BIZENGRI- zenocutuzumab injection
BIZENGRI by
Drug Labeling and Warnings
BIZENGRI by is a Prescription medication manufactured, distributed, or labeled by Partner Therapeutics, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BIZENGRI® safely and effectively. See full prescribing information for BIZENGRI.
BIZENGRI (zenocutuzumab-zbco) injection, for intravenous use
Initial U.S. Approval: 2024WARNING: EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
Embryo-Fetal Toxicity: Exposure to BIZENGRI during pregnancy can cause embryo-fetal harm. Advise patients of this risk and the need for effective contraception [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
RECENT MAJOR CHANGES
Indications and Usage (1.3) 05/2026 INDICATIONS AND USAGE
BIZENGRI® is a bispecific HER2- and HER3-directed antibody indicated for the treatment of:
- Adults with advanced, unresectable or metastatic non-small cell lung cancer (NSCLC) harboring a neuregulin 1 (NRG1) gene fusion with disease progression on or after prior systemic therapy.* (1.1)
- Adults with advanced, unresectable or metastatic pancreatic adenocarcinoma harboring a neuregulin 1 (NRG1) gene fusion with disease progression on or after prior systemic therapy.* (1.2)
- Adults with advanced, unresectable or metastatic cholangiocarcinoma harboring a neuregulin 1 (NRG1) gene fusion with disease progression on or after prior systemic therapy. (1.3)
*This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
DOSAGE AND ADMINISTRATION
- Select patients for treatment with BIZENGRI based on the presence of an NRG1 gene fusion. (2.1)
- Evaluate left ventricular ejection fraction (LVEF) before initiating BIZENGRI. (2.2)
- The recommended dosage of BIZENGRI is 750 mg every 2 weeks until disease progression or unacceptable toxicity. (2.3)
- Administer premedications before each infusion to reduce the risk of infusion-related reactions. (2.4)
- Administer as an intravenous infusion, after dilution, over 4 hours. (2.7)
DOSAGE FORMS AND STRENGTHS
Injection: 375 mg/18.75 mL (20 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Infusion-Related Reactions (IRR)/Hypersensitivity/Anaphylactic Reactions: Administer BIZENGRI in a setting with emergency resuscitation equipment and staff who are trained to monitor for IRRs and to administer emergency medications. Monitor for signs and symptoms of IRR. Interrupt infusion in patients with ≤ Grade 3 IRRs and administer symptomatic treatment as needed. Resume infusion at a reduced rate after resolution of symptoms. Immediately stop the infusion and permanently discontinue BIZENGRI for Grade 4 or life-threatening IRR or hypersensitivity/anaphylaxis. (5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Monitor for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Permanently discontinue BIZENGRI in patients with ≥ Grade 2 ILD/pneumonitis. (5.2)
- Left Ventricular Dysfunction: Assess LVEF before initiating BIZENGRI and at regular intervals during treatment as clinically indicated. Manage through treatment interruption or discontinuation. Permanently discontinue BIZENGRI in patients with symptomatic congestive heart failure (CHF). (5.3)
ADVERSE REACTIONS
- The most common adverse reactions (≥ 10%) in patients were diarrhea musculoskeletal pain, fatigue, nausea, infusion-related reactions (IRR), dyspnea, rash, constipation, vomiting, abdominal pain, and edema. (6.1)
- The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were increased GGT, decreased hemoglobin, decreased sodium, decreased platelets, increased AST, increased ALT, increased alkaline phosphatase, decreased magnesium, decreased phosphate, increased aPTT and increased bilirubin. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Partner Therapeutics, Inc. at 1-888-479-5385 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Females and Males of Reproductive Potential: Verify pregnancy status of females prior to initiation of BIZENGRI. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: EMBRYO-FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Non-Small Cell Lung Cancer
1.2 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Pancreatic Adenocarcinoma
1.3 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Cholangiocarcinoma
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Evaluation Before Initiating BIZENGRI
2.3 Recommended Dosage
2.4 Recommended Premedications
2.5 Dosage Modifications for Adverse Reactions
2.6 Preparation
2.7 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions/Hypersensitivity/Anaphylactic Reactions
5.2 Interstitial Lung Disease/Pneumonitis
5.3 Left Ventricular Dysfunction
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Non-Small Cell Lung Cancer
14.2 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Pancreatic Adenocarcinoma
14.3 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Cholangiocarcinoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: EMBRYO-FETAL TOXICITY
Embryo-Fetal Toxicity: Exposure to BIZENGRI during pregnancy can cause embryo-fetal harm. Advise patients of this risk and the need for effective contraception [see Warnings and Precautions (5.4), Use in Specific Populations (8.1, 8.3)].
-
1 INDICATIONS AND USAGE
1.1 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Non-Small Cell Lung Cancer
BIZENGRI is indicated for the treatment of adults with advanced unresectable or metastatic non-small cell lung cancer (NSCLC) harboring a neuregulin 1 (NRG1) gene fusion with disease progression on or after prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
1.2 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Pancreatic Adenocarcinoma
BIZENGRI is indicated for the treatment of adults with advanced unresectable or metastatic pancreatic adenocarcinoma harboring a neuregulin 1 (NRG1) gene fusion with disease progression on or after prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment with BIZENGRI based on the presence of an NRG1 gene fusion in tumor specimens [see Clinical Studies (14.1, 14.2, 14.3)].
An FDA-approved test for the detection of NRG1 gene fusions is not currently available.
2.2 Recommended Evaluation Before Initiating BIZENGRI
Before initiating BIZENGRI, evaluate left ventricular ejection fraction (LVEF) [see Warnings and Precautions (5.3)].
2.3 Recommended Dosage
- The recommended dosage of BIZENGRI is 750 mg as an intravenous (IV) infusion every 2 weeks until disease progression or unacceptable toxicity [see Dosage and Administration (2.7)].
- Administer premedications before each BIZENGRI infusion as recommended to reduce the risk of infusion-related reactions [see Dosage and Administration (2.4)].
2.4 Recommended Premedications
Prior to each infusion of BIZENGRI, administer premedications to reduce the risk of infusion-related reactions (IRRs) [see Warnings and Precautions (5.1)] (see Table 1).
Table 1: Premedications Prior to BIZENGRI Infusions Medication Dose Route of Administration - * Optional after initial BIZENGRI infusion
Corticosteroid* Dexamethasone (10 mg) Oral or intravenous Antipyretic Acetaminophen (1,000 mg) Oral or intravenous H1 Antihistamine Dexchlorpheniramine (5 mg)
or other anti-H1 equivalentIntravenous
or oral2.5 Dosage Modifications for Adverse Reactions
No dose reduction is recommended for BIZENGRI. The recommended dosage modifications of BIZENGRI for adverse reactions are provided in Table 2.
Table 2: Recommended BIZENGRI Dosage Modifications and Management for Adverse Reactions Adverse Reaction Severity Dose Modifications and Management Infusion-related reactions (IRRs)/Hypersensitivity/Anaphylactic Reactions
[see Warnings and Precautions (5.1)]≤ Grade 3 IRR - Interrupt BIZENGRI infusion if IRR is suspected and monitor patient until reaction symptoms resolve.
- Provide symptomatic treatment as needed.
- Resume the infusion at 50% of the infusion rate at which the reaction occurred. The infusion rate may be escalated if there are no additional symptoms.
- Corticosteroid premedication can be used as necessary for subsequent BIZENGRI infusions [see Recommended Premedications (2.4)].
Grade 4 IRR or any grade hypersensitivity/anaphylactic reaction - Permanently discontinue BIZENGRI.
Interstitial Lung Disease (ILD)/Pneumonitis
[see Warnings and Precautions (5.2)]Grade 1 - Interrupt BIZENGRI until recovery.
- Consider prompt initiation of corticosteroids when the diagnosis is suspected.
- Resume treatment after resolution.
≥ Grade 2 - Permanently discontinue BIZENGRI.
- Promptly treat with corticosteroids.
Left Ventricular Dysfunction
[see Warnings and Precautions (5.3)]LVEF is 45-49% and absolute decrease from baseline ≥10%
or
LVEF less than 45%- Interrupt BIZENGRI.
- Repeat LVEF assessment within 3 weeks.
- If LVEF is less than 45% or LVEF has not recovered to within 10% from baseline, permanently discontinue BIZENGRI.
- If LVEF is 50% or greater or LVEF is 45-49% and recovered to within 10% of baseline, resume BIZENGRI and monitor LVEF every 12 weeks while on treatment and as clinically indicated.
Symptomatic congestive heart failure (CHF) - Permanently discontinue BIZENGRI.
Other Clinically Relevant Adverse Reactions [see Adverse Reactions (6.1)] Grade 3 or 4 - Withhold BIZENGRI until recovery to ≤ Grade 1 or baseline.
- Provide symptomatic treatment as needed.
- Resume treatment after resolution of symptoms.
2.6 Preparation
Dilute and prepare BIZENGRI for intravenous infusion before administration.
For the initial infusion, prepare BIZENGRI as close to administration time as possible to allow for the possibility of extended infusion time in the event of an infusion-related reaction.
- Check that the BIZENGRI solution is clear to slightly opalescent, colorless to slightly yellow. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if discoloration or visible particles are present.
- Withdraw and then discard 37.5 mL 0.9% Sodium Chloride Injection from the 250 mL infusion bag. Only use infusion bags made of polyvinylchloride (PVC), polyolefin or polyolefin/polyamide coextruded plastic.
- Withdraw a total of 37.5 mL of BIZENGRI from 2 vials and add it to the infusion bag. The final volume in the infusion bag should be 250 mL. Discard any unused portion left in the vial.
- Gently invert the bag to mix the solution. Do not shake.
- If not used immediately, store the diluted solution refrigerated at 2°C to 8°C (36°F to 46°F) and protect from light after preparation unless the infusion is initiated within 2 hours of preparation.
2.7 Administration
- If the infusion time exceeds the recommended storage time, the infusion bag must be discarded and a new infusion bag prepared to continue the infusion. Diluted BIZENGRI solution must by administered within:
- 6 hours from end of preparation of infusion solution stored at room temperature [15°C to 25°C (59°F to 77°F)]
- 28 hours from end of preparation of infusion solution stored refrigerated [2°C to 8°C (36°F to 46°F)]
- If the diluted BIZENGRI solution has been refrigerated, allow it to reach room temperature (approximately 30 minutes) prior to administration.
- Administer diluted BIZENGRI solution [see Dosage and Administration (2.6)] by intravenous infusion using an infusion set made of either PVC, polyethylene (PE), polyurethane (PUR) or polybutadiene (PB) with an in-line, sterile, non-pyrogenic, low protein-binding polyethersulfone (PES) filter (pore size 0.2 micrometer).
- Do not infuse BIZENGRI concomitantly in the same IV line with other agents.
- Administer BIZENGRI infusion via a peripheral or central line.
- Monitor patients closely for signs and symptoms of infusion-related reactions during BIZENGRI infusion and monitor patients for at least 1 hour following completion of the first BIZENGRI infusion and as clinically indicated [see Warnings and Precautions (5.1)].
- Administer intravenous infusion over 4 hours.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions/Hypersensitivity/Anaphylactic Reactions
BIZENGRI can cause serious and life-threatening infusion-related reactions (IRRs), hypersensitivity and anaphylactic reactions. Signs and symptoms of IRR may include chills, nausea, fever, and cough.
In the eNRGy study, 13% of patients experienced IRRs, all were Grade 1 or 2; 91% occurred during the first infusion. The median time to onset was 63 minutes (range: 13 minutes to 240 minutes) from the start of infusion.
Administer BIZENGRI in a setting with emergency resuscitation equipment and staff who are trained to monitor for IRRs and to administer emergency medications. Monitor patients closely for signs and symptoms of infusion reactions during infusion and for at least 1 hour following completion of first BIZENGRI infusion and as clinically indicated. Prior to the first BIZENGRI infusion, premedicate with a corticosteroid, an H1 antihistamine and acetaminophen to reduce the risk of IRRs [see Dosage and Administration (2.4)]. Corticosteroid premedication can be used as necessary for subsequent BIZENGRI infusions.
Interrupt BIZENGRI infusion in patients with ≤ Grade 3 IRRs and administer symptomatic treatment as needed. Resume infusion at a reduced rate after resolution of symptoms [see Dosage and Administration (2.5)]. Immediately stop the infusion and permanently discontinue BIZENGRI for Grade 4 or life-threatening IRR or hypersensitivity/anaphylaxis reactions.
5.2 Interstitial Lung Disease/Pneumonitis
BIZENGRI can cause serious and life-threatening interstitial lung disease (ILD)/pneumonitis.
In the eNRGy study [see Adverse Reactions (6.1)], ILD/pneumonitis occurred in 2 (1.1%) patients treated with BIZENGRI. Grade 2 ILD/pneumonitis (Grade 2) resulting in permanent discontinuation of BIZENGRI occurred in 1 (0.6%) patient.
Monitor for new or worsening pulmonary symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Immediately withhold BIZENGRI in patients with suspected ILD/pneumonitis and administer corticosteroids as clinically indicated. Permanently discontinue BIZENGRI if ILD/pneumonitis ≥ Grade 2 is confirmed [see Dosage and Administration (2.5)].
5.3 Left Ventricular Dysfunction
BIZENGRI can cause left ventricular dysfunction.
Left ventricular ejection fraction (LVEF) decrease occurred with anti-HER2 therapies, including BIZENGRI. Treatment with BIZENGRI has not been studied in patients with a history of clinically significant cardiac disease or LVEF less than 50% prior to initiation of treatment.
In the eNRGy study [see Adverse Reactions (6.1)], Grade 2 LVEF decrease [Grade 2 LVEF decrease (40%-50%; 10 - 19% drop from baseline)] occurred in 2% of evaluable patients. Cardiac failure without LVEF decrease occurred in 1.7% of patients including 1 (0.6%) fatal event.
Before initiating BIZENGRI, evaluate LVEF and monitor at regular intervals during treatment as clinically indicated. For LVEF of less than 45% or less than 50% with absolute decrease from baseline of 10% or greater is confirmed, permanently discontinue BIZENGRI. Permanently discontinue BIZENGRI in patients with symptomatic congestive heart failure (CHF) [see Dosage and Administration (2.5)].
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, BIZENGRI can cause fetal harm when administered to a pregnant woman. In literature reports, use of a HER2-directed antibody during pregnancy resulted in cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Animal studies have demonstrated that inhibition of HER2 and/or HER3 results in impaired embryo-fetal development, including effects on cardiac, vascular and neuronal development, and embryolethality. Advise patients of the potential risk to a fetus. Verify the pregnancy status of females of reproductive potential prior to the initiation of BIZENGRI. Advise females of reproductive potential to use effective contraception during treatment with BIZENGRI and for 2 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infusion-Related Reactions/Hypersensitivity/Anaphylaxis [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.2)]
- Left Ventricular Dysfunction [see Warnings and Precautions (5.3)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflects exposure to BIZENGRI as a single agent at 750 mg administered intravenously every 2 weeks until disease progression or unacceptable toxicity in 175 patients with NRG1 gene fusion positive tumors in the eNRGy study. Of these, there were 99 patients with NSCLC, 39 patients with pancreatic adenocarcinoma and 37 patients with other solid tumors [see Clinical Studies (14.1, 14.2)]. Among the 175 patients who received BIZENGRI, the median duration of exposure to BIZENGRI was 5.3 months (range: 0.1 to 36), including 45% of patients exposed for at least 6 months and 15% of patients exposed for at least 1 year. In this pooled safety population, the most common (≥ 10%) adverse reactions were diarrhea, musculoskeletal pain, fatigue, nausea, infusion-related reactions (IRR), dyspnea, rash, constipation, vomiting, abdominal pain, and edema. The most common Grade 3 or 4 laboratory abnormalities (≥ 2%) were increased GGT, decreased hemoglobin, decreased sodium, decreased platelets, increased AST, increased ALT, increased alkaline phosphatase, decreased magnesium, decreased phosphate, increased aPTT, and increased bilirubin.
NRG1 Gene Fusion Positive Unresectable or Metastatic NSCLC
eNRGy Study
The safety of BIZENGRI was evaluated in the eNRGy study in 99 patients with unresectable or metastatic NSCLC with NRG1 gene fusions [see Clinical Studies (14.1)]. Patients received BIZENGRI as a single agent at 750 mg intravenously every 2 weeks until disease progression or unacceptable toxicity. Among patients who received BIZENGRI, 47% were exposed for 6 months or longer and 17% were exposed for greater than one year.
The median age was 66 years (range: 27 to 88), 54% were 65 years or older; 62% were female; 37% were White, 53% were Asian, 2% were Black or African American; and 1% were Hispanic or Latino.
Serious adverse reactions occurred in 25% of patients who received BIZENGRI. Serious adverse reactions in ≥ 2% of patients included pneumonia (n=4) dyspnea and fatigue (n=2 each). Serious adverse reactions occurring in one patient each were: abdominal pain, acute kidney injury, ascites, bradycardia, carotid artery stenosis, cellulitis, acute cholecystitis, COVID-19, decreased appetite, dehydration, dizziness, dysphagia, hyponatremia, ileus, lymphadenitis, nausea, gastric obstruction, pericardial effusion, pneumonitis, pulmonary hypertension, sepsis, staphylococcal infection, tumor pain, urinary tract infection, viral infection and vomiting. Fatal adverse reactions occurred in 3 (3%) patients and included respiratory failure (n=2) and cardiac failure (n=1).
Permanent discontinuation of BIZENGRI due to an adverse reaction occurred in 3% of patients. Adverse reactions resulting in permanent discontinuation of BIZENGRI included dyspnea, pneumonitis and sepsis (n=1 each).
Dosage interruptions of BIZENGRI due to an adverse reaction, excluding temporary interruptions of BIZENGRI due to infusion-related reactions, occurred in 29% of patients. Adverse reactions leading to dosage interruptions in ≥2% of patients included dyspnea, COVID-19, arrhythmia, increased ALT, increased AST, and pneumonia.
Table 3 summarizes the adverse reactions in the eNRGy study in patients with NRG1 gene fusion positive unresectable or metastatic NSCLC.
Table 3: Adverse Reactions (≥10%) in Patients with NRG1 Gene Fusion Positive NSCLC Who Received BIZENGRI in the eNRGy Study Adverse Reaction* BIZENGRI
(N=99)All Grades
%Grade 3 or 4
%- * Based on NCI CTCAE v4.03 and MedDRA v26.0
- † Includes post-procedural diarrhea
- ‡ Includes back pain, pain in extremity, musculoskeletal chest pain, myalgia, arthralgia, non-cardiac chest pain, bone pain, musculoskeletal stiffness, neck pain, spinal pain.
- § Includes dyspnea exertional
- ¶ Includes productive cough
- # Includes asthenia
- Þ Includes breast edema, peripheral edema, face edema
- ß Includes eczema, erythema, dermatitis, dermatitis contact, rash maculopapular, rash erythematous.
- à Includes chills, IRR, nausea, cough, diarrhea, back pain, body temperature increased, dyspnea, face edema, fatigue, non-cardiac chest pain, oropharyngeal discomfort, paresthesia, pyrexia, and vomiting. AEs that were considered IRRs were counted under the composite term 'IRR', irrespective of the reported PT.
Gastrointestinal disorders Diarrhea† 25 2 Nausea 10 1 Musculoskeletal and connective tissue disorders Musculoskeletal pain‡ 23 1 Respiratory, thoracic and mediastinal disorders Dyspnea§ 18 5 Cough¶ 15 1 General disorders and administration site conditions Fatigue# 17 2 EdemaÞ 11 0 Skin and subcutaneous tissue disorders Rashß 14 0 Injury, poisoning and procedural complications Infusion-related reactions à 12 0 Metabolism and nutrition disorders Decreased appetite 11 1 Clinically relevant adverse reactions in <10% of patients receiving BIZENGRI were stomatitis (7%), vomiting (8%), cardiac failure and pneumonitis (2% each).
Table 4 summarizes the laboratory abnormalities in the eNRGy study in patients with NRG1 gene fusion positive unresectable or metastatic NSCLC.
Table 4: Select Laboratory Abnormalities ≥ 20% that Worsened from Baseline in Patients with NRG1 Gene Fusion Positive NSCLC Who Received BIZENGRI in the eNRGy Study Laboratory Abnormality BIZENGRI* All Grades
%Grade 3 or 4
%- * The denominator used to calculate the rate varied from 93 to 96 based on the number of patients with a baseline value and at least one post-treatment value.
Hematology Decreased hemoglobin 35 4.2 Chemistry Increased alanine aminotransferase 30 3.1 Decreased magnesium 28 4.3 Increased alkaline phosphatase 27 0 Decreased phosphate 26 1.1 Increased gamma-glutamyl transferase 23 5 Increased aspartate aminotransferase 22 3.1 Decreased potassium 21 2.1 NRG1 Gene Fusion Positive Unresectable or Metastatic Pancreatic Adenocarcinoma
eNRGy Study
The safety of BIZENGRI was evaluated in the eNRGy study in 39 patients with unresectable or metastatic pancreatic adenocarcinoma with NRG1 gene fusions [see Clinical Studies (14.2)]. Patients received BIZENGRI as a single agent at 750 mg intravenously every 2 weeks until disease progression or unacceptable toxicity. Among patients who received BIZENGRI, 50% were exposed for 6 months or longer and 13% were exposed for greater than one year.
The median age was 51 years (range: 21 to 74), 23% were 65 years or older; 49% were female; 82% were White, 13% were Asian, 2.6% were Black or African American; and 5% were Hispanic or Latino.
Serious adverse reactions occurred in 23% of patients who received BIZENGRI. Serious adverse reactions occurring in one patient each were: anemia, thrombocytopenia, tachycardia, abdominal pain, hemorrhoidal hemorrhage, nausea, cholestatic jaundice, COVID-19, liver abscess, traumatic fracture, blood creatinine increased, back pain, myelodysplastic syndrome, and respiratory disorder. There were 2 fatal adverse reactions, one due to COVID-19 and one due to respiratory failure.
Dosage interruptions of BIZENGRI due to an adverse reaction, excluding temporary interruptions of BIZENGRI due to infusion-related reactions, occurred in 33% of patients. Adverse reactions leading to dosage interruptions in ≥2% of patients included COVID-19, pneumonia, increased AST, neutropenia, abdominal pain, agitation, increased blood alkaline phosphatase, increased blood bilirubin, constipation, increased creatinine, hemorrhage, hyperbilirubinemia, cholestatic jaundice, tachycardia, traumatic fracture, and upper respiratory infection.
Table 5 summarizes the adverse reactions in the eNRGy study in patients with NRG1 gene fusion positive pancreatic adenocarcinoma.
Table 5: Adverse Reactions (≥10%) in Patients with NRG1 Gene Fusion Positive Pancreatic Adenocarcinoma Who Received BIZENGRI in the eNRGy Study Adverse Reaction* BIZENGRI
(N=39)All Grades
(%)Grade 3 or 4
(%)- * Based on NCI CTCAE v4.03 and MedDRA v26.0
- † Includes back pain, pain in extremity, musculoskeletal chest pain, myalgia, arthralgia, non-cardiac chest pain, bone pain, musculoskeletal stiffness, neck pain, spinal pain
- ‡ Includes asthenia
- § Includes peripheral edema, face edema, localized edema, peripheral swelling
- ¶ Includes chills, IRR, nausea, cough, diarrhea, back pain, body temperature increased, dyspnea, face edema, fatigue, non-cardiac chest pain, oropharyngeal discomfort, paresthesia, pyrexia, and vomiting
- # Includes epistaxis, hematochezia, hematuria, hemorrhoidal hemorrhage
Gastrointestinal disorders Diarrhea 36 5 Nausea 23 5 Vomiting 23 2.6 Abdominal pain 18 5 Constipation 15 0 Abdominal distension 13 0 Stomatitis 10 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain† 28 2.6 General disorders and administration site conditions Fatigue‡ 21 5 Edema§ 13 0 Pyrexia 10 0 Infections and infestations COVID-19 18 0 Injury, poisoning and procedural complications Infusion-related reactions¶ 15 0 Vascular disorders Hemorrhage# 13 5 Psychiatric disorders Anxiety 10 0 Skin and subcutaneous tissue disorders Dry skin 10 0 Clinically relevant adverse reactions in <10% of patients receiving BIZENGRI were decreased appetite (5%), and rash (8%) [including dermatitis acneiform, erythema, dermatitis, dermatitis contact, rash maculopapular, rash erythematous].
Table 6 summarizes the laboratory abnormalities in the eNRGy study in patients with NRG1 gene fusion positive pancreatic adenocarcinoma.
Table 6: Select Laboratory Abnormalities ≥ 20% That Worsened from Baseline in Patients with NRG1 Gene Fusion Positive Pancreatic Adenocarcinoma Who Received BIZENGRI in the eNRGy Study Laboratory Abnormality BIZENGRI*
(N=39)All Grades
(%)Grade 3 or 4
(%)- * The denominator used to calculate the rate varied from 35 to 39, based on the number of patients with a baseline value and at least one post-treatment value.
Chemistry Increased alanine aminotransferase 51 5 Increased aspartate aminotransferase 31 5 Increased bilirubin 31 5 Decreased phosphate 31 2.9 Increased alkaline phosphatase 28 8 Decreased sodium 28 10 Decreased albumin 26 0 Decreased potassium 26 2.6 Decreased magnesium 24 2.6 Increased gamma-glutamyl transferase 23 15 Hematology Decreased platelets 26 10 Decreased hemoglobin 23 10 Decreased leukocytes 21 2.6 NRG1 Gene Fusion Positive Advanced, Unresectable or Metastatic Cholangiocarcinoma
eNRGy Study
The safety of BIZENGRI was evaluated in the eNRGy study in 22 patients with unresectable or metastatic cholangiocarcinoma with NRG1 gene fusions [see Clinical Studies (14.3)]. Patients received BIZENGRI as a single agent at 750 mg intravenously every 2 weeks until disease progression or unacceptable toxicity. Among patients who received BIZENGRI, 64% were exposed for 6 months or longer and 36% were exposed for greater than one year.
Serious adverse reactions occurred in 23% of patients who received BIZENGRI. Dosage interruptions of BIZENGRI due to an adverse reaction, excluding temporary interruptions of BIZENGRI due to infusion-related reactions, occurred in 32% of patients. Adverse reactions leading to dosing interruptions or delays in at least 2 patients were increased ALT and increased AST.
Table 7 summarizes the adverse reactions in the eNRGy study in patients with NRG1 gene fusion positive advanced, unresectable or metastatic cholangiocarcinoma.
Table 7 Adverse Reactions (≥10%) in Patients with NRG1 Gene Fusion Positive Cholangiocarcinoma Who Received BIZENGRI in the eNRGy Study Adverse Reaction* BIZENGRI
(N=22)All Grades
%Grade 3 or 4
%- * Based on NCI CTCAE v4.03 and MedDRA v26.0
- † Includes abdominal pain, abdominal pain upper and abdominal discomfort
- ‡ Includes fatigue and asthenia
- § Includes IRR, nausea, diarrhea. AEs that were considered IRRs were counted under the composite term "IRR" irrespective of the reported Preferred Term
- ¶ Includes dyspnea, dyspnea exertional
- # Includes arthralgia, back pain, muscle spasms, myalgia, pain in extremity, muscular weakness.
Gastrointestinal disorders Diarrhea 41 0 Abdominal pain† 36 9 Nausea 27 0 Vomiting 14 0 Ascites 14 0 General disorders and administration site conditions Fatigue‡ 46 5 Injury, poisoning and procedural complications Infusion related reaction§ 14 0 Respiratory, thoracic and mediastinal disorders Dyspnea¶ 23 5 Cough 27 0 Metabolism and nutrition disorders Decreased appetite 23 0 Musculoskeletal and connective tissue disorders Musculoskeletal pain# 36 0 Skin and subcutaneous disorders Dry skin 14 0 Table 8 summarizes the laboratory abnormalities in the eNRGy study in patients with NRG1 gene fusion positive advanced, unresectable or metastatic cholangiocarcinoma.
Table 8 Select Laboratory Abnormalities ≥ 20% that Worsened from Baseline in Patients with NRG1 Gene Fusion Positive Cholangiocarcinoma Who Received BIZENGRI in the eNRGy Study Laboratory Abnormality BIZENGRI
(N=22)All Grades
%Grade 3 or 4
%Hematology Decreased platelets 46 0 Decreased hemoglobin 41 14 Chemistry Decreased magnesium 59 5 Increased alanine aminotransferase 50 9 Increased aspartate aminotransferase 41 14 Increased alkaline phosphatase 41 14 Decreased phosphate 41 14 Increased gamma-glutamyl transferase 36 23 Increased bilirubin 32 5 Decreased potassium 32 0 Decreased sodium 32 0 Decreased albumin 23 0 Coagulation Increased activated partial thromboplastin time (aPTT) 27 5 -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, BIZENGRI can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on the use of BIZENGRI in pregnant women to inform a drug-associated risk.
Animal studies have demonstrated that HER2 and/or HER3 deficiency results in embryo-fetal malformation, including effects on cardiac, vascular and neuronal development, and embryolethality (see Data).
Human IgG1 is known to cross the placenta; therefore, BIZENGRI has the potential to be transmitted from the mother to the developing fetus. Advise patients of the potential risk to a fetus.
There are clinical considerations if BIZENGRI is used in pregnant women, or if a patient becomes pregnant within 2 months after the last dose of BIZENGRI (see Clinical Considerations).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Fetal/Neonatal Adverse Reactions
Monitor women who received BIZENGRI during pregnancy or within 2 months prior to conception for oligohydramnios. If oligohydramnios occurs, perform fetal testing that is appropriate for gestational age and consistent with community standards of care.
Human Data
There are no available data on the use of BIZENGRI in pregnant women. In literature reports in pregnant women receiving a HER2-directed antibody, cases of oligohydramnios manifesting as fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death have been reported. These case reports described oligohydramnios in pregnant women who received HER2-directed antibody alone or in combination with chemotherapy. In some case reports, amniotic fluid index increased after use of a HER2-directed antibody was stopped.
Animal Data
There were no animal reproductive or developmental toxicity studies conducted with zenocutuzumab-zbco. A literature-based assessment of the effects on reproduction demonstrated that HER2 and HER3 are critically important in embryo-fetal development. HER2 knockout mice or mice expressing catalytically inactive HER2 die at mid-gestation due to cardiac dysfunction. HER2 knockout mice have also shown abnormal sympathetic nervous system development. In HER3-deficient mice, embryolethality occurred on embryonic day 13.5 due to cardiac and vascular defects, as well as abnormalities in other organs (neural crest, pancreas, stomach, and adrenal). In addition, HER3 is shown to be involved in mammary gland ductal morphogenesis in mice. Zenocutuzumab-zbco can cause embryo-fetal toxicity based on its mechanism of action.
8.2 Lactation
Risk Summary
There are no data on the presence of zenocutuzumab-zbco in human milk, the effects on the breastfed child, or the effects on milk production. Maternal IgG1 is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed child to BIZENGRI are unknown. Consider the developmental and health benefits of breast feeding along with the mother's clinical need for BIZENGRI treatment and any potential adverse effects on the breastfed child from BIZENGRI or from the underlying maternal condition. This consideration should also take into account the elimination half-life of zenocutuzumab-zbco and washout period of 2 months.
8.3 Females and Males of Reproductive Potential
BIZENGRI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BIZENGRI [see Use in Specific Populations (8.1)].
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment with BIZENGRI and for 2 months after the last dose.
8.4 Pediatric Use
The safety and effectiveness of BIZENGRI have not been established in pediatric patients.
8.5 Geriatric Use
Of the 175 patients with NRG1 gene fusion positive tumors in the eNRGy study treated with BIZENGRI at 750 mg every 2 weeks, 75 patients (43%) were 65 years of age or older and 26 patients (15%) were 75 years of age and older. No clinically important differences in safety or efficacy were observed between patients who were ≥65 years of age and younger patients.
-
11 DESCRIPTION
Zenocutuzumab-zbco is a low-fucose humanized full-length immunoglobulin G1 (IgG1) bispecific HER2- and HER3-directed antibody. It has a molecular weight of approximately 146 kDa and is produced in a mammalian cell line (Chinese Hamster Ovary [CHO]) using recombinant DNA technology.
BIZENGRI is a sterile, clear to slightly opalescent, colorless to slightly yellow, preservative-free injection for intravenous infusion in single-dose vials. The pH is 6.0. Each BIZENGRI vial contains 375 mg/18.75 mL zenocutuzumab-zbco at a concentration of 20 mg/mL. Each vial also contains the following inactive ingredients: histidine (34.9 mg), L-histidine hydrochloride monohydrate (51.1 mg), polysorbate 20 (3.7 mg), trehalose (1412 mg), and water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Zenocutuzumab-zbco is a bispecific antibody that binds to the extracellular domains of HER2 and HER3 expressed on the surface of cells, including tumor cells, inhibiting HER2:HER3 dimerization and preventing NRG1 binding to HER3. Zenocutuzumab-zbco decreased cell proliferation and signaling through the phosphoinositide 3-kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) pathway. In addition, zenocutuzumab-zbco mediates antibody-dependent cellular cytotoxicity (ADCC). Zenocutuzumab-zbco showed antitumor activity in mouse models of NRG1 fusion-positive lung and pancreatic cancers.
12.2 Pharmacodynamics
The exposure-response relationship and time-course of pharmacodynamic response for zenocutuzumab-zbco have not been fully characterized.
12.3 Pharmacokinetics
Zenocutuzumab-zbco pharmacokinetic parameters are expressed as mean unless otherwise specified. Zenocutuzumab-zbco exposure increases proportionally over a dose range from 480 mg (0.6 times the approved recommended dosage) to 900 mg (1.2 times the approved recommended dosage). The median time to steady state of zenocutuzumab-zbco concentrations is 8 weeks and the median accumulation ratio is 1.6-fold at the approved recommended dosage.
Distribution
Zenocutuzumab-zbco volume of distribution is 6 L (CV 18%).
Elimination
The steady state zenocutuzumab-zbco half-life is 8 days (SD ±1.3 days) with a clearance of 22 mL/h (CV 37%).
Metabolism
Zenocutuzumab-zbco is expected to be metabolized into small peptides by catabolic pathways.
Specific Populations
No clinically significant differences in the pharmacokinetics of zenocutuzumab-zbco were observed based on age (22 to 88 years), sex, race [White or Asian], body weight (38 to 126 kg), albumin level (20 to 49 g/L), mild or moderate renal impairment (creatinine clearance (CLcr) 30 to 89 mL/min), and mild hepatic impairment (total bilirubin >1 to 1.5 times ULN or AST > ULN).
The pharmacokinetics of zenocutuzumab-zbco in patients with moderate to severe hepatic impairment (total bilirubin > 1.5 to 3 times ULN with any AST) or severe renal impairment (CLcr < 30 mL/min) is unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparison of the incidence of anti-drug antibodies in the studies below with the incidence of anti-drug antibodies in other studies, including those of zenocutuzumab-zbco or of other zenocutuzumab products.
In patients who received zenocutuzumab-zbco at the approved recommended dosage for up to 30 months, 8% (15/188) of patients developed treatment-emergent anti-zenocutuzumab antibodies. Of the patients who developed treatment-emergent anti-zenocutuzumab antibodies, 60% (9/15) patients also developed neutralizing antibodies. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and efficacy of zenocutuzumab is unknown.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Non-Small Cell Lung Cancer
The efficacy of BIZENGRI was evaluated in the eNRGy study (NCT02912949) a multicenter, open-label, multi-cohort clinical study. The study enrolled adult patients with advanced or metastatic NRG1 fusion-positive NSCLC who had disease progression following standard of care treatment for their disease. Identification of positive NRG1 gene fusion status was prospectively determined based on next generation sequencing (NGS) assays performed at local laboratories or central laboratories. Patients received BIZENGRI as an intravenous infusion, 750 mg every 2 weeks, until unacceptable toxicity or disease progression. Tumor assessments were performed every 8 weeks. The major efficacy outcome measures were confirmed overall response rate (ORR) and duration of response (DOR) as determined by blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Efficacy was evaluated in 64 patients with NRG1 fusion-positive NSCLC previously treated with systemic therapy enrolled in eNRGy.
The trial population characteristics were: median age 63.5 years (range: 32 to 86) with 47% of patients ≥ 65 years of age; 64% female; 56% Asian, 33% White, and 11% other races or not reported; none were Hispanic or Latino; baseline ECOG performance status of 0 or 1 (97%) or 2 (3%) and 98% of patients had metastatic disease. Patients received a median of 2 prior systemic therapies (range 1 to 6); 95% had prior platinum chemotherapy and 64% had prior anti-PD-1/PD-L1 therapy. A total of 54 patients (84%) had an NRG1 gene fusion detected by RNA-based NGS that may include DNA sequencing and 9 (14%) had an NRG1 gene fusion detected by DNA-based NGS.
Efficacy results are summarized in Table 9 and Table 10
Table 9: Efficacy Results for Advanced Unresectable or Metastatic NRG1 Fusion-Positive NSCLC in the eNRGy Study Efficacy Parameter BIZENGRI
Previously Treated with Systemic Therapy
(n = 64)- * Confirmed overall response rate assessed by BICR
- † Based on observed duration of response
Overall response rate* (95% CI) 33% (22%, 46%) Complete response rate 1.6% Partial response rate 31% Duration of response Median (95% CI) (months) 7.4 (4.0, 16.6) Patients with DOR ≥6 months† 43% Table 10: Efficacy Results by NRG1 Gene Fusion Partner in NRG1 Fusion-Positive NSCLC Patients in the eNRGy Study NRG1 Partner* BIZENGRI
(n = 64)ORR DOR n (%) 95% CI Range (Months) PR=partial response; PD=progressive disease; NA=not applicable; "+" indicates ongoing response - * Fusion partners identified in this primary analysis set (n=64) may not represent all potential fusion partners.
CD74 37 12 (32) (18, 50) 1.8+; 20.3+ SLC3A2 14 5 (36) (13, 65) 3.6; 20.8+ SDC4 7 2 (29) (3.7, 71) 7.4; 16.6 CDH1 2 1 (50) (1.3, 99) 1.9+ FUT10 1 PD NA NA PVALB 1 PD NA NA ST14 1 PD NA NA VAMP2 1 PR NA 5.6 14.2 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Pancreatic Adenocarcinoma
The efficacy of BIZENGRI was evaluated in the eNRGy study (NCT02912949), a multicenter, open-label, multi-cohort clinical study. The study enrolled 30 adult patients with advanced or metastatic NRG1 fusion-positive pancreatic adenocarcinoma who had disease progression following standard of care treatment. Identification of an NRG1 gene fusion was prospectively determined in local laboratories using next generation sequencing (NGS). Patients received BIZENGRI as an intravenous infusion, 750 mg every 2 weeks, until unacceptable toxicity or disease progression. Tumor assessments were performed every 8 weeks. The major efficacy outcome measures were confirmed overall response rate (ORR) and duration of response (DOR) as determined by a blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
The trial population characteristics were: median age 49 years (range: 21 to 72) with 10% of patients ≥ 65 years of age; 43% female; 87% White, 7% Asian, 3.3% Black or African American, and 3.3% other races or not reported; 3.3% were Hispanic or Latino; baseline ECOG performance status of 0 (53%) or 1 (47%) and all patients had metastatic disease. Patients received a median of 2 prior systemic therapies (range 0 to 5); 97% had prior systemic therapy with FOLFIRINOX, gemcitabine/taxane-based therapy, or both. A total of 27 patients (90%) had an NRG1 gene fusion detected by RNA-based NGS that may include DNA sequencing and 3 (10%) had an NRG1 gene fusion detected by DNA-based NGS.
Efficacy results are summarized in Table 11 and Table 12
Table 11: Efficacy Results for Advanced Unresectable or Metastatic NRG1 Fusion-Positive Pancreatic Adenocarcinoma in the eNRGy Study Efficacy Parameter BIZENGRI
(n = 30)- * Confirmed overall response rate assessed by BICR
- † Based on observed duration of response
Overall response rate* (95% CI) 40% (23%, 59%) Complete response rate 3.3% Partial response rate 37% Duration of response Range (months) 3.7, 16.6 Patients with DOR ≥6 months† 67% Table 12: Efficacy Results by NRG1 Gene Fusion Partner in NRG1 Fusion-Positive Pancreatic Adenocarcinoma Patients in the eNRGy Study NRG1 Partner* BIZENGRI
(n = 30)ORR DOR n (%) 95% CI Range (Months) PR=partial response; PD=progressive disease; SD=stable disease; NA=not applicable; "+" indicates ongoing response - * Fusion partners identified in this primary analysis set (n=30) may not represent all potential fusion partners.
ATP1B1 14 7 (50) (23, 77) 3.7, 16.6 CD44 3 0 (0, 71) NA NOTCH2 3 1 (33) (0.8, 91) 7.4+ SLC4A4 3 2 (67) (9, 99) 7.5+, 15.2+ AGRN 1 PR NA 9.1+ APP 1 PR NA 3.7 CDH1 2 SD, SD NA NA SDC4 1 SD NA NA THBS1 1 PD NA NA VTCN1 1 SD NA NA 14.3 Advanced Unresectable or Metastatic NRG1 Fusion-Positive Cholangiocarcinoma
The efficacy of BIZENGRI was evaluated in the eNRGy study (NCT02912949), a multicenter, open-label, multi-cohort clinical study. The study enrolled adult patients with advanced, unresectable or metastatic NRG1 fusion-positive cholangiocarcinoma. Identification of an NRG1 gene fusion was prospectively determined in local laboratories using next generation sequencing (NGS). Patients received BIZENGRI as an intravenous infusion, 750 mg every 2 weeks, until unacceptable toxicity or disease progression. Tumor assessments were performed every 8 weeks. The major efficacy outcome measures were confirmed overall response rate (ORR) and duration of response (DOR) as determined by a blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Efficacy was evaluated in 19 patients with NRG1 fusion positive cholangiocarcinoma.
The trial population characteristics were: median age 55 years (range 23-82 years), with 26% patients ≥65 years of age, 53% female, 68% White, 11% Asian, 5% Other and 16% not reported; 5% were Hispanic or Latino. Baseline ECOG performance status was 0 (58%), 1 (37%) or 2 (5%), 18 of 19 patients had metastatic disease, and disease site of origin was intrahepatic cholangiocarcinoma in 100% of 16 patients with reported information. Seventeen patients received prior systemic therapies (median 1, range 0 to 4); 14 received cisplatin/gemcitabine (with or without immunotherapy). A total of 17 patients (89%) had an NRG1 gene fusion detected by RNA-based NGS that may include DNA sequencing, 3 patients (16%) had an NRG1 gene fusion detected by whole transcriptome sequencing and 1 patient (5%) had an NRG1 gene fusion detected by DNA-based NGS.
Efficacy results are summarized in Table 13 and Table 14
Table 13 Efficacy Results for Advanced Unresectable or Metastatic NRG1 Fusion-Positive Cholangiocarcinoma in the eNRGy Study Efficacy Parameter BIZENGRI
(n = 19)- * Confirmed overall response rate assessed by BICR
- † Based on observed duration of response
Overall response rate* (95% CI) 36.8% (16.3, 61.6%) Complete response rate 5.3% Partial response rate 31.6% Duration of response Range (months) 2.8, 12.9 Patients with DOR ≥6 months† 28.6% Table 14 Efficacy Results by NRG1 Gene Fusion Partner in NRG1 Fusion-Positive Cholangiocarcinoma Patients in the eNRGy Study NRG1 Partner* BIZENGRI
(n= 19)ORR† DOR n (%) 95% CI Months CR= complete response; PR=partial response; NE=not evaluable; SD=stable disease; NA=not applicable; "+" indicates ongoing response - * Fusion partners identified in this primary analysis set (N=19) may not represent all potential fusion partners.
- † Confirmed overall response rate assessed by BICR.
RBPMS 5 1 (20.0) (PR) 0.5, 71.6 3.7 ATP1B1 4 1 (25.0) (PR) 0.6, 80.6 2.8 VTCN1 2 1 (50.0) (PR) 1.3, 98.7 3.9 ALB 1 CR NA 12.9 CFH 1 NE NA NA FBLN2 1 PR NA 9.1+ FN1 1 PR NA 5.9+ NOTCH2 1 SD NA NA SDC4 1 SD NA NA TNC 1 PR NA 3.8+ TNFSF15 1 SD NA NA -
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
BIZENGRI (zenocutuzumab-zbco) injection is a sterile, clear to slightly opalescent, colorless to slightly yellow, preservative-free solution for intravenous infusion. Each single-dose vial contains 375 mg/18.75 mL (20 mg/mL) BIZENGRI. Two vials (equivalent to 1 dose) are packed in a single carton. (NDC: 71837-1000-1 for individual vial and NDC: 71837-1000-2 for a single carton).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Infusion-Related Reactions/Hypersensitivity/Anaphylaxis
Advise patients that BIZENGRI can cause serious and life-threatening infusion-related reactions (IRRs). Advise patients to alert their healthcare provider immediately for any signs or symptoms of IRRs during and following the infusion [see Warnings and Precautions (5.1)].
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients that BIZENGRI can cause serious and life threatening ILD/pneumonitis. Advise patients to immediately contact their healthcare provider for new or worsening respiratory symptoms [see Warnings and Precautions (5.2)].
Left Ventricular Dysfunction
Inform patients that BIZENGRI can cause serious and life threatening left ventricular dysfunction. Advise patients to immediately contact their healthcare provider for new or worsening cardiovascular symptoms [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- Inform female patients of the potential risk to a fetus. Advise female patients to contact their healthcare provider with a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with BIZENGRI and for 2 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with BIZENGRI and for 2 months after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 05/2026 PATIENT INFORMATION
BIZENGRI® (bi zen gree)
(zenocutuzumab-zbco)
injection, for intravenous useWhat is the most important information I should know about BIZENGRI?
BIZENGRI can cause serious side effects, including:- Infusion-related, allergic and anaphylactic reactions. BIZENGRI can cause serious infusion-related and allergic reactions that can be life-threatening. Infusion-related reactions are also common during BIZENGRI treatment. Before each BIZENGRI infusion, your healthcare provider will give you medicines to help reduce your chance of getting infusion-related reactions. Your healthcare provider will monitor you for signs and symptoms during your infusion and for at least 1 hour after your first infusion and as needed. Tell your healthcare provider right away if you develop any of the following signs or symptoms during or after your BIZENGRI infusion:
- chills or shaking
- nausea, vomiting, or diarrhea
- fever
- cough
- sudden swelling of your face, tongue, throat, or troubled swallowing
- throat tightness or discomfort
- itching or rash
- shortness of breath or wheezing
- chest discomfort
- feeling light-headed
- dizziness
- back or neck pain
- feeling of numbness or tingling
- Lung problems. BIZENGRI can cause serious lung problems that may be life-threatening. If you develop lung problems, your healthcare provider may treat you with corticosteroid medicines. Tell your healthcare provider right away if you develop any new or worsening symptoms of lung problems, including:
- trouble breathing
- shortness of breath
- cough
- fever
- Heart problems that may affect your heart's ability to pump blood. BIZENGRI can cause serious and life-threatening heart problems that may lead to death. Your healthcare provider will check your heart function before you start treatment with BIZENGRI and as needed during your treatment. Tell your healthcare provider right away if you develop any new or worsening symptoms of heart problems, including:
- shortness of breath
- coughing
- tiredness
- swelling of your feet, ankles or legs
- irregular heartbeat
- sudden weight gain
- dizziness or feeling light-headed
- loss of consciousness
Your healthcare provider will check you for these side effects during your treatment with BIZENGRI and may delay your treatment, slow the infusion rate, or completely stop your treatment with BIZENGRI if you develop severe side effects. -
Harm to your unborn baby. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with BIZENGRI.
Females who are able to become pregnant:- Your healthcare provider will do a pregnancy test before you start treatment with BIZENGRI.
- Use effective birth control (contraception) during treatment and for 2 months after your last dose of BIZENGRI.
What is BIZENGRI?
BIZENGRI is a prescription medicine used to treat adults who have:- lung cancer called non-small cell lung cancer (NSCLC):
- that has a neuregulin 1 (NRG1) gene fusion and cannot be removed by surgery or has spread to other parts of the body (advanced unresectable or metastatic), and
- whose disease has worsened on or after prior cancer treatment.
- pancreatic cancer called pancreatic adenocarcinoma:
- that has a neuregulin 1 (NRG1) gene fusion and cannot be removed by surgery or has spread to other parts of the body (advanced unresectable or metastatic), and
- whose disease has worsened on or after prior cancer treatment.
- bile duct cancer called cholangiocarcinoma:
- that has a neuregulin 1 (NRG1) gene fusion and cannot be removed by surgery or has spread to other parts of the body (advanced unresectable or metastatic), and
- whose disease has worsened on or after prior cancer treatment.
Before receiving BIZENGRI, tell your healthcare provider about all of your medical conditions, including if you: - have lung or breathing problems other than your lung cancer.
- have or have had any heart problems.
- are breastfeeding or plan to breastfeed. It is not known if BIZENGRI passes into your breast milk. Do not breastfeed during treatment and for 2 months after your last dose of BIZENGRI.
How will I receive BIZENGRI? - BIZENGRI will be given to you by your healthcare provider as an intravenous (IV) infusion into your vein, usually over 4 hours.
- BIZENGRI is usually given 1 time every 2 weeks.
- Your healthcare provider will decide how many treatments you will need.
What are the possible side effects of BIZENGRI?
BIZENGRI can cause serious side effects, including: The most common side effects of BIZENGRI include:- diarrhea
- muscle or bone pain
- tiredness
- nausea
- shortness of breath
- rash
- constipation
- vomiting
- stomach-area (abdominal) pain
- swelling of your breast, face, ankles or legs
The most common severe abnormal blood test results with BIZENGRI include: - increased blood levels of liver enzymes and bilirubin
- decreased red blood cell counts and platelet counts
- decreased blood level of sodium, magnesium, and phosphate
- increase in the time that it takes your blood to clot
These are not all of the possible side effects of BIZENGRI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about safe and effective use of BIZENGRI:
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about BIZENGRI that is written for health professionals.What are the ingredients in BIZENGRI?
Active ingredient: zenocutuzumab-zbco
Inactive ingredients: histidine, L-histidine hydrochloride monohydrate, polysorbate 20, trehalose, and water for injection.
Manufactured by: Partner Therapeutics, Inc. Lexington, MA 02421
BIZENGRI® is a registered trademark of Merus B.V., a wholly owned subsidiary of Genmab A/S.
U.S. License Number 2087
© 2026 Partner Therapeutics, Inc.
For more information, go to www.BIZENGRI.com or call 1-888-479-5385
AW029-02 -
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 750 mg Carton Label
NDC 71837-1000-2
Rx only
Bizengri®
zenocutuzumab-zbco
Injection 750 mg375 mg/18.75 mL
(20 mg/mL) per vialFor Intravenous Infusion After Dilution
Each carton contains:
Two 375 mg/18.75 mL single-dose vials. Discard unused portion.
 +
+  = one 750 mg dose
= one 750 mg dose
PTx
PARTNER
THERAPEUTICS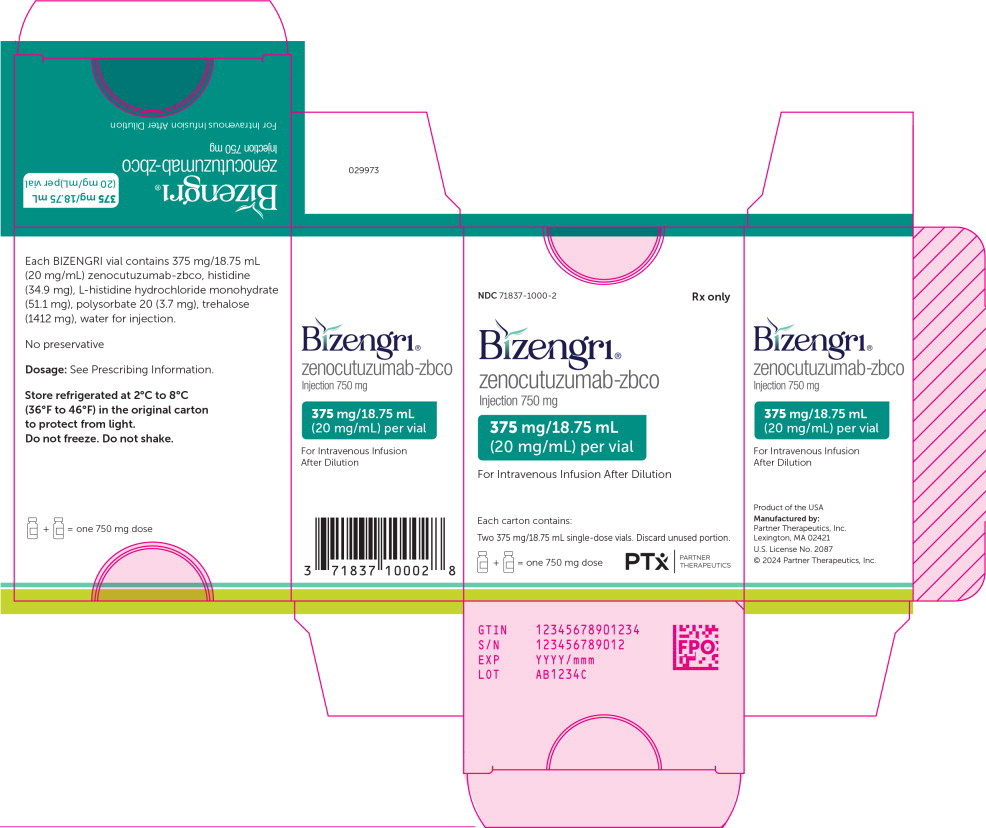
-
INGREDIENTS AND APPEARANCE
BIZENGRI
zenocutuzumab injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71837-1000 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Zenocutuzumab (UNII: AE72RB1W1X) (Zenocutuzumab - UNII:AE72RB1W1X) Zenocutuzumab 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength histidine monohydrochloride monohydrate (UNII: X573657P6P) histidine (UNII: 4QD397987E) trehalose dihydrate (UNII: 7YIN7J07X4) polysorbate 20 (UNII: 7T1F30V5YH) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71837-1000-2 2 in 1 CARTON 12/16/2024 1 NDC: 71837-1000-1 18.75 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761352 12/04/2024 Labeler - Partner Therapeutics, Inc. (080709490)
Trademark Results [BIZENGRI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BIZENGRI 79396591 not registered Live/Pending |
Merus N.V. 2024-04-18 |
 BIZENGRI 79337753 not registered Live/Pending |
Merus N.V. 2022-02-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.