm Flavor Imprint Code 300 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 82928-300-04 4 in 1 CARTON 07/07/2025 1 NDC: 82928-300-01 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219301 07/07/2025 Labeler - KalVista Pharmaceuticals Ltd (156762366) Establishment Name Address ID/FEI Business Operations Catalent Nottingham Limited 520311192 manufacture(82928-300) Establishment Name Address ID/FEI Business Operations Almac Pharma Services Limited 233170864 pack(82928-300) , label(82928-300) Establishment Name Address ID/FEI Business Operations Zach System S.A. 552044943 api manufacture(82928-300)


Trademark Results [EKTERLY]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 EKTERLY 97832368 not registered Live/Pending |
Kalvista Pharmaceuticals Limited 2023-03-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.