Digoxin by NCS HealthCare of KY, Inc dba Vangard Labs DIGOXIN tablet
Digoxin by
Drug Labeling and Warnings
Digoxin by is a Prescription medication manufactured, distributed, or labeled by NCS HealthCare of KY, Inc dba Vangard Labs. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION:
Digoxin Tablets, USP are one of the cardiac (or digitalis) glycosides, a closely related group of drugs having in common specific effects on the myocardium. These drugs are found in a number of plants. Digoxin is extracted from the leaves of Digitalis lanata. The term “digitalis” is used to designate the whole group of glycosides. The glycosides are composed of two portions: a sugar and a cardenolide (hence “glycosides”).
Digoxin is described chemically as (3β,5β,12β)-3-[(O-2,6-dideoxy-β-D-ribo-hexopyranosyl-(1→4)-O-2,6-dideoxy-β-D-ribo-hexopyranosyl-(1→4)-2,6-dideoxy-β-D-ribo-hexopyranosyl)oxy]-12,14-dihydroxy-card-20(22)-enolide. lts molecular formula is C41H64O14, its molecular weight is 780.95, and its structural formula is:
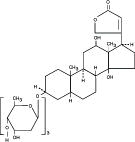
Digoxin exists as odorless white crystals that melt with decomposition above 230°C. The drug is practically insoluble in water and in ether; slightly soluble in diluted (50%) alcohol and in chloroform; and freely soluble in pyridine.
Digoxin Tablets, USP are supplied as 125 mcg (0.125 mg) or 250 mcg (0.25 mg) tablets for oral administration. Each tablet contains the labeled amount of digoxin USP and the following inactive ingredients:
125 mcg (0.125 mg): anhydrous lactose, colloidal silicon dioxide, corn starch, D&C yellow #10 aluminum lake, FD&C yellow #6 aluminum lake, lactose monohydrate, magnesium stearate and starch 1500.
250 mcg (0.25 mg): anhydrous lactose, colloidal silicon dioxide, corn starch, lactose monohydrate, magnesium stearate and starch 1500.
-
CLINICAL PHARMACOLOGY:
Mechanism of Action
Digoxin inhibits sodium-potassium ATPase, an enzyme that regulates the quantity of sodium and potassium inside cells. Inhibition of the enzyme leads to an increase in the intracellular concentration of sodium and thus (by stimulation of sodium-calcium exchange) an increase in the intracellular concentration of calcium. The beneficial effects of digoxin result from direct actions on cardiac muscle, as well as indirect actions on the cardiovascular system mediated by effects on the autonomic nervous system. The autonomic effects include: (1) a vagomimetic action, which is responsible for the effects of digoxin on the sinoatrial and atrioventricular (AV) nodes; and (2) baroreceptor sensitization, which results in increased afferent inhibitory activity and reduced activity of the sympathetic nervous system and renin-angiotensin system for any given increment in mean arterial pressure. The pharmacologic consequences of these direct and indirect effects are: (1) an increase in the force and velocity of myocardial systolic contraction (positive inotropic action); (2) a decrease in the degree of activation of the sympathetic nervous system and renin-angiotensin system (neurohormonal deactivating effect); and (3) slowing of the heart rate and decreased conduction velocity through the AV node (vagomimetic effect). The effects of digoxin in heart failure are mediated by its positive inotropic and neurohormonal deactivating effects, whereas the effects of the drug in atrial arrhythmias are related to its vagomimetic actions. In high doses, digoxin increases sympathetic outflow from the central nervous system (CNS). This increase in sympathetic activity may be an important factor in digitalis toxicity.
Pharmacokinetics
Absorption: Following oral administration, peak serum concentrations of digoxin occur at 1 to 3 hours. Absorption of digoxin from digoxin tablets has been demonstrated to be 60% to 80% complete compared to an identical intravenous dose of digoxin (absolute bioavailability). When digoxin tablets are taken after meals, the rate of absorption is slowed, but the total amount of digoxin absorbed is usually unchanged. When taken with meals high in bran fiber, however, the amount absorbed from an oral dose may be reduced. Comparisons of the systemic availability and equivalent doses for oral preparations of digoxin tablets are shown in Table 1.
Table 1: Comparisons of the Systemic Availability and Equivalent Doses for Oral Preparations of Digoxin Tablets * For example, 125 mcg Digoxin Tablets equivalent to 100 mcg Digoxin Injection/IV.
Product Absolute Equivalent Doses (mcg)* Bioavailability Among Dosage Forms Digoxin Tablets 60 - 80% 62.5 125 250 500 Digoxin Injection/IV 100% 50 100 200 400 In some patients, orally administered digoxin is converted to inactive reduction products (e.g., dihydrodigoxin) by colonic bacteria in the gut. Data suggest that one in ten patients treated with digoxin tablets will degrade 40% or more of the ingested dose. As a result, certain antibiotics may increase the absorption of digoxin in such patients. Although inactivation of these bacteria by antibiotics is rapid, the serum digoxin concentration will rise at a rate consistent with the elimination half-life of digoxin. The magnitude of rise in serum digoxin concentration relates to the extent of bacterial inactivation, and may be as much as two-fold in some cases.
Distribution
Following drug administration, a 6- to 8-hour tissue distribution phase is observed. This is followed by a much more gradual decline in the serum concentration of the drug, which is dependent on the elimination of digoxin from the body. The peak height and slope of the early portion (absorption/distribution phases) of the serum concentration-time curve are dependent upon the route of administration and the absorption characteristics of the formulation. Clinical evidence indicates that the early high serum concentrations do not reflect the concentration of digoxin at its site of action, but that with chronic use, the steady-state post-distribution serum concentrations are in equilibrium with tissue concentrations and correlate with pharmacologic effects. In individual patients, these post-distribution serum concentrations may be useful in evaluating therapeutic and toxic effects (see DOSAGE AND ADMINISTRATION: Serum Digoxin Concentrations).
Digoxin is concentrated in tissues and therefore has a large apparent volume of distribution. Digoxin crosses both the blood-brain barrier and the placenta. At delivery, the serum digoxin concentration in the newborn is similar to the serum concentration in the mother. Approximately 25% of digoxin in the plasma is bound to protein. Serum digoxin concentrations are not significantly altered by large changes in fat tissue weight, so that its distribution space correlates best with lean (i.e., ideal) body weight, not total body weight.
Metabolism
Only a small percentage (16%) of a dose of digoxin is metabolized. The end metabolites, which include 3β-digoxigenin, 3-keto-digoxigenin, and their glucuronide and sulfate conjugates, are polar in nature and are postulated to be formed via hydrolysis, oxidation, and conjugation. The metabolism of digoxin is not dependent upon the cytochrome P-450 system, and digoxin is not known to induce or inhibit the cytochrome P-450 system.
Excretion
Elimination of digoxin follows first-order kinetics (that is, the quantity of digoxin eliminated at any time is proportional to the total body content). Following intravenous administration to healthy volunteers, 50% to 70% of a digoxin dose is excreted unchanged in the urine. Renal excretion of digoxin is proportional to glomerular filtration rate and is largely independent of urine flow. In healthy volunteers with normal renal function, digoxin has a half-life of 1.5 to 2.0 days. The half-life in anuric patients is prolonged to 3.5 to 5 days. Digoxin is not effectively removed from the body by dialysis, exchange transfusion, or during cardiopulmonary bypass because most of the drug is bound to tissue and does not circulate in the blood.
Special Populations
Race differences in digoxin pharmacokinetics have not been formally studied. Because digoxin is primarily eliminated as unchanged drug via the kidney and because there are no important differences in creatinine clearance among races, pharmacokinetic differences due to race are not expected.
The clearance of digoxin can be primarily correlated with renal function as indicated by creatinine clearance. The Cockcroft and Gault formula for estimation of creatinine clearance includes age, body weight, and gender. Table 5 that provides the usual daily maintenance dose requirements of digoxin tablets based on creatinine clearance (per 70 kg) is presented in the DOSAGE AND ADMINISTRATION section.
Plasma digoxin concentration profiles in patients with acute hepatitis generally fell within the range of profiles in a group of healthy subjects.
Pharmacodynamic and Clinical Effects
The times to onset of pharmacologic effect and to peak effect of preparations of digoxin tablets are shown in Table 2.
Table 2: Times to Onset of Pharmacologic Effect and to Peak Effect of Preparations of Digoxin Tablets * Documented for ventricular response rate in atrial fibrillation, inotropic effects and electrocardiographic changes.
† Depending upon rate of infusion.
Product Time to Time to Onset of Effect* Peak Effect* Digoxin Tablets 0.5 - 2 hours 2 - 6 hours Digoxin Injection/IV 5-30 minutes† 1 - 4 hours Hemodynamic Effects
Digoxin produces hemodynamic improvement in patients with heart failure. Short- and long-term therapy with the drug increases cardiac output and lowers pulmonary artery pressure, pulmonary capillary wedge pressure, and systemic vascular resistance. These hemodynamic effects are accompanied by an increase in the left ventricular ejection fraction and a decrease in end-systolic and end-diastolic dimensions.
Chronic Heart Failure
Two 12-week, double-blind placebo-controlled studies enrolled 178 (RADIANCE trial) and 88 (PROVED trial) patients with NYHA class II or III heart failure previously treated with digoxin, a diuretic, and an ACE inhibitor (RADIANCE only) and randomized them to placebo or treatment with digoxin tablets. Both trials demonstrated better preservation of exercise capacity in patients randomized to digoxin tablets. Continued treatment with digoxin tablets reduced the risk of developing worsening heart failure, as evidenced by heart failure-related hospitalizations and emergency care and the need for concomitant heart failure therapy. The larger study also showed treatment-related benefits in NYHA class and patients' global assessment. In the smaller trial, these trended in favor of a treatment benefit.
The Digitalis Investigation Group (DIG) main trial was a multicenter, randomized, double-blind, placebo-controlled mortality study of 6801 patients with heart failure and left ventricular ejection fraction ≤0.45. At randomization, 67% were NYHA class I or II, 71% had heart failure of ischemic etiology, 44% had been receiving digoxin, and most were receiving concomitant ACE inhibitor (94%) and diuretic (82%). Patients were randomized to placebo or digoxin tablets, the dose of which was adjusted for the patient's age, sex, lean body weight, and serum creatinine (see DOSAGE AND ADMINISTRATION), and followed for up to 58 months (median 37 months). The median daily dose prescribed was 0.25 mg. Overall all-cause mortality was 35% with no difference between groups (95% confidence limits for relative risk of 0.91 to 1.07). Digoxin was associated with a 25% reduction in the number of hospitalizations for heart failure, a 28% reduction in the risk of a patient having at least one hospitalization for heart failure, and a 6.5% reduction in total hospitalizations (for any cause).
Use of digoxin tablets was associated with a trend to increase in time to all-cause death or hospitalization. The trend was evident in subgroups of patients with mild heart failure as well as more severe disease, as shown in Table 3. Although the effect on all-cause death or hospitalization was not statistically significant, much of the apparent benefit derived from effects on mortality and hospitalization attributed to heart failure.
Table 3: Subgroup Analyses of Mortality and Hospitalization During the First Two Years Following Randomization * Number of patients with an event during the first 2 years per 1000 randomized patients.
† Relative-risk (95% confidence interval).
‡ DIG Ancillary Study.
Risk of All-Cause Mortality or All-Cause Hospitalization* n Placebo Digoxin Tablets Relative risk† All patients 0.94 (EF ≤0.45) 6801 604 593 (0.88-1.00) 0.96 NYHA I/II 4571 549 541 (0.89-1.04) 0.99 EF 0.25-0.45 4543 568 571 (0.91-1.07) 0.98 CTR ≤0.55 4455 561 563 (0.91-1.06) 0.88 NYHA III/IV 2224 719 696 (0.80-0.97) 0.84 EF <0.25 2258 677 637 (0.76-0.93) 0.85 CTR >0.55 2346 687 650 (0.77-0.94) 1.04 EF >0.45‡ 987 571 585 (0.88-1.23) Risk of HF-Related Mortality or HF-Related Hospitalization* n Placebo Digoxin Tablets Relative risk† All patients 0.69 (EF ≤0.45) 6801 294 217 (0.63-0.76) 0.70 NYHA I/II 4571 242 178 (0.62-0.80) 0.74 EF 0.25-0.45 4543 244 190 (0.66-0.84) 0.71 CTR ≤0.55 4455 239 180 (0.63-0.81) 0.65 NYHA III/IV 2224 402 295 (0.57-0.75) 0.61 EF <0.25 2258 394 270 (0.53-0.71) 0.65 CTR >0.55 2346 398 287 (0.57-0.75) 0.72 EF >0.45‡ 987 179 136 (0.53-0.99) In situations where there is no statistically significant benefit of treatment evident from a trial's primary endpoint, results pertaining to a secondary endpoint should be interpreted cautiously.
-
INDICATIONS AND USAGE:
Heart Failure
Digoxin Tablets are indicated for the treatment of mild to moderate heart failure. Digoxin Tablets increase left ventricular ejection fraction and improve heart failure symptoms as evidenced by exercise capacity and heart failure-related hospitalizations and emergency care, while having no effect on mortality. Where possible, Digoxin Tablets should be used with a diuretic and an angiotensin-converting enzyme inhibitor, but an optimal order for starting these three drugs cannot be specified.
- CONTRAINDICATIONS:
-
WARNINGS:
Sinus Node Disease and AV Block
Because digoxin slows sinoatrial and AV conduction, the drug commonly prolongs the PR interval. The drug may cause severe sinus bradycardia or sinoatrial block in patients with preexisting sinus node disease and may cause advanced or complete heart block in patients with preexisting incomplete AV block. In such patients consideration should be given to the insertion of a pacemaker before treatment with digoxin.
Accessory AV Pathway (Wolff-Parkinson-White Syndrome)
After intravenous digoxin therapy, some patients with paroxysmal atrial fibrillation or flutter and a coexisting accessory AV pathway have developed increased antegrade conduction across the accessory pathway bypassing the AV node, leading to a very rapid ventricular response or ventricular fibrillation. Unless conduction down the accessory pathway has been blocked (either pharmacologically or by surgery), digoxin should not be used in such patients. The treatment of paroxysmal supraventricular tachycardia in such patients is usually direct-current cardioversion.
Use in Patients with Preserved Left Ventricular Systolic Function
Patients with certain disorders involving heart failure associated with preserved left ventricular ejection fraction may be particularly susceptible to toxicity of the drug. Such disorders include restrictive cardiomyopathy, constrictive pericarditis, amyloid heart disease, and acute cor pulmonale. Patients with idiopathic hypertrophic subaortic stenosis may have worsening of the outflow obstruction due to the inotropic effects of digoxin. Digoxin should generally be avoided in these patients, although it has been used for ventricular rate control in the subgroup of patients with atrial fibrillation.
-
PRECAUTIONS:
Use in Patients with Impaired Renal Function
Digoxin is primarily excreted by the kidneys; therefore, patients with impaired renal function require smaller than usual maintenance doses of digoxin (see DOSAGE AND ADMINISTRATION). Because of the prolonged elimination half-life, a longer period of time is required to achieve an initial or new steady-state serum concentration in patients with renal impairment than in patients with normal renal function. If appropriate care is not taken to reduce the dose of digoxin, such patients are at high risk for toxicity, and toxic effects will last longer in such patients than in patients with normal renal function.
Use in Patients with Electrolyte Disorders
In patients with hypokalemia or hypomagnesemia, toxicity may occur despite serum digoxin concentrations below 2 ng/mL, because potassium or magnesium depletion sensitizes the myocardium to digoxin. Therefore, it is desirable to maintain normal serum potassium and magnesium concentrations in patients being treated with digoxin. Deficiencies of these electrolytes may result from malnutrition, diarrhea, or prolonged vomiting, as well as the use of the following drugs or procedures: diuretics, amphotericin B, corticosteroids, antacids, dialysis, and mechanical suction of gastrointestinal secretions.
Hypercalcemia from any cause predisposes the patient to digitalis toxicity. Calcium, particularly when administered rapidly by the intravenous route, may produce serious arrhythmias in digitalized patients. On the other hand, hypocalcemia can nullity the effects of digoxin in humans; thus, digoxin may be ineffective until serum calcium is restored to normal. These interactions are related to the fact that digoxin affects contractility and excitability of the heart in a manner similar to that of calcium.
Use in Thyroid Disorders and Hypermetabolic States
Hypothyroidism may reduce the requirements for digoxin. Heart failure and/or atrial arrhythmias resulting from hypermetabolic or hyperdynamic states (e.g., hyperthyroidism, hypoxia or arteriovenous shunt) are best, treated by addressing the underlying condition. Atrial arrhythmias associated with hypermetabolic states are particularly resistant to digoxin treatment. Care must be taken to avoid toxicity if digoxin is used.
Use in Patients with Acute Myocardial Infarction
Digoxin should be used with caution in patients with acute myocardial infarction. The use of inotropic drugs in some patients in this setting may result in undesirable increases in myocardial oxygen demand and ischemia.
Use During Electrical Cardioversion
It may be desirable to reduce the dose of digoxin for 1 to 2 days prior to electrical cardioversion of atrial fibrillation to avoid the induction of ventricular arrhythmias, but physicians must consider the consequences of increasing the ventricular response if digoxin is withdrawn. If digitalis toxicity is suspected, elective cardioversion should be delayed. If it is not prudent to delay cardioversion, the lowest possible energy level should be selected to avoid provoking ventricular arrhythmias.
Use in Patients with Myocarditis
Digoxin can rarely precipitate vasoconstriction and therefore should be avoided in patients with myocarditis.
Use in Patients with Beriberi Heart Disease
Patients with beriberi heart disease may fail to respond adequately to digoxin if the underlying thiamine deficiency is not treated concomitantly.
Laboratory Test Monitoring
Patients receiving digoxin should have their serum electrolytes and renal function (serum creatinine concentrations) assessed periodically; the frequency of assessments will depend on the clinical setting. For discussion of serum digoxin concentrations; see DOSAGE AND ADMINISTRATION section.
Drug Interactions
Potassium-depleting diuretics are a major contributing factor to digitalis toxicity. Calcium, particularly if administered rapidly by the intravenous route, may produce serious arrhythmias in digitalized patients. Quinidine, verapamil, amiodarone, propafenone, indomethacin, itraconazole, alprazolam, and spironolactone raise the serum digoxin concentration due to a reduction in clearance and/or in volume of distribution of the drug, with the implication that digitalis intoxication may result. Erythromycin and clarithromycin (and possibly other macrolide antibiotics) and tetracycline may increase digoxin absorption in patients who inactivate digoxin by bacterial metabolism in the lower intestine, so that digitalis intoxication may result (see CLINICAL PHARMACOLOGY: Absorption). Propantheline and diphenoxylate, by decreasing gut motility, may increase digoxin absorption. Antacids, kaolin-pectin, sulfasalazine, neomycin, cholestyramine; certain anticancer drugs, and metoclopramide may interfere with intestinal digoxin absorption, resulting in unexpectedly low serum concentrations. Rifampin may decrease serum digoxin concentration, especially in patients with renal dysfunction, by increasing the non-renal clearance of digoxin. There have been inconsistent reports regarding the effects of other drugs [e.g., quinine, penicillamine] on serum digoxin concentration. Thyroid administration to a digitalized, hypothyroid patient may increase the dose requirement of digoxin. Concomitant use of digoxin and sympathomimetics increases the risk of cardiac arrhythmias. Succinylcholine may cause a sudden extrusion of potassium from muscle cells, and may thereby cause arrhythmias in digitalized patients. Although calcium channel blockers and digoxin may be useful in combination to control atrial fibrillation, their additive effects on AV node conduction can result in advanced or complete heart block. Both digitalis glycosides and beta-blockers slow atrioventricular conduction and decrease heart rate. Concomitant use can increase the risk of bradycardia. Digoxin concentrations are increased by about 15% when digoxin and cervedilol are administered concomitantly. Therefore, increased monitoring of digoxin is recommended when initiating, adjusting, or discontinuing carvedilol.
Due to the considerable variability of these interactions; the dosage of digoxin should be individualized when patients receive these medications concurrently. Furthermore, caution should be exercised when combining digoxin with any drug that may cause a significant deterioration in renal function, since a decline in glomerular filtration or tubular secretion may impair the excretion of digoxin.
Drug/Laboratory Test Interactions
The use of therapeutic doses of digoxin may cause prolongation of the PR interval and depression of the ST segment on the electrocardiogram. Digoxin may produce false positive ST-T changes on the electrocardiogram during exercise testing. These electrophysiologic effects reflect an expected effect of the drug and are not indicative of toxicity.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Digoxin showed no genotoxic potential in vitro studies (Ames test and mouse lymphoma). No data are available on the carcinogenic potential of digoxin, nor have studies been conducted to assess its potential to affect fertility.
Pregnancy: Teratogenic Effects. Pregnancy Category C
Animal reproduction studies have not been conducted with Digoxin. It is also not known whether digoxin can cause fetal harm when administered to a pregnant woman or can affect reproductive capacity. Digoxin should be given to a pregnant woman only if clearly needed.
Nursing Mothers
Studies have shown that digoxin concentrations in the mother's serum and milk are similar. However, the estimated exposure of a nursing infant to digoxin via breastfeeding will be far below the usual infant maintenance dose. Therefore, this amount should have no pharmacologic effect upon the infant. Nevertheless, caution should be exercised when digoxin is administered to a nursing woman.
Pediatric Use
Newborn infants display considerable variability in their tolerance to digoxin. Premature and immature infants are particularly sensitive to the effects of digoxin, and the dosage of the drug must not only be reduced but must be individualized according to their degree of maturity. Digitalis glycosides can cause poisoning in children due to accidental ingestion.
Geriatric Use
The majority of clinical experience gained with digoxin has been in the elderly population. This experience has not identified differences in response or adverse effects between the elderly and younger patients. However, this drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, which should be based on renal function, and it may be useful to monitor renal function (see DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS:
In general, the adverse reactions of digoxin are dose-dependent and occur at doses higher than those needed to achieve a therapeutic effect. Hence, adverse reactions are less common when digoxin is used within the recommended dose range or therapeutic serum concentration range and when there is careful attention to concurrent medications and conditions.
Because some patients may be particularly susceptible to side effects with digoxin, the dosage of the drug should always be selected carefully and adjusted as the clinical condition of the patient warrants. In the past, when high doses of digoxin were used and little attention was paid to clinical status or concurrent medications, adverse reactions to digoxin were more frequent and severe. Cardiac adverse reactions accounted for about one-half, gastrointestinal disturbances for about one-fourth, and CNS and other toxicity for about one-fourth of these adverse reactions. However, available evidence suggests that the incidence and severity of digoxin toxicity has decreased substantially in recent years. In recent controlled clinical trials, in patients with predominantly mild to moderate heart failure, the incidence of adverse experiences was comparable in patients taking digoxin and in those taking placebo. In a large mortality trial, the incidence of hospitalization for suspected digoxin toxicity was 2% in patients taking Digoxin Tablets compared to 0.9% in patients taking placebo. In this trial, the most common manifestations of digoxin toxicity included gastrointestinal and cardiac disturbances; CNS manifestations were less common.
Adults: Cardiac
Therapeutic doses of digoxin may cause heart block in patients with pre-existing sinoatrial or AV conduction disorders; heart block can be avoided by adjusting the dose of digoxin. Prophylactic use of a cardiac pacemaker may be considered if the risk of heart block is considered unacceptable. High doses of digoxin may produce a variety of rhythm disturbances, such as first-degree, second-degree (Wenckebach), or third-degree heart block (including asystole); atrial tachycardia with block; AV dissociation; accelerated junctional (nodal) rhythm; unifocal or multiform ventricular premature contractions (especially bigeminy or trigeminy); ventricular tachycardia; and ventricular fibrillation. Digoxin produces PR prolongation and ST segment depression which should not by themselves be considered digoxin toxicity. Cardiac toxicity can also occur at therapeutic doses in patients who have conditions which may alter their sensitivity to digoxin (see WARNINGS and PRECAUTIONS).
Gastrointestinal
Digoxin may cause anorexia, nausea, vomiting, and diarrhea. Rarely, the use of digoxin has been associated with abdominal pain, intestinal ischemia, and hemorrhagic necrosis of the intestines.
CNS
Digoxin can produce visual disturbances (blurred or yellow vision), headache, weakness, dizziness, apathy, confusion, and mental disturbances (such as anxiety, depression, delirium; and hallucination).
Other
Gynecomastia has been occasionally observed following the prolonged use of digoxin. Thrombocytopenia and maculopapular rash and other skin reactions have been rarely observed.
Table 4 summarizes the incidence of those adverse experiences listed above for patients treated with Digoxin Tablets or placebo from two randomized, double-blind, placebo-controlled withdrawal trials. Patients in these trials were also receiving diuretics with or without angiotensin-converting enzyme inhibitors. These patients had been stable on digoxin, and were randomized to digoxin or placebo. The results shown in Table 4 reflect the experience in patients following dosage titration with the use of serum digoxin concentrations and careful follow-up. These adverse experiences are consistent with results from a large, placebo-controlled mortality trial (DIG trial) wherein over half the patients were not receiving digoxin prior to enrollment.
Table 4: Adverse Experiences in Two Parallel, Double-Blind, Placebo-Controlled Withdrawal Trials (Number of Patients Reporting) Adverse Experience Digoxin Patients Placebo Patients (n=123) (n=125) Cardiac Palpitation 1 4 Ventricular extrasystole 1 1 Tachycardia 2 1 Heart arrest 1 1 Gastrointestinal Anorexia 1 4 Nausea 4 2 Vomiting 2 1 Diarrhea 4 1 Abdominal pain 0 6 CNS Headache 4 4 Dizziness 6 5 Mental disturbances 5 1 Other Rash 2 1 Death 4 3 Infants and Children
The side effects of digoxin in infants and children differ from those seen in adults in several respects. Although digoxin may produce anorexia, nausea, vomiting, diarrhea, and CNS disturbances in young patients, these are rarely the initial symptoms of overdosage. Rather, the earliest and most frequent manifestation of excessive dosing with digoxin in infants and children is the appearance of cardiac arrhythmias, including sinus bradycardia. In children, the use of digoxin may produce any arrhythmia. The most common are conduction disturbances or supraventricular tachyarrhythmias, such as atrial tachycardia (with or without block) and junctional (nodal) tachycardia. Ventricular arrhythmias are less common. Sinus bradycardia may be a sign of impending digoxin intoxication, especially in infants, even in the absence of first-degree heart block. Any arrhythmia or alteration in cardiac conduction that develops in a child taking digoxin should be assumed to be caused by digoxin, until further evaluation proves otherwise.
To report SUSPECTED ADVERSE REACTIONS, contact West-ward Pharmaceutical Corp. at 1-877-233-2001, or the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
OVERDOSAGE:
Signs and Symptoms
The signs and symptoms of toxicity are generally similar to those described in the ADVERSE REACTIONS section but may be more frequent and can be more severe. Signs and symptoms of digoxin toxicity become more frequent with levels above 2 ng/mL. However, in deciding whether a patient's symptoms are due to digoxin, the clinical state together with serum electrolyte levels and thyroid function are important factors (see DOSAGE AND ADMINISTRATION).
Adults
In adults without heart disease, clinical observation suggests that an overdose of digoxin of 10 to 15 mg was the dose resulting in death of half of the patients. If more than 25 mg of digoxin was ingested by an adult without heart disease, death or progressive toxicity responsive only to digoxin-binding Fab antibody fragments resulted. Cardiac manifestations are the most frequent and serious sign of both acute and chronic toxicity. Peak cardiac effects generally occur 3 to 6 hours following overdosage and may persist for the ensuing 24 hours or longer. Digoxin toxicity may result in almost any type of arrhythmia (see ADVERSE REACTIONS). Multiple rhythm disturbances in the same patient are common. Cardiac arrest from asystole or ventricular fibrillation due to digoxin toxicity is usually fatal.
Among the extra-cardiac manifestations, gastrointestinal symptoms (e.g., nausea, vomiting, anorexia) are very common (up to 80% incidence) and precede cardiac manifestations in approximately half of the patients in most literature reports. Neurologic manifestations (e.g., dizziness, various CNS disturbances), fatigue, and malaise are very common. Visual manifestations may also occur with aberration in color vision (predominance of yellow green) the most frequent. Neurological and visual symptoms may persist after other signs of toxicity have resolved. In chronic toxicity, non-specific extra-cardiac symptoms, such as malaise and weakness, may predominate.
Children
In children aged 1 to 3 years without heart disease, clinical observation suggests that an overdose of digoxin of 6 to 10 mg was the dose resulting in death in half of the patients. If more than 10 mg of digoxin was ingested by a child aged 1 to 3 years without heart disease, the outcome was uniformly fatal when Fab fragment treatment was not given. Most manifestations of toxicity in children occur during or shortly after the loading phase with digoxin. The same arrhythmias or combination of arrhythmias that occur in adults can occur in pediatrics. Sinus tachycardia, supraventricular tachycardia, and rapid atrial fibrillation are seen less frequently in the pediatric population. Pediatric patients are more likely to present with an AV conduction disturbance or a sinus bradycardia. Any arrhythmia or alteration in cardiac conduction that develops in a child taking digoxin should be assumed to be caused by digoxin, until further evaluation proves otherwise.
The frequent extracardiac manifestations similar to those seen in adults are gastrointestinal, CNS, and visual. However, nausea and vomiting are not frequent in infants and small children.
In addition to the undesirable effects seen with recommended doses, weight loss in older age groups and failure to thrive in infants, abdominal pain due to mesenteric artery ischemia, drowsiness, and behavioral disturbances including psychotic manifestations have been reported in overdose.
Treatment
In addition to cardiac monitoring, digoxin should be temporarily discontinued until the adverse reaction resolves and may be all that is required to treat the adverse reaction such as in asymptomatic bradycardia or digoxin-related heart block. Every effort should also be made to correct factors that may contribute to the adverse reaction (such as electrolyte disturbances, thyroid function, or concurrent medications) (see WARNINGS and PRECAUTIONS: Drug Interactions). Once the adverse reaction has resolved, therapy with digoxin may be reinstituted, following a careful reassessment of dose.
When the primary manifestation of digoxin overdosage is a cardiac arrhythmia, additional therapy may be needed.
If the rhythm disturbance is a symptomatic bradyarrhythmia or heart block, consideration should be given to the reversal of toxicity with Digoxin Immune Fab (Ovine) [DIGIBIND® or DigiFab®] (see Massive Digitalis Overdosage subsection), the use of atropine, or the insertion of a temporary cardiac pacemaker. Digoxin Immune Fab (Ovine) is a specific antidote for digoxin and may be used to reverse potentially life-threatening ventricular arrhythmias due to digoxin overdosage.
If the rhythm disturbance is a ventricular arrhythmia, consideration should be given to the correction of electrolyte disorders, particularly if hypokalemia (see Administration of Potassium subsection) or hypomagnesemia is present. Ventricular arrhythmias may respond to lidocaine or phenytoin.
Administration of Potassium
Before administering potassium in digoxin overdose for hypokalemia, the serum potassium must be known and every effort should be made to maintain the serum potassium concentration between 5 and 5.5 mmol/L. Potassium salts should be avoided as they may be dangerous in patients who manifest bradycardia or heart block due to digoxin (unless primarily related to supraventricular tachycardia) and in the setting of massive digitalis overdosage. Potassium is usually administered orally, but when correction of the arrhythmia is urgent and the serum potassium concentration is low, potassium may be administered cautiously by the intravenous route. The electrocardiogram should be monitored for any evidence of potassium toxicity (e.g., peaking of T waves) and to observe the effect on the arrhythmia.
Massive Digitalis Overdosage
Manifestations of life-threatening toxicity include ventricular tachycardia or ventricular fibrillation, or progressive bradyarrhythmias, or heart block. Digoxin Immune Fab (Ovine) should be used to reverse the toxic effects of ingestion of a massive overdose. The decision to administer Digoxin Immune Fab (Ovine) to a patient who has ingested a massive dose of digoxin but who has not yet manifested life-threatening toxicity should depend on the likelihood that life-threatening toxicity will occur (see above).
Digoxin is not effectively removed from the body by dialysis due to its large extravascular volume of distribution. Patients with massive digitalis ingestion should receive large doses of activated charcoal to prevent absorption and bind digoxin in the gut during enteroenteric recirculation. Emesis may be indicated especially if ingestion has occurred within 30 minutes of the patient's presentation at the hospital. Emesis should not be induced in patients who are obtunded. If a patient presents more than 2 hours after ingestion or already has toxic manifestations, it may be unsafe to induce vomiting because such maneuvers may induce an acute vagal episode that can worsen digitalis-related arrhythmias.
In cases where a large amount of digoxin has been ingested, hyperkalemia may be present due to release of potassium from skeletal muscle. Hyperkalemia caused by massive digitalis toxicity is best treated with Digoxin Immune Fab (Ovine); initial treatment with glucose and insulin may also be required if hyperkalemia itself is acutely life-threatening.
-
DOSAGE AND ADMINISTRATION:
General
Recommended dosages of digoxin may require considerable modification because of individual sensitivity of the patient to the drug, the presence of associated conditions, or the use of concurrent medications. In selecting a dose of digoxin, the following factors must be considered:
- The body weight of the patient. Doses should be calculated based upon lean (i.e., ideal) body weight.
- The patient's renal function, preferably evaluated on the basis of estimated creatinine clearance.
- The patient's age. Infants and children require different doses of digoxin than adults. Also, advanced age may be indicative of diminished renal function even in patients with normal serum creatinine concentration (i.e., below 1.5 mg/dL).
- Concomitant disease states, concurrent medications, or other factors likely to alter the pharmacokinetic or pharmacodynamic profile of digoxin (see PRECAUTIONS).
Serum Digoxin Concentrations
In general, the dose of digoxin used should be determined on clinical grounds. However, measurement of serum digoxin concentrations can be helpful to the clinician in determining the adequacy of digoxin therapy and in assigning certain probabilities to the likelihood of digoxin intoxication. About two-thirds of adults considered adequately digitalized (without evidence of toxicity) have serum digoxin concentrations ranging from 0.8 to 2 ng/mL (lower serum trough concentrations of 0.5 to 1 ng/mL may be appropriate in some adult patients, see Maintenance Dosing.) However, digoxin may produce clinical benefits even at serum concentrations below this range. About two-thirds of adult patients with clinical toxicity have serum digoxin concentrations greater than 2 ng/mL. However, since one-third of patients with clinical toxicity have concentrations less than 2 ng/mL, values below 2 ng/mL do not rule out the possibility that a certain sign or symptom is related to digoxin therapy. Rarely, there are patients who are unable to tolerate digoxin at serum concentrations below 0.8 ng/mL. Consequently, the serum concentration of digoxin should always be interpreted in the overall clinical context, and an isolated measurement should not be used alone as the basis for increasing or decreasing the dose of the drug.
To allow adequate time for equilibration of digoxin between serum and tissue, sampling of serum concentrations should be done just before the next scheduled dose of the drug. If this is not possible, sampling should be done at least 6 to 8 hours after the last dose, regardless of the route of administration or the formulation used. On a once-daily dosing schedule, the concentration of digoxin will be 10% to 25% lower when sampled at 24 versus 8 hours, depending upon the patient's renal function. On a twice-daily dosing schedule, there will be only minor differences in serum digoxin concentrations whether sampling is done at 8 or 12 hours after a dose.
If a discrepancy exists between the reported serum concentration and the observed clinical response, the clinician should consider the following possibilities:
- Analytical problems in the assay procedure.
- Inappropriate serum sampling time.
- Administration of a digitalis glycoside other than digoxin.
- Conditions (described in WARNINGS and PRECAUTIONS) causing an alteration in the sensitivity of the patient to digoxin.
- Serum digoxin concentration may decrease acutely during periods of exercise without any associated change in clinical efficacy due to increased binding of digoxin to skeletal muscle.
Heart Failure: Adults
Digitalization may be accomplished by either of two general approaches that vary in dosage and frequency of administration, but reach the same endpoint in terms of total amount of digoxin accumulated in the body.
- If rapid digitalization is considered medically appropriate, it may be achieved by administering a loading dose based upon projected peak digoxin body stores. Maintenance dose can be calculated as a percentage of the loading dose.
- More gradual digitalization may be obtained by beginning an appropriate maintenance dose, thus allowing digoxin body stores to accumulate slowly. Steady-state serum digoxin concentrations will be achieved in approximately five half-lives of the drug for the individual patient. Depending upon the patient's renal function, this will take between 1 and 3 weeks.
Rapid Digitalization with a Loading Dose
Peak digoxin body stores of 8 to 12 mcg/kg should provide therapeutic effect with minimum risk of toxicity in most patients with heart failure and normal sinus rhythm. Because of altered digoxin distribution and elimination, projected peak body stores for patients with renal insufficiency should be conservative (i.e., 6 to 10 mcg/kg) (see PRECAUTIONS).
The loading dose should be administered in several portions, with roughly half the total given as the first dose. Additional fractions of this planned total dose may be given at 6- to 8-hour intervals, with careful assessment of clinical response before each additional dose.
If the patient's clinical response necessitates a change from the calculated loading dose of digoxin, then calculation of the maintenance dose should be based upon the amount actually given.
A single initial dose of 500 to 750 mcg (0.5 to 0.75 mg) of Digoxin Tablets usually produces a detectable effect in 0.5 to 2 hours that becomes maximal in 2 to 6 hours. Additional doses of 125 to 375 mcg (0.125 to 0.375 mg) may be given cautiously at 6- to 8-hour intervals until clinical evidence of an adequate effect is noted. The usual amount of Digoxin Tablets that a 70 kg patient requires to achieve 8 to 12 mcg/kg peak body stores is 750 to 1250 mcg (0.75 to 1.25 mg).
Digoxin Injection is frequently used to achieve rapid digitalization, with conversion to Digoxin Tablets for maintenance therapy. If patients are switched from intravenous to oral digoxin formulations, allowances must be made for differences in bioavailability when calculating maintenance dosages (see Table 1, CLINICAL PHARMACOLOGY).
Maintenance Dosing
The doses of digoxin used in controlled trials in patients with heart failure have ranged from 125 to 500 mcg (0.125 to 0.5 mg) once daily. In these studies, the digoxin dose has been generally titrated according to the patient's age, lean body weight, and renal function. Therapy is generally initiated at a dose of 250 mcg (0.25 mg) once daily in patients under age 70 with good renal function, at a dose of 125 mcg (0.125 mg) once daily in patients over age 70 or with impaired renal function, and at a dose of 62.5 mcg (0.0625 mg) in patients with marked renal impairment. Doses may be increased every 2 weeks according to clinical response.
In a subset of approximately 1800 patients enrolled in the DIG trial (wherein dosing was based on an algorithm similar to that in Table 5) the mean (± SD) serum digoxin concentrations at 1 month and 12 months were 1.01 ± 0.47 ng/mL and 0.97 ± 0.43 ng/mL, respectively. There are no rigid guidelines as to the range of serum concentrations that are most efficacious. Several post hoc analyses of heart failure patients in the DIG trial suggest that the optimal trough digoxin serum level may be 0.5 ng/mL to 1 ng/mL.
The maintenance dose should be based upon the percentage of the peak body stores lost each day through elimination. The following formula has had wide clinical use:
Maintenance Dose = Peak Body Stores (i.e., Loading Dose) x % Daily Loss 100 Where: % Daily Loss = 14 + Ccr/5 (Ccr is creatinine clearance, corrected to 70 kg body weight or 1.73 m2 body surface area.) Table 5 provides average daily maintenance dose requirements of Digoxin Tablets for patients with heart failure based upon lean body weight and renal function:
Table 5: Usual Daily Maintenance Dose Requirements (mcg) of Digoxin Tablets for Estimated Peak Body Stores of 10 mcg/kg * Ccr is creatinine clearance, corrected to 70 kg body weight or 1.73 m2 body surface area. For adults, if only serum creatinine concentrations (Scr) are available, a Ccr (corrected to 70 kg body weight) may be estimated in men as (140 - Age)/Scr. For women, this result should be multiplied by 0.85. Note: This equation cannot be used for estimating creatinine clearance in infants or children.
† lf no loading dose administered.
‡ 62.5 mcg = 0.0625 mg
Corrected Ccr Lean Body Weight Number of (mL/min kg 50 60 70 80 90 100 Days Before per 70 kg)* lb 110 132 154 176 198 220 Steady State Achieved† 0 62.5‡ 125 125 125 187.5 187.5 22 10 125 125 125 187.5 187.5 187.5 19 20 125 125 187.5 187.5 187.5 250 16 30 125 187.5 187.5 187.5 250 250 14 40 125 187.5 187.5 250 250 250 13 50 187.5 187.5 250 250 250 250 12 60 187.5 187.5 250 250 250 375 11 70 187.5 250 250 250 250 375 10 80 187.5 250 250 250 375 375 9 90 187.5 250 250 250 375 500 8 100 250 250 250 375 375 500 7 Example
Based on Table 5, a patient in heart failure with an estimated lean body weight of 70 kg and a Ccr of 60 mL/min should be given a dose of 250 mcg (0.25 mg) daily of Digoxin Tablets, usually taken after the morning meal. If no loading dose is administered, steady-state serum concentrations in this patient should be anticipated at approximately 11 days.
Infants and Children
In general, divided daily dosing is recommended for infants and young children (under age 10). In the newborn period, renal clearance of digoxin is diminished and suitable dosage adjustments must be observed. This is especially pronounced in the premature infant. Beyond the immediate newborn period, children generally require proportionally larger doses than adults on the basis of body weight or body surface area. Children over 10 years of age require adult dosages in proportion to their body weight. Some researchers have suggested that infants and young children tolerate slightly higher serum concentrations than do adults.
Daily maintenance doses for each age group are given in Table 6 and should provide therapeutic effects with minimum risk of toxicity in most patients with heart failure and normal sinus rhythm. These recommendations assume the presence of normal renal function:
Table 6: Daily Maintenance Doses in Children with Normal Renal Function Age Daily Maintenance Dose (mcg/kg) 2 to 5 Years 10 to 15 5 to 10 Years 7 to 10 Over 10 Years 3 to 5 In children with renal disease, digoxin must be carefully titrated, based upon clinical response.
It cannot be overemphasized that both the adult and pediatric dosage guidelines provided are based upon average patient response and substantial individual variation can be expected. Accordingly, ultimate dosage selection must be based upon clinical assessment of the patient.
Atrial Fibrillation
Peak digoxin body stores larger than the 8 to 12 mcg/kg required for most patients with heart failure and normal sinus rhythm have been used for control of ventricular rate in patients with atrial fibrillation. Doses of digoxin used for the treatment of chronic atrial fibrillation should be titrated to the minimum dose that achieves the desired ventricular rate control without causing undesirable side effects. Data are not available to establish the appropriate resting or exercise target rates that should be achieved.
Dosage Adjustment When Changing Preparations
The difference in bioavailability between Digoxin Injection or Digoxin Tablets must be considered when changing patients from one dosage form to another.
Doses of 100 mcg (0.1. mg) and 200 mcg (0.2 mg) of digoxin solution in capsules are approximately equivalent to 125 mcg (0.125 mg) and 250 mcg (0.25 mg) doses of digoxin tablets and digoxin pediatric elixir, respectively (see Table 1 in CLINICAL PHARMACOLOGY: Pharmacokinetics).
-
HOW SUPPLIED:
Digoxin Tablets, USP 0.125 mg are Yellow, Round, Scored Tablets, Debossed "W 40" on Scored Side.
Digoxin Tablets, USP 0.25 mg are White, Round, Scored Tablets, Debossed "WW 41" on Scored Side and are available in:
- Blistercards of 30 tablets.
- Blistercards of 31 tablets.
Store at 20-25ºC (68-77ºF) [See USP Controlled Room Temperature] in a dry place.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
Digibind® is a registered trademark of GlaxoSmithKline.
DigiFab is a registered trademark of Prostherics Inc.Manufactured By:
West-ward Pharmaceutical Corp.
Eatontown, NJ 07724
Revised December 2011 - PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
DIGOXIN
digoxin tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0615-0518(NDC:0143-1241) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DIGOXIN (UNII: 73K4184T59) (DIGOXIN - UNII:73K4184T59) DIGOXIN 250 ug Inactive Ingredients Ingredient Name Strength ANHYDROUS LACTOSE (UNII: 3SY5LH9PMK) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, CORN (UNII: O8232NY3SJ) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) Product Characteristics Color WHITE (WHITE) Score 2 pieces Shape ROUND (ROUND) Size 7mm Flavor Imprint Code WW41 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0615-0518-31 31 in 1 BLISTER PACK 2 NDC: 0615-0518-39 30 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077002 10/30/2007 Labeler - NCS HealthCare of KY, Inc dba Vangard Labs (050052943) Establishment Name Address ID/FEI Business Operations NCS HealthCare of KY, Inc dba Vangard Labs 050052943 RELABEL(0615-0518) , REPACK(0615-0518)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.