IMDELLTRA (AMG757)- tarlatamab-dlle kit
IMDELLTRA (AMG757) by
Drug Labeling and Warnings
IMDELLTRA (AMG757) by is a Prescription medication manufactured, distributed, or labeled by Amgen Inc, Amgen, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IMDELLTRA® safely and effectively. See full prescribing information for IMDELLTRA.
IMDELLTRA® (tarlatamab-dlle) for injection, for intravenous use
Initial U.S. Approval: 2024WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
See full prescribing information for complete boxed warning.
Cytokine release syndrome (CRS), including life-threatening or fatal reactions, can occur in patients receiving IMDELLTRA. Initiate treatment with the IMDELLTRA using step-up dosing schedule to reduce the incidence and severity of CRS. Withhold IMDELLTRA until CRS resolves or permanently discontinue based on severity. (2.5, 5.1)
Neurologic toxicity and Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), including life-threatening or fatal reactions, can occur in patients receiving IMDELLTRA. Monitor patients for signs or symptoms of neurologic toxicity, including ICANS, during treatment and treat promptly. Withhold IMDELLTRA until ICANS resolves or permanently discontinue based on severity. (2.5, 5.2)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
IMDELLTRA is a bispecific delta-like ligand 3 (DLL3)-directed CD3 T- cell engager indicated for the treatment of adult patients with extensive stage small cell lung cancer (ES-SCLC) with disease progression on or after platinum-based chemotherapy. (1)
DOSAGE AND ADMINISTRATION
Administer as an intravenous infusion over 1 hour. (2.2)
- Administer IMDELLTRA according to the step-up dosing schedule in Table 1 to reduce the risk of cytokine release syndrome. (2.2)
- Administer concomitant medications as recommended. (2.3)
- Monitor patients from the start of the IMDELLTRA infusion for 22 to 24 hours on Cycle 1 Day 1 and Cycle 1 Day 8 in an appropriate healthcare setting.
- Recommend patients to remain within 1 hour of an appropriate healthcare setting for a total of 48 hours from the start of the infusion with IMDELLTRA following Cycle 1 Day 1 and Cycle 1 Day 8 doses, accompanied by a caregiver. (2.2)
- See Full Prescribing Information for instructions on preparation and administration. (2.2, 2.6)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Cytopenias: Monitor complete blood counts prior to administration of all doses of IMDELLTRA up through Cycle 5 Day 15 and then prior to administration of IMDELLTRA on Day 1 of each cycle starting with Cycle 6. More frequent evaluation may be necessary as clinically indicated. Withhold or permanently discontinue based on severity. (5.3)
- Infections: Monitor for signs and symptoms of infection; treat appropriately. Withhold or permanently discontinue based on severity. (5.4)
- Hepatotoxicity: Monitor liver enzymes and bilirubin prior to administration of all doses of IMDELLTRA up through Cycle 5 Day 15 and then prior to administration of IMDELLTRA on Day 1 of each cycle starting with Cycle 6. More frequent evaluation may be necessary as clinically indicated. Withhold or permanently discontinue based on severity. (5.5)
-
Hypersensitivity: Monitor for signs and symptoms of hypersensitivity and treat accordingly. Withhold or permanently discontinue based on severity. (5.6)
Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception (5.7, 8.1, 8.3)
ADVERSE REACTIONS
- The most common adverse reactions (> 20%) were cytokine release syndrome, fatigue, decreased appetite, anemia, dysgeusia, pyrexia, constipation, musculoskeletal pain, and nausea.
- The most common (≥ 5%) Grade 3 or 4 laboratory abnormalities were decreased lymphocytes, decreased sodium, decreased total neutrophils, and increased uric acid.
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
USE IN SPECIFIC POPULATIONS
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Information
2.2 Recommended Dosage and Administration
2.3 Recommended Concomitant Medications for IMDELLTRA Administration for Cycle 1 Day 1 and Cycle 1 Day 8
2.4 Restarting IMDELLTRA After Dosage Delay
2.5 IMDELLTRA Dosage Modifications and Adverse Reaction Management
2.6 Preparation
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
5.2 Neurologic Toxicity Including ICANS
5.3 Cytopenias
5.4 Infections
5.5 Hepatotoxicity
5.6 Hypersensitivity
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Small Cell Lung Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CYTOKINE RELEASE SYNDROME and NEUROLOGIC TOXICITY including IMMUNE EFFECTOR CELL-ASSOCIATED NEUROTOXICITY SYNDROME
Cytokine release syndrome (CRS), including life-threatening or fatal reactions, can occur in patients receiving IMDELLTRA. Initiate IMDELLTRA using the step-up dosing schedule to reduce the incidence and severity of CRS. Withhold IMDELLTRA until CRS resolves or permanently discontinue based on severity [see Dosage and Administration (2.5) and Warnings and Precautions (5.1)].
Neurologic toxicity and immune effector cell-associated neurotoxicity syndrome (ICANS), including life-threatening or fatal reactions, can occur in patients receiving IMDELLTRA. Monitor patients for signs and symptoms of neurologic toxicity, including ICANS, during treatment and treat promptly. Withhold IMDELLTRA until ICANS resolves or permanently discontinue based on severity [see Dosage and Administration (2.5) and Warnings and Precautions (5.2)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Information
- Administer IMDELLTRA according to the step-up dose and schedule in Table 1 to reduce the incidence and severity of cytokine release syndrome (CRS) [see Dosage and Administration (2.2)].
- Evaluate complete blood count, liver enzymes and bilirubin prior to administration of all doses of IMDELLTRA up through Cycle 5 Day 15 and then prior to administration of IMDELLTRA on Day 1 of each cycle starting with Cycle 6. More frequent evaluation may be necessary if clinically indicated [see Warnings and Precautions (5.3, 5.5)].
- Ensure patients are well hydrated prior to administration of IMDELLTRA [see Warnings and Precautions (5.1)].
- For Cycle 1, administer recommended concomitant medications in Table 3 before and after Cycle 1 Day 1 and Cycle 1 Day 8 IMDELLTRA infusions to reduce the risk of CRS reactions [see Dosage and Administration (2.3)].
- IMDELLTRA should only be administered by a qualified healthcare professional with appropriate medical support to manage severe reactions, such as CRS and neurologic toxicity, including immune effector cell-associated neurotoxicity syndrome (ICANS) [see Warnings and Precautions (5.1, 5.2)].
- Due to the risk of CRS and neurologic toxicity, including ICANS, monitor patients from the start of the IMDELLTRA infusion for 22 to 24 hours on Cycle 1 Day 1 and Cycle 1 Day 8 in an appropriate healthcare setting [see Dosage and Administration (2.5) and Warnings and Precautions (5.1, 5.2)].
- Recommend patients to remain within 1 hour of an appropriate healthcare setting for a total of 48 hours from start of the infusion with IMDELLTRA following Cycle 1 Day 1 and Cycle 1 Day 8 doses, accompanied by a caregiver.
- Inform both the patient and the caregiver on the signs and symptoms of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) prior to discharge.
2.2 Recommended Dosage and Administration
- Administer IMDELLTRA as an intravenous infusion for one hour.
- The recommended step-up dose and schedule for IMDELLTRA is provided in Table 1. Administer step-up dose and schedule on Cycle 1 Day 1 to reduce the incidence and severity of CRS.
- After step-up dose and schedule on Cycle 1 Day 1, administer IMDELLTRA every 2 weeks until disease progression or unacceptable toxicity.
Table 1. Recommended Dose and Schedule of IMDELLTRA Dosing Schedule Day Dose of IMDELLTRA Administration Instructions Recommended Monitoring Note: See Table 4 for recommendation on restarting IMDELLTRA after dose delays. - * Administer recommended concomitant medications before and after Cycle 1 Day 1 and Cycle 1 Day 8 IMDELLTRA infusions as described in Table 3.
- † Extended monitoring in a healthcare setting is not required unless the patient experiences Grade ≥ 2 CRS, ICANS or neurological toxicity during prior treatments. See Tables 5 and 6 for monitoring recommendations.
Step-up Dose and Schedule Cycle 1 Day 1* Step-up dose* 1 mg Administer IMDELLTRA as a 1-hour intravenous infusion in an appropriate healthcare setting. Monitor patients from the start of the IMDELLTRA infusion for 22 to 24
hours on Cycle 1 Day 1 and Cycle 1 Day 8 in an appropriate healthcare setting.
Recommend that patients remain within 1 hour of an appropriate healthcare setting for a total of 48 hours from start of the IMDELLTRA infusion accompanied by a caregiver.Day 8* 10 mg* Day 15 10 mg Observe patients for 6-8 hours post IMDELLTRA infusion† Cycle 2 Day 1 and 15 10 mg Observe patients for 6-8 hours post IMDELLTRA infusion†. Cycles 3 and 4 Day 1 and 15 10 mg Observe patients for 3-4 hours post IMDELLTRA infusion†. Cycle 5 and subsequent infusions Day 1 and 15 10 mg Observe patients for 2 hours post IMDELLTRA infusion†. Administration
- The intravenous (IV) catheter for concomitant medications administration can be used to administer the IMDELLTRA infusion.
- To ensure patency, flush the IV catheter over 3 to 5 minutes using 0.9% Sodium Chloride for Injection.
- Administer the reconstituted and diluted IMDELLTRA as a 1-hour intravenous infusion at a constant flow rate using an infusion pump. The pump should be programmable, lockable, non-elastomeric, and have an alarm. Flush the IV-line upon completion of the IMDELLTRA infusion.
Table 2 provides the infusion duration and rate.
Table 2. IMDELLTRA Infusion Duration and Rate Infusion Duration for 250 mL IV Preparation Infusion Rate 1 hour 250 mL/hour 2.3 Recommended Concomitant Medications for IMDELLTRA Administration for Cycle 1 Day 1 and Cycle 1 Day 8
Administer recommended concomitant medications for IMDELLTRA during Cycle 1 Day 1 and Cycle 1 Day 8 as presented in Table 3 to reduce the risk of CRS [see Warnings and Precautions (5.1)].
Table 3. Recommended Concomitant Medications for IMDELLTRA Administration for Cycle 1 Day 1 and Cycle 1 Day 8 Treatment Day Medication Administration Cycle 1 Day 1 and Cycle 1 Day 8 Administer dexamethasone 8 mg intravenously (or equivalent) Within 1 hour prior to IMDELLTRA administration Administer 1 liter of normal saline intravenously over 2 to 4 hours Immediately after completion of IMDELLTRA infusion 2.4 Restarting IMDELLTRA After Dosage Delay
If a dose of IMDELLTRA is delayed, restart based on the recommendation as listed in Table 4 and resume the dose and schedule accordingly [see Dosage and Administration (2.2)].
Administer recommended concomitant medications as indicated in Table 3.
Table 4. Recommendations for Restarting IMDELLTRA After Dosage Delay Last Dose Administered Time Since the Last Dose Administered Action* - * Administer recommended concomitant medications before and after Cycle 1 Day 1 and Cycle 1 Day 8 IMDELLTRA infusions and monitor patients accordingly [see Dosage and Administration (2.1, 2.2 and 2.3)].
1 mg on Cycle 1 Day 1 2 weeks or less (≤ 14 days) Administer IMDELLTRA 10 mg, then resume with the planned dose and schedule. Greater than 2 weeks (> 14 days) Administer IMDELLTRA step-up dose 1 mg. If tolerated, increase to 10 mg 1 week later. Then resume with the planned dose and schedule. 10 mg on Cycle 1 Day 8 3 weeks or less (≤ 21 days) Administer IMDELLTRA 10 mg, then resume with the planned dose and schedule. Greater than 3 weeks (> 21 days) Administer IMDELLTRA step-up dose 1 mg. If tolerated, increase to 10 mg 1
week later. Then resume with the planned dose and schedule.10 mg on Cycle 1 Day 15 and subsequent Cycles every 2 weeks thereafter 4 weeks or less (≤ 28 days) Administer IMDELLTRA 10 mg, then resume with the planned dose and schedule. Greater than 4 weeks (> 28 days) Administer IMDELLTRA step-up dose 1 mg. If tolerated, increase to 10 mg 1
week later. Then resume with the planned dose and schedule.2.5 IMDELLTRA Dosage Modifications and Adverse Reaction Management
No dose reduction for IMDELLTRA is recommended. See Table 5 and Table 6 for recommended management of CRS, neurologic toxicity including ICANS respectively and Table 7 for cytopenias, infections and other adverse reactions.
Cytokine Release Syndrome (CRS)
Diagnose CRS based on clinical presentation [see Warnings and Precautions (5.1)]. Evaluate for and treat other causes of fever, hypoxia, and hypotension.
If CRS is suspected, manage according to the recommendations in Table 5. Monitor patients who experience Grade 2 or higher CRS (e.g., hypotension not responsive to fluids, or hypoxia requiring supplemental oxygen) with continuous cardiac telemetry and pulse oximetry.
For severe or life-threatening CRS, recommend administering tocilizumab or equivalent therapy and intensive monitoring (e.g., ICU) for supportive therapy. Perform laboratory testing to monitor for disseminated intravascular coagulation (DIC), hematology parameters, as well as pulmonary, cardiac, renal, and hepatic function. Table 5 provides the guidelines for grading and dosage modification and management of cytokine release syndrome.
Table 5. Guidelines for Grading and Dosage Modification and Management of Cytokine Release Syndrome* CRS Grade Defining Symptoms IMDELLTRA Dosage Modification Management - * CRS based on American Society for Transplantation and Cellular Therapy (ASTCT) Consensus
Grading (2019).- † See Table 4 for recommendations on restarting IMDELLTRA after dose delays [see Dosage and Administration (2.4)].
- ‡ Taper steroids per standard of care guidelines.
Grade 1 Symptoms require symptomatic treatment only (e.g., fever ≥ 100.4°F without hypotension or hypoxia). Withhold IMDELLTRA until event resolves, then resume IMDELLTRA at the next scheduled dose†. - Administer symptomatic treatment (e.g., acetaminophen) for fever.
- Consider dexamethasone 4 mg to 10 mg oral or IV (or equivalent)‡.
Grade 2 Symptoms require and respond to moderate intervention. Fever ≥ 100.4°F, - Hypotension responsive to fluids not requiring vasopressors and/or
- Hypoxia requiring low flow nasal cannula or blow-by.
Withhold IMDELLTRA until event resolves, then resume IMDELLTRA at the next scheduled dose†. - Recommend hospitalization for a minimum of 24 hours with cardiac telemetry and pulse oximetry.
- Administer symptomatic treatment (e.g., acetaminophen) for fever.
- Administer supplemental oxygen and intravenous fluids when indicated.
- Consider dexamethasone‡ (or equivalent) 8 mg oral or IV.
- Consider tocilizumab (or equivalent).
Grade 3 Severe symptoms defined as temperature ≥ 100.4°F with: - Hemodynamic instability requiring a vasopressor (with or without vasopressin) and/or
- Worsening hypoxia or respiratory distress requiring high flow nasal cannula (> 6 L/min oxygen) or face mask.
Withhold IMDELLTRA until the event resolves, then resume IMDELLTRA at the next scheduled dose†.
For recurrent Grade 3 events, permanently discontinue IMDELLTRA.In addition to Grade 2 treatment: - Recommend intensive monitoring, e.g., ICU care.
- Administer dexamethasonec (or equivalent) 8 mg IV every 8 hours up to 3 doses.
- Vasopressor support as needed.
- High flow oxygen support as needed.
- Recommend tocilizumab (or equivalent).
When resuming the next planned dose, monitor patients from the start of the IMDELLTRA infusion for 22 to 24 hours in an appropriate healthcare setting†.Grade 4 Life-threatening symptoms defined as temperature ≥ 100.4°F with: - Hemodynamic instability requiring multiple vasopressors (excluding vasopressin).
and/or
Worsening hypoxia or respiratory distress despite oxygen administration requiring positive pressure.
Permanently discontinue IMDELLTRA. - ICU care.
- Per Grade 3 treatment. Recommend tocilizumab (or equivalent)
Neurologic Toxicity including ICANS
At the first sign of neurologic toxicity, including ICANS, withhold IMDELLTRA and consider neurology evaluation. Rule out other causes of neurologic symptoms. Provide supportive therapy, which may include intensive care, for severe or life-threatening neurologic toxicities, including ICANS [see Warnings and Precautions (5.2)]. Manage ICANS and neurologic toxicity according to the recommendations in Table 6 and consider further management per current practice guidelines.
Table 6. Guidelines for Management of Neurologic Toxicity including Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS)* ICANS Grade* Defining Symptoms IMDELLTRA Dosage Modifications Management - * ICANS based on American Society for Transplantation and Cellular Therapy (ASTCT) Consensus Grading (2019)
- † If patient is arousable and able to perform Immune Effector Cell-Associated Encephalopathy (ICE) Assessment, assess: Orientation (oriented to year, month, city, hospital = 4 points); Naming (names 3 objects, e.g., point to clock, pen, button = 3 points); Following commands (e.g., "show me 2 fingers" or "close your eyes and stick out your tongue" = 1 point); Writing (ability to write a standard sentence = 1 point); and Attention (count backwards from 100 by ten = 1 point). If patient is unarousable and unable to perform ICE Assessment (Grade 4 ICANS) = 0 points
- ‡ See Table 4 for recommendations on restarting IMDELLTRA after dose delays [see Dosage and Administration (2.4)]
- § Taper steroids per standard of care guidelines
Grade 1 ICE score 7-9† with no depressed level of consciousness. - Withhold IMDELLTRA until ICANS resolves, then resume IMDELLTRA at the next scheduled dose‡.
- Supportive care.
Grade 2 ICE score 3-6† and/or mild somnolence awaking to voice. - Withhold IMDELLTRA until ICANS resolves, then resume IMDELLTRA at the next scheduled dose‡.
- Supportive care.
- Dexamethasone§ (or equivalent) 8 to 10 mg oral or IV. Can repeat every 12 hours or methylprednisolone§ (or equivalent) 1 mg/kg IV every 12 hours if symptoms worsen.
- Monitor neurologic symptoms and consider consultation with neurologist and other specialists for further evaluation and management.
- Monitor patients from the start of the IMDELLTRA infusion for 22 to 24 hours following the next dose of IMDELLTRA.
Grade 3 ICE score 0-2† and/or depressed level of consciousness awakening only to tactile stimulus and/or any clinical seizure focal or generalized that resolves rapidly or nonconvulsive seizures on EEG that resolve with intervention and/or
Focal or local edema on neuroimaging.- Withhold IMDELLTRA until the ICANS resolves, then resume IMDELLTRA at the next scheduled dose‡.
- If there is no improvement to Grade ≤ 1 within 7 days permanently discontinue IMDELLTRA.
- For recurrent Grade 3 events, permanently discontinue IMDELLTRA.
- Recommend intensive monitoring, e.g., ICU care.
- Consider mechanical ventilation for airway protection. Dexamethasone§ (or equivalent) 10 mg IV every 6 hours or methylprednisolone§ (or equivalent) 1 mg/kg IV every 12 hours.
- Consider repeat neuroimaging (CT or MRI) every 2-3 days if patient has persistent Grade ≥ 3 neurotoxicity.
Monitor patients from the start of the IMDELLTRA infusion for 22 to 24 hours following the next dose of IMDELLTRA.
Grade 4 ICE score 0† (patient is unarousable and unable to perform ICE) and/or Stupor or coma and/or Life-threatening prolonged seizure (> 5 minutes) or repetitive clinical or electrical seizures without return to baseline in between and/or diffuse cerebral edema on neuroimaging, decerebrate or decorticate posturing or papilledema, cranial nerve VI palsy, or Cushing's triad. - Permanently discontinue IMDELLTRA.
- ICU care.
- Consider mechanical ventilation for airway protection.
- High dose corticosteroids (e.g., methylprednisolone§ 1000 mg/day in divided doses IV for 3 days).
- Consider repeat neuroimaging (CT or MRI) every 2-3 days if patient has persistent Grade ≥ 3 neurotoxicity.
- Treat convulsive status epilepticus per institutional guidelines.
Table 7. Recommended Treatment Interruptions of IMDELLTRA for the Management of Cytopenias, Infections, and Other Adverse Reactions Adverse Reactions Severity* Dosage Modification† - * Severity based on National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 5.0.
- † Refer to Table 4 for dose restarting guidance.
Cytopenias [see Warnings and Precautions (5.3)] Grade 3 Neutropenia Withhold IMDELLTRA until recovery to Grade ≤ 2.
Consider administration of granulocyte colony stimulating factor (G-CSF).
Permanently discontinue if recovery to Grade ≤ 2 does not occur within 3 weeks.Grade 4 Neutropenia Withhold IMDELLTRA until recovery to Grade ≤ 2.
Consider administration of granulocyte colony stimulating factor (G-CSF).
Permanently discontinue if recovery to Grade ≤ 2 does not occur within 1 week.Recurrent Grade 4 Neutropenia Permanently discontinue IMDELLTRA Febrile neutropenia Withhold IMDELLTRA until neutropenia recovers to Grade ≤ 2 and fever resolves. Hemoglobin <8 g/dL Withhold IMDELLTRA until hemoglobin is ≥8 g/dL. Grade 3 or Grade 4 Decreased platelet count Withhold IMDELLTRA until platelet count is Grade ≤ 2 and no evidence of bleeding.
Permanently discontinue if recovery to Grade ≤ 2 does not occur within 3 weeks.Recurrent Grade 4 Decreased platelet count Permanently discontinue IMDELLTRA. Infections [see Warnings and Precautions (5.4)] All Grades Withhold IMDELLTRA in the step-up phase in patients until infection resolves. Grade 3 Withhold IMDELLTRA during the treatment phase until infection improves to Grade ≤ 1†. Grade 4 Permanently discontinue IMDELLTRA. Hepatotoxicity [see Warnings and Precautions (5.5)] Grade 3
Increased ALT or AST or bilirubinWithhold IMDELLTRA until improved to Grade ≤ 1. Grade 4
Increased ALT or AST or bilirubinPermanently discontinue IMDELLTRA. AST or ALT > 3 × ULN with total bilirubin > 2 × ULN in the absence of alternative causes Permanently discontinue IMDELLTRA. Other Adverse Reactions [see Adverse Reactions (6.1)] Grade 3 or 4 Withhold IMDELLTRA until recovery to Grade ≤ 1 or baseline.
Consider permanently discontinuing if adverse reaction does not resolve within 28 days.
Consider permanent discontinuation for Grade 4 events.2.6 Preparation
Material Compatibility Information
- IV bags composed of ethyl vinyl acetate (EVA), polyolefin, and polyvinyl chloride (PVC) have been shown to be compatible with IMDELLTRA at the specified administration conditions.
- IV line and catheter materials composed of polyolefin, PVC, and polyurethane have been shown to be compatible with IMDELLTRA at the specified administration conditions.
- The use of Closed System Transfer Device (CSTD) is not recommended due to potential wrong dose medication error risk. Amgen has not performed compatibility testing of vial adaptor CSTDs with IMDELLTRA.
Step 1: Reconstitute IMDELLTRA with Sterile Water for Injection
- Table 8 provides the required amount of sterile water for injection required to reconstitute IMDELLTRA 1 mg and 10 mg vials.
Do not use IV Solution Stabilizer (IVSS) to reconstitute IMDELLTRA.
The IV Solution Stabilizer (IVSS) is used to coat the intravenous bag prior to addition of reconstituted IMDELLTRA to prevent adsorption of IMDELLTRA to IV bags and IV tubing.
Table 8. Required Amount of Sterile Water for Injection to Reconstitute IMDELLTRA* IMDELLTRA Vial Strength Amount of Sterile Water for Injection Needed to Reconstitute IMDELLTRA Resulting Concentration - * Each vial contains overfill to allow for withdrawal of 1.1 mL (1 mg vial) or 4.2 mL (10 mg vial) after reconstitution to ensure delivery at the stated concentration of labeled vial strength.
1 mg 1.3 mL 0.9 mg/mL 10 mg 4.4 mL 2.4 mg/mL - Using a needle and syringe filled with the required amount of sterile water, inject the sterile water against the glass vial. Avoid injecting the water directly onto the powder to prevent foaming.
- Gently swirl the contents to mix. Do not shake.
- Inspect parenteral drug products for particulate matter and discoloration prior to administration. Inspect that the solution is clear to opalescent, colorless to slightly yellow. Do not use if the solution is cloudy or has particulates.
- Further dilute reconstituted IMDELLTRA.
- The reconstituted IMDELLTRA must be further diluted within 4 hours of reconstitution or discarded.
Prepare the infusion bag: Steps 2 to 5
Step 2 : Withdraw 0.9% Sodium Chloride for Injection
- Using a 250 mL prefilled bag of 0.9% Sodium Chloride for Injection, withdraw the amount of sodium chloride specified in Table 9 and discard.
Table 9. Required Amount of 0.9% Sodium Chloride to Withdraw from 250 mL IV Bag IMDELLTRA Vial Strength IMDELLTRA Dose Volume of 0.9% Sodium Chloride to Withdraw From 250 mL IV Bag 1 mg 1 mg 14 mL 10 mg 10 mg 17 mL Step 3: Add IV Solution Stabilizer to the infusion bag
- Inject 13 mL of IV Solution Stabilizer (IVSS) into the 250 mL 0.9% Sodium Chloride infusion bag, see Table 10.
- Gently mix the contents of the infusion bag to avoid foaming. Do not shake.
Table 10. Required Amount of IV Solution Stabilizer (IVSS) to Add to IV Bag IMDELLTRA Vial Strength IMDELLTRA Dose Volume of IV Solution Stabilizer (IVSS) to Add to IV Bag 1 mg 1 mg 13 mL 10 mg 10 mg 13 mL Step 4: Dilute the reconstituted IMDELLTRA into the infusion bag
- Transfer the required volume of reconstituted IMDELLTRA listed in Table 11 to the infusion bag (containing IV Solution Stabilizer).
NOTE: The final concentrations for the different strength vials are NOT the same following reconstitution and further dilution.
Table 11. Required Amount of Reconstituted IMDELLTRA to Add to 250 mL IV Bag IMDELLTRA Vial Strength IMDELLTRA Dose Volume of Reconstituted IMDELLTRA to Add to 250 mL IV Bag 1 mg 1 mg 1.1 mL 10 mg 10 mg 4.2 mL - Gently mix the contents of the bag. Do not shake.
Step 5: Remove air from IV bag
Remove air from the prepared IV bag using an empty syringe to avoid foaming.
Step 6: Prime IV tubing
- Prime intravenous tubing with either 0.9% Sodium Chloride for Injection or with the final prepared product.
- See Table 12 for maximum storage time of prepared IMDELLTRA infusion.
Prepared IMDELLTRA Infusion Bag Storage Requirements
- Administer reconstituted and diluted IMDELLTRA immediately.
- Table 12 displays the maximum storage time for the prepared IMDELLTRA infusion bag.
- Maximum storage time includes total duration from the time of reconstitution of the vial of IMDELLTRA to the end of the infusion.
Table 12. Maximum Storage Time for Prepared IMDELLTRA Infusion Bag Room Temperature 20°C to 25°C (68°F to 77°F) Refrigerated 2°C to 8°C (36°F to 46°F) Prepared IMDELLTRA Infusion Bag 8 hours 7 days - Discard the prepared IMDELLTRA infusion bag after maximum storage time (from time of reconstitution).
- If refrigerated, allow the prepared IMDELLTRA infusion bag to come room temperature prior to administration, and complete the infusion within 8 hours (including preparation and infusion time).
- Do not re-refrigerate prepared infusion bag.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
IMDELLTRA can cause cytokine release syndrome (CRS) including life-threatening or fatal reactions.
In the pooled safety population [see Adverse Reactions (6.1)], CRS occurred in 57% (268/473) of patients who received IMDELLTRA, including 39% Grade 1, 15% Grade 2, 1.7% Grade 3 and 0.2% Grade 4. Recurrent CRS occurred in 24% of IMDELLTRA-treated patients including 20% Grade 1 and 3.4% Grade 2; one patient experienced recurrent Grade 3.
Among the 268 patients who experienced CRS, 73% had CRS after the first dose, 60% had CRS after the second dose, and 15% had CRS following the third or later dose. Following the Cycle 1 Day 1, Day 8, Day 15 infusions, 24%, 8%, and 1% of patients experienced Grade ≥ 2 CRS, respectively. From Cycle 2 onwards, 1.5% of patients experienced Grade ≥ 2 CRS. Of the patients who experienced CRS, 31% received steroids and 10% required tocilizumab. The median time to onset of all grade CRS from most recent dose of IMDELLTRA was 16 hours (range: start of infusion to 15 days). The median time to onset of Grade ≥ 2 CRS from most recent dose of IMDELLTRA was 15 hours (range: start of infusion to 15 days).
Clinical signs and symptoms of CRS included pyrexia, hypotension, fatigue, tachycardia, headache, hypoxia, nausea and vomiting. Potentially life-threatening complications of CRS may include cardiac dysfunction, acute respiratory distress syndrome, neurologic toxicity, renal and/or hepatic failure, and disseminated intravascular coagulation (DIC).
Administer IMDELLTRA following the recommended step-up dosing and administer concomitant medications before and after Cycle 1 Day 1 and Cycle 1 Day 8 IMDELLTRA infusions as described in Table 3 to reduce the risk of CRS [see Dosage and Administration (2.3)]. Administer IMDELLTRA in an appropriate healthcare facility equipped to monitor and manage CRS. Ensure patients are well hydrated prior to administration of IMDELLTRA.
Closely monitor patients for signs and symptoms of CRS during treatment with IMDELLTRA. At the first sign of CRS, immediately discontinue IMDELLTRA infusion, evaluate the patient for hospitalization and institute supportive care based on severity. Withhold or permanently discontinue IMDELLTRA based on severity [see Dosage and Administration (2.5)]. Counsel patients and caregivers to seek medical attention should signs or symptoms of CRS occur.
5.2 Neurologic Toxicity Including ICANS
IMDELLTRA can cause life-threatening or fatal neurologic toxicity including ICANS.
In the pooled safety population [see Adverse Reactions (6.1)], neurologic toxicity occurred in 65% of patients who received IMDELLTRA, with Grade 3 or higher events in 7% of patients including fatal events in 0.2%. The most frequent neurologic toxicities were dysgeusia (34%), headache (17%), peripheral neuropathy (9%), dizziness (9%), and insomnia (8%).
The incidence of signs and symptoms consistent with ICANS was 10% in IMDELLTRA- treated patients, including events with the preferred terms: ICANs (4.7%), muscular weakness (3.2%), cognitive disorder (0.6%), aphasia (0.6%), depressed level of consciousness (0.4%), seizures (0.4%), encephalopathy (0.4%), and leukoencephalopathy (0.2%). There was one fatal reaction of ICANS [see Adverse Reactions (6.1)]. Recurrent ICANS occurred in 1.5% of patients. Of the patients who experienced ICANS, most experienced the event following Cycle 1 Day 1 (2.5%) and Cycle 1 Day 8 (3.6%). Following Day 1, Day 8, and Day 15 infusions, 1.3%, 1.3% and 0.4% of patients experienced Grade ≥ 2 ICANS, respectively. ICANS can occur several weeks following administration of IMDELLTRA. The median time to onset of ICANS from the first dose of IMDELLTRA was 16 days (range: 1 to 862 days). The median time to resolution of ICANS was 4 days (range: 1 to 40 days).
The onset of ICANS can be concurrent with CRS, following resolution of CRS, or in the absence of CRS. Clinical signs and symptoms of ICANS may include but are not limited to confusional state, depressed level of consciousness, disorientation, somnolence, lethargy, and bradyphrenia.
Patients receiving IMDELLTRA are at risk of neurologic adverse reactions and ICANS resulting in depressed level of consciousness. Advise patients to refrain from driving and engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery, until neurologic symptoms resolve.
Closely monitor patients for signs and symptoms of neurologic toxicity and ICANS during treatment with IMDELLTRA. At the first sign of ICANS, immediately discontinue the infusion, evaluate the patient and provide supportive therapy based on severity.
Withhold IMDELLTRA or permanently discontinue based on severity [see Dosage and Administration (2.5)].
5.3 Cytopenias
IMDELLTRA can cause cytopenias including neutropenia, thrombocytopenia, and anemia.
In the pooled safety population, [see Adverse Reactions (6.1)] based on laboratory data, decreased neutrophils occurred in 16% of patients, including 9% Grade 3 or 4. The median time to onset for Grade 3 or 4 decreased neutrophil count was 41 days (range: 2 to 306 days). Decreased platelets occurred in 30%, including 2.2% Grade 3 or 4. The median time to onset for Grade 3 or 4 decreased platelets was 67 days (range: 3 to 420 days). Decreased hemoglobin occurred in 56% of patients, including 4.7% Grade 3 or 4.
Febrile neutropenia was reported as an adverse event in 1.5% of patients treated with IMDELLTRA.
Monitor patients for signs and symptoms of cytopenias. Perform complete blood counts prior to treatment with all doses of IMDELLTRA, up through Cycle 5 Day 15 and then prior to administration of IMDELLTRA on Day 1 of each cycle starting with Cycle 6.
Based on the severity of cytopenias, temporarily withhold or permanently discontinue IMDELLTRA [see Dosage and Administration (2.5)].
5.4 Infections
IMDELLTRA can cause serious infections, including life-threatening and fatal infections.
In the pooled safety population, [see Adverse Reactions (6.1)], infections including opportunistic infections occurred in 43% of patients who received IMDELLTRA, including 14% Grade 3 or 4. The most frequent infections were pneumonia (11%), urinary tract infection (9%), COVID-19 (6%), upper respiratory tract infection (4.7%), respiratory tract infection (4%), candida infection (2.1%), oral candidiasis (2.1%) and nasopharyngitis (2.1%).
Monitor patients for signs and symptoms of infection prior to and during treatment with IMDELLTRA and treat as clinically indicated. Withhold or permanently discontinue IMDELLTRA based on severity [see Dosage and Administration (2.5)].
5.5 Hepatotoxicity
IMDELLTRA can cause hepatotoxicity.
In the pooled safety population [see Adverse Reactions (6.1)], based on laboratory data, elevated ALT occurred in 39% of patients who received IMDELLTRA, including 2.5% Grade 3 or 4 ALT. Elevated AST occurred in 43% of patients, including 3.2% Grade 3 or 4. Elevated bilirubin occurred in 16% of patients, including 1.3% Grade 3 or 4 [see Adverse Reactions (6.1)]. Liver enzyme elevation can occur with or without concurrent CRS.
Monitor liver enzymes and bilirubin prior to treatment with IMDELLTRA, and as clinically indicated. Withhold IMDELLTRA or permanently discontinue based on severity [see Dosage and Administration (2.5)].
5.6 Hypersensitivity
IMDELLTRA can cause severe hypersensitivity reactions.
Clinical signs and symptoms of hypersensitivity may include, but are not limited to, rash and bronchospasm.
Monitor patients for signs and symptoms of hypersensitivity during treatment with IMDELLTRA and manage as clinically indicated. Withhold or consider permanent discontinuation of IMDELLTRA based on severity [see Dosage and Administration (2.5)].
5.7 Embryo-Fetal Toxicity
Based on its mechanism of action, IMDELLTRA may cause fetal harm when administered to a pregnant woman. Advise patients of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with IMDELLTRA and for 2 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Cytokine Release Syndrome (CRS) [see Warnings and Precautions (5.1)]
- Neurologic Toxicity Including ICANS [see Warnings and Precautions (5.2)]
- Cytopenias [see Warnings and Precautions (5.3)]
- Infections [see Warnings and Precautions (5.4)]
- Hepatotoxicity [see Warnings and Precautions (5.5)]
- Hypersensitivity [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflects exposure to intravenous IMDELLTRA, as a single agent, at the recommended dosage of IMDELLTRA 1 mg on Cycle 1 Day 1 followed by 10 mg on Days 8 and 15, and then every 2 weeks until disease progression or intolerable toxicity in 473 patients with small cell lung cancer enrolled in three clinical trials: DeLLphi-300, DeLLphi-301 and DeLLphi-304. Among 473 patients who received IMDELLTRA, 40% were exposed for 6 months or longer and 19% were exposed for greater than one year. The most common (≥ 20%) adverse reactions were CRS (57%), fatigue (48%), decreased appetite (38%), dysgeusia (34%), pyrexia (33%), constipation (31%), musculoskeletal pain (31%), and nausea (25%). The most common (≥ 5%) Grade 3 or 4 laboratory abnormalities were decreased lymphocytes (43%), decreased sodium (12%), decreased total neutrophils (9%), and increased uric acid (6%).
Extensive Stage Small Cell Lung Cancer
The safety of IMDELLTRA was evaluated in 252 patients in DeLLphi-304, a multicenter, randomized, open label trial in patients with extensive stage small cell lung cancer (ES- SCLC) with disease progression following treatment with platinum-based chemotherapy with or without an anti-PD-(L)1 antibody [see Clinical Studies (14.1)]. Patients received IMDELLTRA (n=252) or investigator's choice or investigator's choice of topotecan [n=176], lurbinectedin [n=45] or amrubicin [n=23].
Among patients who received IMDELLTRA, 41% were exposed for 6 months or longer and 18% were exposed for greater than one year.
The demographic characteristics of patients who received IMDELLTRA were: median age 64 years (range: 20 to 86); 71% male; 60% White, 38 % Asian, 0.8% Black or African American; and 4.8% were of Hispanic or Latino ethnicity.
Serious adverse reactions occurred in 52% of patients who received IMDELLTRA. Serious adverse reactions in >3% of patients included CRS (17%), pyrexia (6%), pneumonia (5%) and ICANS (3.6%). Fatal adverse reactions occurred in 8% of patients who received IMDELLTRA, including one fatal adverse reaction of ICANS (0.4%). Fatal adverse reactions occurring in more than one patient included pneumonia (1.6%), cardio-respiratory arrest (1.6%), and sepsis (0.8%).
Permanent discontinuation of IMDELLTRA due to an adverse reaction occurred in 6% of patients. Adverse reactions which resulted in permanent discontinuation of IMDELLTRA in > 1% of patients included pneumonia (1.2%).
Dosage interruptions of IMDELLTRA due to an adverse reaction occurred in 38% of patients. Adverse reactions which required dosage interruption in ≥ 2% of patients included neutropenia (5%), fatigue (4.4%), pneumonia (4%), decreased appetite (2.8%), COVID-19 (2%).
Table 13 summarizes adverse reactions observed in DeLLphi-304.
Table 13. Adverse Reactions (≥ 15%) in Patients with SCLC Who Received IMDELLTRA in DeLLphi-304 Adverse Reaction IMDELLTRA*
(N = 252)Standard of Care
(N = 244)Any Grade
(%)Grade 3 or 4
(%)Any Grade
(%)Grade 3 or 4
(%)- * Graded using CTCAE Version 4.0 and Version 5.0.
- † Based on American Society for Transplantation and Cellular Therapy (ASTCT) 2019.
- ‡ Includes fatigue and asthenia
- § Includes body temperature increased, hyperthermia, pyrexia
- ¶ Includes ageusia, dysgeusia, hypogeusia
- # Includes headache and tension headache
- Þ Includes arthralgia, back pain, bone pain, musculoskeletal pain, myalgia, neck pain, non-cardiac chest pain, pain in extremity, spinal pain
- ß Includes cough and productive cough
Immune system disorders Cytokine release syndrome† 56 1.2 1.2 0 General disorders and administration site conditions Fatigue‡ 39 6 43 10 Pyrexia§ 29 1.2 11 1.2 Metabolism and nutrition disorders Decreased appetite 37 2 23 1.6 Gastrointestinal disorders Constipation 30 0.4 22 0 Nausea 25 0.4 32 0 Nervous system disorders Dysgeusia¶ 28 0 2.5 0 Headache# 16 0 9 0 Musculoskeletal and connective tissue disorders Musculoskeletal painÞ 27 1.6 21 2.5 Respiratory, thoracic and mediastinal disorders Coughß 17 0 17 0 Clinically relevant adverse reactions occurring in < 15% of patients who received IMDELLTRA were immune effector cell-associated neurotoxicity syndrome, neurotoxicity, tremor, seizure, ataxia, confusional state, delirium, dyspnea, encephalopathy and weight decreased.
Table 14 summarizes laboratory abnormalities in DeLLphi-304.
Table 14. Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients with SCLC in DeLLphi-304 Laboratory Abnormality IMDELLTRA*
N=252Standard of care
N=244All Grades
(%)Grade 3 or 4
(%)All Grades
(%)Grade 3 or 4
(%)- * The denominator used to calculate the rate varied for IMDELLTRA (Range: 229 to 250) and SOC (Range: 205 to 226) based on the number of patients with a baseline value and at least one post-treatment value.
- † All Grade lab abnormalities occurring at a frequency less than 20% included decreased neutrophils.
Hematology Lymphocytes decreased 65 27 62 27 Hemoglobin decreased 51 4.5 86 29 White blood cells decreased 50 7 70 29 Platelets decreased 25 0.4 55 20 Neutrophils decreased† 15 10 44 36 Chemistry Sodium decreased 57 8 38 7 Potassium decreased 41 4.8 34 4 Aspartate amino transferase increased 40 2.8 29 0.4 Sodium increased 35 0.4 27 0 Alanine aminotransferase increased 32 2 25 0.9 Activated Partial Thromboplastin Time (sec) increased 26 1.3 16 0.9 Creatinine increased 23 0.8 19 0.4 Alkaline phosphate increased 22 0.4 26 1.4 Magnesium decreased 21 0.8 15 1.8 Potassium increased 21 0.8 12 1.8 Creatine Phosphokinase increased 21 1.7 11 0 DeLLphi-300 and DeLLphi-301
The safety of IMDELLTRA, as a single agent, at the recommended dosage was evaluated in patients with extensive stage small cell lung cancer enrolled in DeLLphi-300 and DeLLphi-301 [see Clinical Studies (14.1)]. Among 187 patients who received IMDELLTRA, 31% were exposed for 6 months or longer and 14% were exposed for greater than one year.
The demographic characteristics of patients who received IMDELLTRA were: median age 66 years (range: 35 to 82); 65% male; 70% White, 26% Asian, 2.1% Black or African American; and 2.1% Hispanic or Latino.
Serious adverse reactions occurred in 58% of patients who received IMDELLTRA. Serious adverse reactions in >3% of patients included cytokine release syndrome (24%), pneumonia (6%), pyrexia (3.7%) and hyponatremia (3.6%). Fatal adverse reactions occurred in 2.7% of patients who received IMDELLTRA including pneumonia 0.5%, aspiration (0.5%), pulmonary embolism (0.5%), respiratory acidosis (0.5%), and respiratory failure (0.5%).
Permanent discontinuation of IMDELLTRA due to an adverse reaction occurred in 7% of patients. Adverse reactions which resulted in permanent discontinuation of IMDELLTRA in >1% of patients included cytokine release syndrome (1.6%) and tumor lysis syndrome (1.1%).
Dosage interruptions of IMDELLTRA due to an adverse reaction occurred in 27% of patients. Adverse reactions which required dosage interruption in ≥ 2% of patients included fatigue (3.2%), cytokine release syndrome (2 .7%) and respiratory tract infection (2.1%).
Table 15 summarizes adverse reactions observed in DeLLphi-300 and DeLLphi-301.
Table 15. Adverse Reactions (≥ 15%) in Patients with ES-SCLC Who Received IMDELLTRA in DeLLphi-300 and DeLLphi-301 Adverse Reaction IMDELLTRA*
(N = 187)Any Grade
(%)Grade 3 or 4
(%)- * Graded using CTCAE Version 4.0 and Version 5.0.
- † Based on American Society for Transplantation and Cellular Therapy (ASTCT) 2019.
- ‡ Includes fatigue and asthenia.
- § Includes myalgia, arthralgia, back pain, pain in extremity, neck pain, musculoskeletal chest pain, non- cardiac chest pain and bone pain.
- ¶ Includes dyspnea and exertional dyspnea.
Immune system disorders Cytokine release syndrome† 55 1.6 General disorders and administration site conditions Fatigue‡ 51 10 Pyrexia 36 0 Nervous system disorders Dysgeusia 36 0 Metabolism and nutrition disorders Decreased appetite 34 2.7 Nausea 22 1.6 Gastrointestinal disorders Constipation 30 0.5 Musculoskeletal and connective tissue disorders Musculoskeletal pain§ 30 1.1 Respiratory, thoracic and mediastinal disorders Dyspnea¶ 17 2.1 Cough 17 0 Table 16 summarizes laboratory abnormalities in DeLLphi-300 and DeLLphi-301 IMDELLTRA* All Grades (%) Grade 3 or 4 (%) Laboratory Abnormality - * The denominator used to calculate the rate varied from 41 to 187 based on the number of patients with a baseline value and at least one post-treatment value.
- † All Grade lab abnormalities occurring at a frequency less than 20% included decreased neutrophils.
Hematology Lymphocytes decreased 84 57 Hemoglobin decreased 58 5 White blood cells decreased 44 3.8 Platelets decreased 33 3.2 Neutrophils decreased† 12 6 Chemistry Sodium decreased 68 16 Potassium decreased 50 5 Aspartate amino transferase increased 44 3.2 Alanine aminotransferase increased 42 2.1 Magnesium decreased 33 1.6 Creatinine increased 29 0.5 Sodium increased 26 0 Alkaline phosphate increased 22 0 -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, IMDELLTRA may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on the use of IMDELLTRA in pregnant women to inform a drug-associated risk.
In an animal reproduction study, a murine surrogate molecule administered intravenously to pregnant mice crossed the placental barrier.
Tarlatamab-dlle causes T-cell activation and cytokine release; immune activation may compromise pregnancy maintenance.
Human immunoglobulin G (IgG) and proteins comprising IgG-derived fragment crystallizable (Fc) domains are known to cross the placental barrier; therefore, IMDELLTRA has the potential to be transmitted from the mother to the developing fetus. Advise women of the potential risk to the fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% - 4% and 15% - 20%, respectively.
Data
Animal Data
Animal reproduction studies have not been conducted with tarlatamab-dlle. In an embryo-fetal developmental toxicity study, a murine surrogate molecule was administered intravenously to pregnant mice during the period of organogenesis. The surrogate molecule crossed the placental barrier and did not cause maternal toxicity, embryo-fetal toxicity or teratogenicity.
8.2 Lactation
Risk Summary
There are no data on the presence of tarlatamab-dlle in human milk or the effects on the breastfed child or on milk production. Maternal IgG is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed child to IMDELLTRA are unknown. Because of the potential for serious adverse reactions in a breastfed child, advise patients not to breastfeed during treatment with IMDELLTRA and for 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
IMDELLTRA may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status of females of reproductive potential prior to initiating IMDELLTRA.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with IMDELLTRA and for 2 months after the last dose.
8.4 Pediatric Use
The safety and effectiveness of IMDELLTRA have not been established in pediatric patients.
8.5 Geriatric Use
Of the 473 patients with SCLC who received IMDELLTRA 10 mg as a single agent, 51% were 65 years of age or older and 11% were 75 years of age or older. No overall differences in IMDELLTRA pharmacokinetics, safety or efficacy were observed between older patients (≥ 65 years of age) and younger patients.
-
11 DESCRIPTION
Tarlatamab-dlle is a bispecific DLL3-directed CD3 T-cell engager that binds to DLL3 expressed on the surface of cells, including tumor cells, and CD3 expressed on the surface of T cells. Tarlatamab-dlle is produced using recombinant DNA technology in Chinese hamster ovary cells. It consists of 982 amino acids and has a molecular weight of approximately 105 kilodaltons.
IMDELLTRA (tarlatamab-dlle) for injection is supplied as a sterile, preservative-free, white to slightly yellow, lyophilized powder in a single-dose vial for reconstitution and further dilution.
Each 1 mg vial contains tarlatamab-dlle (1 mg), glutamic acid (0.72 mg), polysorbate 80 (0.04 mg), sucrose (37.1 mg), and sodium hydroxide to adjust pH to 4.2. After reconstitution with 1.3 mL of Sterile Water for Injection the resulting concentration is 0.9 mg/mL IMDELLTRA.
Each 10 mg vial contains tarlatamab-dlle (10 mg), glutamic acid (3.7 mg), polysorbate 80 (0.2 mg), sucrose (194.4 mg), and sodium hydroxide to adjust pH to 4.2. After reconstitution with 4.4 mL of Sterile Water for Injection the resulting concentration is 2.4 mg/mL IMDELLTRA.
IV Solution Stabilizer is supplied as a sterile, preservative-free, colorless to slightly yellow, clear solution. Each vial of IV Solution Stabilizer contains citric acid monohydrate (36.75 mg), lysine hydrochloride (1598.8 mg), polysorbate 80 (7 mg), sodium hydroxide to adjust pH to 7.0, and water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tarlatamab-dlle is a bispecific T-cell engager that binds to DLL3 expressed on the surface of cells, including tumor cells, and CD3 expressed on the surface of T cells. Tarlatamab-dlle causes T-cell activation, release of inflammatory cytokines, and lysis of DLL3-expressing cells. Tarlatamab-dlle had anti-tumor activity in mouse models of SCLC.
12.2 Pharmacodynamics
Exposure-Response Relationships
There are no clinically significant exposure-response relationships for efficacy over the exposure range observed between tarlatamab-dlle 10 mg and 100 mg (10 times the highest approved recommended dosage).
There is an exposure-response relationship between tarlatamab-dlle exposure and neutropenia or neurologic toxicity including ICANS with a higher risk of any grade neutropenia or neurologic toxicity including ICANS at higher exposure.
Serum Cytokines
Transient elevation of serum cytokines IL-2, IL-6, IL-8, IL-10, and IFN-γ were observed at a tarlatamab-dlle dosage of 0.3 mg and above. Peak elevation of cytokines was generally observed 24 hours following the initial dose of IMDELLTRA at 1 mg on Cycle 1 Day 1 and generally returned to baseline levels prior to the next infusion on Cycle 1 Day 8.
12.3 Pharmacokinetics
Tarlatamab-dlle pharmacokinetic data in patients with SCLC at the approved recommended dosage are presented as mean (CV%) unless otherwise specified. The exposure of tarlatamab-dlle increases in a dose proportional manner over the dosage range of 1 mg to 100 mg (10 times the highest approved recommended dosage) every 2 weeks. Tarlatamab-dlle steady state is achieved by Cycle 2 Day 15. Pharmacokinetic parameters are summarized for the recommended dosage of IMDELLTRA in Table 17.
Table 17. Pharmacokinetic Parameters of Tarlatamab-dlle Parameter Cavg
(ng/mL)Cmax
(ng/mL)Ctrough
(ng/mL)First step-up dose 1 mg 106 (26%) 314 (35%) 49 (35%) First treatment dose 10 mg 1,100 (26%) 3,190 (35%) 517 (36%) Steady state 10 mg every 2 weeks 1,040 (37%) 3,640 (35%) 472 (62%) Distribution
Tarlatamab-dlle steady state volume of distribution is 8.5 L (33%).
Metabolism
Tarlatamab-dlle is expected to be metabolized into small peptides by catabolic pathways.
Elimination
Tarlatamab-dlle terminal elimination half-life is 11 days (31%) with an estimated systemic clearance of 0.7 L/day (34%).
Specific Populations
No clinically significant differences in the pharmacokinetics of tarlatamab-dlle were observed based on age (20 to 86 years), body weight (35 to 149 kg), sex, race (68% White and 27% Asian), mild or moderate renal impairment (eGFR 30 to < 90 mL/min), or mild hepatic impairment (total bilirubin ≤ upper limit of normal (ULN) and AST > ULN).
The effects of severe renal impairment (eGFR 15 to < 30 mL/min), end-stage renal disease (eGFR <15 mL/min), or moderate to severe hepatic impairment (total bilirubin > 1.5 × ULN and any AST) on the pharmacokinetics of tarlatamab-dlle are unknown.
Effects of Tarlatamab-dlle on CYP450 Substrates
Tarlatamab-dlle causes transient release of cytokines that may suppress CYP450 enzymes and may result in an increased exposure of concomitant CYP substrates during and up to 14 days after occurrence of cytokine release syndrome [see Clinical Pharmacology (12.2)].
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of tarlatamab-dlle or of other tarlatamab products.
During the maximum 3-year treatment period during which the presence of ADA was evaluated in DeLLphi-300, DeLLphi-301, and DeLLphi-304, 8% (36/445) of patients who received the recommended step-up and full dose of IMDELLTRA developed treatment- emergent ADA. In DeLLphi-301 and DeLLphi-304, which included neutralizing antibody assessments, 38% (11/29) of the patients who developed treatment-emergent ADA also developed neutralizing antibodies. ADA resulted in a 14% increase in the clearance of tarlatamab-dlle. Because of the low occurrence of ADA, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and effectiveness of tarlatamab-dlle is unknown.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Small Cell Lung Cancer
DeLLphi-304
The efficacy of IMDELLTRA was evaluated in DeLLphi-304 (NCT05740566), a multicenter, randomized, open-label trial. Eligible patients were required to have SCLC with disease progression following treatment with platinum-based chemotherapy with or without an anti-PD-(L)1 antibody. Patients were required to have an ECOG Performance Status of 0 or 1 and at least one measurable lesion per Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Patients with symptomatic brain metastases or active immunodeficiency were ineligible.
A total of 509 patients were randomized 1:1 to receive either IMDELLTRA (N=254) at an initial dose of 1 mg on Cycle 1 Day 1 followed by 10 mg on Days 8, 15, and every 2 weeks thereafter until disease progression or unacceptable toxicity or Investigator's choice of standard of care (SOC) chemotherapy (N=255) (topotecan [73%], lurbinectedin [18%] or amrubicin [9%]) until unacceptable toxicity or disease progression. Randomization was stratified by prior anti-PD-(L)1 exposure (yes versus no), platinum sensitivity status (chemotherapy-free interval (CFI) ≥ 180 days, < 180 to ≥ 90 days, or < 90 days), presence (previous or current) of brain metastases (yes versus no) and investigator's choice of standard of care (topotecan/amrubicin versus lurbinectedin).
The median age was 65 years (range: 20 to 86); 52% age 65 or older; 69% male; 57% White, 40% Asian, 1.4% were other races or had race not reported, 1% Black or African American, 0.4% American Indian or Alaska Native; 32% had ECOG PS of 0 and 67% ECOG PS of 1; 100% had extensive stage disease at baseline of whom 91% had metastatic disease; 45% had brain metastases at baseline; 35% had liver metastases at baseline. Sixty-nine percent (69%) of patients were former smokers, 21% were current smokers, 11% were never smokers. All patients received prior platinum therapy; 71% received prior anti-PD-(L)1 therapy; 223 patients (44%) had chemotherapy-free interval < 90 days after end of first line platinum therapy, while 286 patients (56%) had chemotherapy-free interval ≥ 90 days.
The major efficacy outcome measure was overall survival (OS). Key secondary efficacy outcome measures included progression-free survival (PFS) based on investigator assessment per Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) and select patient reported outcomes.
Efficacy results are summarized in Table 18 and Figure 1.
Table 18. Efficacy Results for Patients with SCLC who received IMDELLTRA Efficacy Parameter IMDELLTRA (N = 254) Standard of Care (N = 255) - * per Kaplan-Meier estimates
- † Hazard ratio based on the stratified Cox proportional hazard model
- ‡ p-value based on the stratified log-rank test
- § PFS based on investigator assessment per RECIST 1.1
Overall Survival (OS) Deaths (%) 111 (43.7) 152 (59.6) Median* in months (95% CI) 13.6 (11.1, NE) 8.3 (7.0, 10.2) Hazard ratio† (95% CI) 0.60 (0.47, 0.77) p-value‡ <0.001 Progression-free Survival (PFS)§ Events (%) 191 (75.2) 205 (80.4) Median* in months (95% CI) 4.2 (3.0, 4.4) 3.2 (2.9, 4.2) Hazard ratio† (95% CI) 0.72 (0.59, 0.88) p-value‡ <0.001 In a pre-specified exploratory subgroup analysis, the HR for OS was similar between patients with a chemotherapy-free interval (CFI) <90 days (n=223) and patients with a CFI ≥90 days (n=286), with HRs of 0.60 (95% CI: 0.43, 0.84) and 0.65 (95% CI: 0.45, 0.93), respectively.
Figure 1: Kaplan-Meier Plot of Overall Survival in ITT on DeLLphi-304

The analysis of mean change from baseline in dyspnea as assessed using the EORTC QLQ-C30 and EORTC QLQ-LC13 at week 18 demonstrated a statistically significant improvement in patients randomized to IMDELLTRA compared to SOC. At week 18, 149 patients (59%) randomized to IMDELLTRA and 116 (45%) patients randomized to SOC were still on treatment, and the compliance rates were 79% and 76% respectively at that timepoint.
Figure 2 shows the change from baseline in dyspnea at week 18 in patients who had a change from baseline score at week 18 (n=116 for IMDELLTRA, n=88 for SOC). Two patients with a missing baseline value, both from the IMDELLTRA arm, are not included in the waterfall plot. Patients with no change in dyspnea score are not graphically represented in Figure 2 (n=38 for IMDELLTRA, n=26 for SOC).
Figure 2: Waterfall plot of Change From Baseline in Dyspnea (Composite Score) at Week 18

DeLLphi-301
The efficacy of IMDELLTRA was evaluated in DeLLphi-301 [NCT05060016], an open- label, multicenter, multi-cohort clinical trial. Eligible patients were required to have relapsed/refractory SCLC with disease progression after receiving previous treatment with platinum-based chemotherapy and at least one other line of prior therapy, an ECOG Performance Status of 0 or 1, and at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST v1.1). The trial excluded patients with symptomatic brain metastases, evidence of interstitial lung disease or non-infectious pneumonitis, and active immunodeficiency.
A total of 99 patients received IMDELLTRA intravenously at an initial dose of 1 mg on Cycle 1 Day 1 followed by 10 mg on Days 8, 15, and every 2 weeks thereafter until disease progression or unacceptable toxicity.
The study population characteristics were: median age 64 years (range: 35 to 82); 48% of patients ≥ 65 years and 10% of patients ≥ 75 years; 72% male; 58% White, 41% Asian; 1% Hispanic or Latino; and 74% have ECOG 1.
Ninety-seven percent of patients had metastatic disease at baseline; 22% had brain metastases at baseline; and 92% were former/current smokers. All patients received prior platinum-based chemotherapy (median two lines); 74% received prior anti-PD-(L)1 therapy (including 59% who received anti-PD[L]1 therapy in combination with platinum-based chemotherapy in the frontline setting); 51% received prior topoisomerase I inhibitor (including 20% who received topotecan). Platinum sensitivity status, defined by time to progression after first line platinum therapy, was known for 69/99 patients. Twenty-seven patients (27%) had platinum-resistant SCLC, defined as time to progression < 90 days after first line platinum therapy, while 42 patients (42%) had platinum-sensitive SCLC.
Tumor assessments were performed every 6 weeks for the first 48 weeks and every 12 weeks thereafter. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) as evaluated by Blinded Independent Central Review (BICR) according to RECIST v1.1.
Efficacy results are presented in Table 19.
Table 19. Efficacy Results for DeLLphi-301 Efficacy Parameter IMDELLTRA (N = 99) - * Assessed by Blinded Independent Central Review, CI = Confidence Interval
- † Median based on Kaplan-Meier estimate.
- ‡ Based on observed duration of response.
Overall Response Rate (ORR) ORR, % (95% CI)* 40 (31, 51) Complete Response, n (%) 2 (2) Partial Response, n (%) 38 (38) Duration of Response (DOR)* Median†, months (range) 9.7 (2.7, 20.7+) Duration ≥ 6 months‡, % 68 Duration ≥ 12 months‡, % 40 Of the 69 patients with available data regarding platinum sensitivity status, the ORR was 52% (95% CI: 32, 71) in 27 patients with platinum-resistant SCLC and 31% (95% CI: 18, 47) in 42 patients with platinum-sensitive SCLC.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
IMDELLTRA (tarlatamab-dlle) for injection is a sterile, preservative-free, white to slightly yellow, lyophilized powder supplied as follows:
- 1 mg package (NDC: 55513-059-01) contains 1 single-dose vial of 1 mg IMDELLTRA and 2 vials of 7 mL IV Solution Stabilizer.
- 10 mg package (NDC: 55513-077-01) contains 1 single-dose vial of 10 mg IMDELLTRA and 2 vials of 7 mL IV Solution Stabilizer.
16.2 Storage and Handling
Store IMDELLTRA and IV Solution Stabilizer (IVSS) vials refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light until time of use. Do not freeze.
IMDELLTRA and IV Solution Stabilizer (IVSS) vials may be kept at room temperature between 20°C to 25°C (68°F to 77°F) for up to 24 hours in the original carton to protect from light.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Cytokine Release Syndrome (CRS)
Inform patients and their caregivers of the risk of CRS, and to immediately contact their healthcare provider for signs and symptoms associated with CRS including pyrexia, hypotension, fatigue, tachycardia, headache, hypoxia, nausea and vomiting [see Warnings and Precautions (5.1)].
Advise patients that they should be monitored from the start of the IMDELLTRA infusion for 22 to 24 hours on Cycle 1 Day 1 and Cycle 1 Day 8 doses in an appropriate healthcare setting [see Warnings and Precautions (5.1)].
Advise patients to remain within 1 hour of an appropriate healthcare setting for a total of 48 hours from start of the infusion with IMDELLTRA following Cycle 1 Day 1 and Cycle 1 Day 8 doses, accompanied by a caregiver.
Neurologic Toxicity Including Immune Effector Cell Associated Neurotoxicity Syndrome (ICANS)
Discuss the signs and symptoms associated with ICANS with patients and their caregivers. Advise patients to immediately contact their healthcare provider if they experience any signs or symptoms of ICANS, such as encephalopathy, confusion, delirium, seizure, ataxia, weakness or numbness of arms and legs, tremor, and headache.
Advise patients who experience neurologic toxicity or symptoms of ICANS to refrain from driving, operating heavy or potentially dangerous machinery, and engaging in hazardous occupations or activities during treatment with IMDELLTRA [see Warnings and Precautions (5.2)].
Cytopenias
Discuss the signs and symptoms associated with cytopenias, including neutropenia and febrile neutropenia, anemia, and thrombocytopenia with patients and their caregivers [see Warnings and Precautions (5.3)]. Inform patients that they will need to undergo lab tests to monitor blood counts. Advise patients to immediately contact their healthcare provider if they experience any signs or symptoms of cytopenias.
Infections
Discuss the signs and symptoms of infections with patients and their caregivers. Advise patients of the risk of serious infections, and to immediately contact their healthcare provider for signs or symptoms of infections [see Warnings and Precautions (5.4)].
Hepatotoxicity
Discuss the signs and symptoms of hepatotoxicity and bilirubin with patients and their caregivers. Inform patients that they will need to undergo lab tests to monitor liver function. Advise patients to immediately contact their healthcare provider for signs and symptoms of liver dysfunction [see Warnings and Precautions (5.5)].
Hypersensitivity
Discuss the signs and symptoms of allergic reactions with patients and their caregivers. Advise patients to immediately seek medical attention for any signs and symptoms of severe reactions [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider if they are pregnant or become pregnant. Advise females of reproductive potential to use effective contraception during treatment with IMDELLTRA and for 2 months after the last dose [see Warnings and Precautions (5.7), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with IMDELLTRA and for 2 months after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
IMDELLTRA® (im del trah) (tarlatamab-dlle)
for injection, for intravenous useThis Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 11/2025 What is the most important information I should know about IMDELLTRA?
IMDELLTRA can cause serious side effects, including:- Cytokine Release Syndrome (CRS). CRS is common during treatment with IMDELLTRA and can also be severe, life-threatening, or cause death. Tell your healthcare provider or get medical help right away if you develop any signs or symptoms of CRS, including:
- fever of 100.4°F (38°C) or higher
- low blood pressure
- tiredness
- fast heartbeat or dizziness
- headache
- shortness of breath or trouble breathing
- nausea and vomiting
- confusion, restlessness, or feeling anxious
- problems with balance and movement, such as trouble walking
- heart, liver, or kidney problems
- blood clots or unusual bleeding or bleeding that lasts a long time
Due to the risk of CRS, you will receive IMDELLTRA on a "step-up dosing schedule": - The step-up dosing schedule is when you receive a smaller dose of IMDELLTRA on Day 1 of your first treatment cycle (Cycle 1).
- You will receive the full treatment dose of IMDELLTRA on Day 8 and Day 15 of Cycle 1. You will receive the full treatment dose 1 time every 2 weeks after Day 15 of Cycle 1.
- If your dose of IMDELLTRA is delayed for any reason, you may need to repeat the step-up dosing schedule.
- Before receiving your Day 1 and Day 8 doses of Cycle 1 of IMDELLTRA, you will be given a medicine to help reduce your risk of CRS. This will be given into your vein by intravenous (IV) infusion. You will also receive IV fluids after each of your Cycle 1 Day 1 and Day 8 doses. Your healthcare provider will decide if you need to receive medicines to help reduce your risk of CRS with future doses.
- See "How will I receive IMDELLTRA?" for more information about how you will receive IMDELLTRA.
- Neurologic Problems. IMDELLTRA can cause neurologic problems that can be severe, life-threatening, or cause death. Neurologic problems may happen days or weeks after you receive IMDELLTRA. Your healthcare provider may refer you to a healthcare provider who specializes in neurologic problems. Tell your healthcare provider right away if you develop any signs or symptoms of neurologic problems, including:
- changes in taste
- headache
- numbness or tingling of your hands or feet
- dizziness
- trouble sleeping
- muscle weakness or numbness of arms or legs
- problems with walking, or loss of balance or coordination
- trouble speaking, memory loss, or personality changes
- confusion, feeling disoriented, slow thinking, or not being able to think clearly
- fainting or loss of consciousness
- seizures
- shaking (tremors)
- sleepiness
- feeling like you have no energy
Due to the risk of CRS and neurologic problems you will receive the following monitoring during treatment with IMDELLTRA: - For Day 1 and Day 8 of Cycle 1 doses, your healthcare provider will monitor you for 22 to 24 hours from the start of the IMDELLTRA infusion in an appropriate healthcare setting that can manage these side effects.
- You should remain within 1 hour of an appropriate healthcare setting for a total of 48 hours from the start of the IMDELLTRA infusion after your Day 1 and Day 8 of Cycle 1 doses and be accompanied by a caregiver.
- For Day 15 of Cycle 1 and Cycle 2 doses, your healthcare provider will watch you for 6 to 8 hours after the IMDELLTRA infusion.
- For Cycle 3 and Cycle 4 doses, your healthcare provider will watch you for 3 to 4 hours after the IMDELLTRA infusion.
- For Cycle 5 and later doses, your healthcare provider will watch you for 2 hours after the IMDELLTRA infusion.
See "What are the possible side effects of IMDELLTRA?" for more information about side effects.What is IMDELLTRA?
IMDELLTRA is a prescription medicine used to treat adults with extensive stage small cell lung cancer (ES-SCLC), which is cancer that has spread throughout the lung or to other parts of the body, and who have received treatment with chemotherapy that contains platinum, and it did not work or is no longer working.
It is not known if IMDELLTRA is safe and effective in children.Before receiving IMDELLTRA, tell your healthcare provider about all of your medical conditions, including if you: - have an infection
- are pregnant or plan to become pregnant. IMDELLTRA may harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider should do a pregnancy test before you start treatment with IMDELLTRA.
- You should use an effective form of birth control (contraception) during treatment with IMDELLTRA, and for 2 months after your last dose of IMDELLTRA.
- Tell your healthcare provider right away if you become pregnant or think that you may be pregnant during treatment with IMDELLTRA.
- are breastfeeding or plan to breastfeed. It is not known if IMDELLTRA passes into your breast milk. Do not breastfeed during treatment with IMDELLTRA and for 2 months after the last dose of IMDELLTRA.
How will I receive IMDELLTRA? - IMDELLTRA will be given to you by your healthcare provider by intravenous (IV) infusion through a needle placed in a vein. The infusion will take about 1 hour.
- Your IMDELLTRA treatment schedule is divided into cycles that are usually 28 days (4 weeks) long.
- Your healthcare provider will decide how many treatment cycles you will receive.
- See "What is the most important information I should know about IMDELLTRA?" for more information about how you will receive IMDELLTRA.
What should I avoid while receiving IMDELLTRA?
Do not drive, operate heavy or potentially dangerous machinery or do other dangerous activities, including work-related activities, during treatment with IMDELLTRA if you develop dizziness, confusion, tremors, sleepiness, or any other symptoms that impair consciousness until your signs and symptoms go away. These may be signs and symptoms of neurologic problems. See "What is the most important information I should know about IMDELLTRA" for more information about signs and symptoms of neurologic problems.What are the possible side effects of IMDELLTRA?
IMDELLTRA can cause serious side effects, including:- See "What is the most important information I should know about IMDELLTRA?"
-
Low blood cell counts (cytopenia). Decreased blood cell counts can be severe and may include the following:
- low white blood cell counts (neutropenia). Low white blood cells can increase your risk for infection.
- low red blood cell counts (anemia). Low red blood cells can cause tiredness and shortness of breath.
- low platelet counts (thrombocytopenia). Low platelet counts can cause bruising or bleeding problems.
- Infections. IMDELLTRA can cause serious infections that can be life-threatening and cause death. Your healthcare provider will check you for signs and symptoms of infection before and during treatment with IMDELLTRA. Tell your healthcare provider right away if you develop any signs or symptoms of infection during treatment with IMDELLTRA, including:
- fever of 100.4°F (38°C) or higher
- cough
- chest pain
- tiredness
- shortness of breath
- painful rash
- sore throat or runny nose
- pain during urination
- feeling weak or generally unwell
- yeast infections in the mouth or other areas
- Liver problems. IMDELLTRA can cause increased liver enzymes and bilirubin in your blood. These increases can happen with or without you also having CRS. Tell your healthcare provider right away if you develop any signs or symptoms of liver problems, including:
- tiredness
- loss of appetite
- pain in your right upper stomach-area (abdomen)
- dark urine
- yellowing of your skin or the white part of your eyes
- Allergic reactions. IMDELLTRA can cause allergic reactions that can be severe. Go to the nearest emergency room or get medical help right away if you develop any signs or symptoms of a severe allergic reaction during treatment with IMDELLTRA, including:
- shortness of breath or trouble breathing
- pain or tightness in your chest and back
- wheezing
- coughing
- feeling lightheaded or dizzy
- rash
Your healthcare provider will do bloodwork before you start and during treatment with IMDELLTRA. Your healthcare provider will monitor you for signs or symptoms of these serious side effects during treatment and may temporarily or completely stop treatment with IMDELLTRA if you develop certain serious side effects.
The most common side effects of IMDELLTRA also include:- tiredness
- decreased appetite
- a bad or metallic taste in your mouth
- fever
- muscle or bone pain
- constipation
- nausea
The most common severe abnormal blood test results with IMDELLTRA include: decreased white blood cells, decreased sodium, and increased uric acid.
These are not all of the possible side effects of IMDELLTRA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of IMDELLTRA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about IMDELLTRA that is written for health professionals.What are the ingredients in IMDELLTRA?
Active ingredients: tarlatamab-dlle
Inactive ingredients: glutamic acid, polysorbate 80, sucrose, and sodium hydroxide.
Inactive ingredients of IV Solution Stabilizer: citric acid monohydrate, lysine hydrochloride, polysorbate 80, sodium hydroxide and water for Injection.
Manufactured by: Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320-1799
U.S. License No. 1080
© 2024-2025 Amgen Inc. All rights reserved. V2
For more information, go to www.imdelltra.com or call Amgen at 1-800-772-6436. -
PRINCIPAL DISPLAY PANEL - Kit Carton - 1 mg/vial
Contains 1 IMDELLTRA Single-Dose Vial
Contains 2 IV Solution Stabilizer VialsNDC: 55513-059-01
AMGEN
IMDELLTRA®
(tarlatamab-dlle)
for injection1
mg/vial1 mg/vial
For Intravenous Infusion after Dilution
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the
original carton to protect from light until time of use.
Do not freeze.
ATTENTION: Dispense the enclosed Medication
Guide to each patient.Must be reconstituted
with sterile water for
injection and further
diluted.No Preservative
Single-Dose Vial –
Discard unused portion.Rx Only

-
PRINCIPAL DISPLAY PANEL - Kit Carton - 10 mg/vial
Contains 1 IMDELLTRA Single-Dose Vial
Contains 2 IV Solution Stabilizer VialsNDC: 55513-077-01
AMGEN
IMDELLTRA®
(tarlatamab-dlle)
for injection10
mg/vial10 mg/vial
For Intravenous Infusion after Dilution
Store refrigerated at 2°C to 8°C (36°F to 46°F) in the
original carton to protect from light until time of use.
Do not freeze.
ATTENTION: Dispense the enclosed Medication
Guide to each patient.Must be reconstituted
with sterile water for
injection and further
diluted.No Preservative
Single-Dose Vial –
Discard unused portion.Rx Only
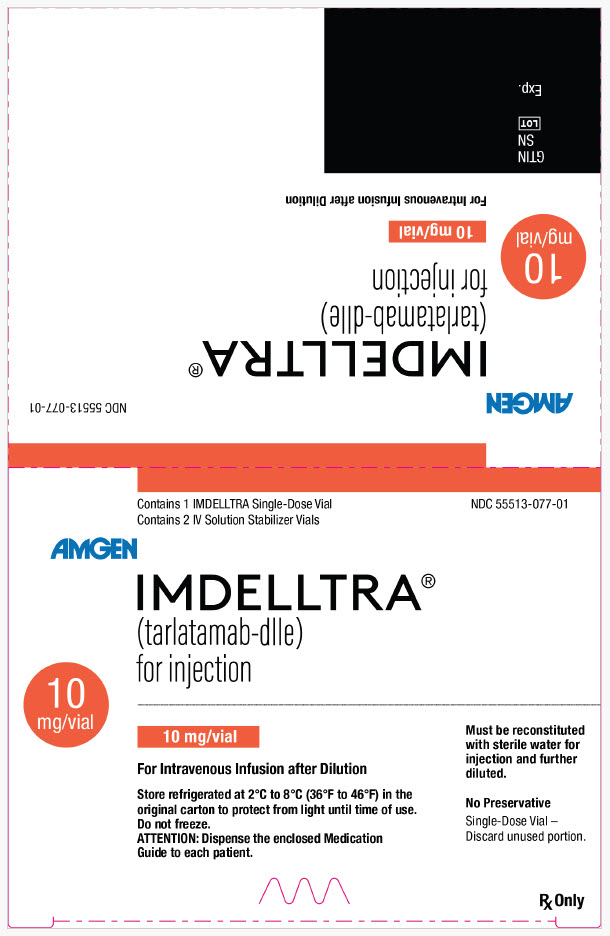
-
INGREDIENTS AND APPEARANCE
IMDELLTRA (AMG757)
tarlatamab-dlle kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-059 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-059-01 1 in 1 PACKAGE 05/16/2024 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 1 mg Part 2 2 VIAL 14 mL Part 1 of 2 IMDELLTRA
tarlatamab-dlle injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 55513-103 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TARLATAMAB (UNII: 74X82ST8Q1) (TARLATAMAB - UNII:74X82ST8Q1) TARLATAMAB 0.9 mg in 1 mg Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 37.1 mg in 1 mg GLUTAMIC ACID (UNII: 3KX376GY7L) 0.72 mg in 1 mg POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.04 mg in 1 mg SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-103-01 1 mg in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 Part 2 of 2 IV STABILIZER
iv stabilizer solutionProduct Information Item Code (Source) NDC: 55513-068 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 36.75 mg in 1 mL LYSINE HYDROCHLORIDE (UNII: JNJ23Q2COM) 1598.8 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 7 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-068-01 7 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 IMDELLTRA (AMG757)
tarlatamab-dlle kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-077 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-077-01 1 in 1 PACKAGE 06/12/2024 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 10 mg Part 2 2 VIAL 14 mL Part 1 of 2 IMDELLTRA (AMG757)
tarlatamab-dlle injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 55513-069 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TARLATAMAB (UNII: 74X82ST8Q1) (TARLATAMAB - UNII:74X82ST8Q1) TARLATAMAB 2.4 mg in 1 mg Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 194.4 mg in 1 mg GLUTAMIC ACID (UNII: 3KX376GY7L) 3.7 mg in 1 mg POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.2 mg in 1 mg SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-069-01 10 mg in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 Part 2 of 2 IV STABILIZER
iv stabilizer solutionProduct Information Item Code (Source) NDC: 55513-068 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 36.75 mg in 1 mL LYSINE HYDROCHLORIDE (UNII: JNJ23Q2COM) 1598.8 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 7 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-068-01 7 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761344 05/16/2024 Labeler - Amgen, Inc (039976196) Registrant - Immunex Rhode Island Corporation (968084785)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.