BRAFTOVI- encorafenib capsule
BRAFTOVI by
Drug Labeling and Warnings
BRAFTOVI by is a Prescription medication manufactured, distributed, or labeled by Array BioPharma Inc., Pfizer Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BRAFTOVI safely and effectively. See full prescribing information for BRAFTOVI.
BRAFTOVI® (encorafenib) capsules, for oral use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BRAFTOVI is a kinase inhibitor indicated:
- in combination with binimetinib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test. (1.1, 2.1)
- in combination with cetuximab, for the treatment of adult patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation, as detected by an FDA-approved test, after prior therapy. (1.2, 2.1)
Limitations of Use
BRAFTOVI is not indicated for treatment of patients with wild-type BRAF melanoma or wild-type BRAF CRC. (1.3, 5.2)
DOSAGE FORMS AND STRENGTHS
Capsules: 75 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- New Primary Malignancies, cutaneous and non-cutaneous: Can occur. Monitor for malignancies and perform dermatologic evaluations prior to, while on therapy, and following discontinuation of treatment. (5.1)
- Tumor Promotion in BRAF Wild-Type Tumors: Increased cell proliferation can occur with BRAF inhibitors. (5.2)
- Hemorrhage: Major hemorrhagic events can occur. (5.3)
- Uveitis: Perform ophthalmologic evaluation at regular intervals and for any visual disturbances. (5.4)
- QT Prolongation: Monitor electrolytes before and during treatment. Correct electrolyte abnormalities and control for cardiac risk factors for QT prolongation. Withhold BRAFTOVI for QTc of 500 ms or greater. (5.5)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females with reproductive potential of potential risk to the fetus and to use effective non-hormonal method of contraception. (5.6, 8.1, 8.3)
ADVERSE REACTIONS
Melanoma: Most common adverse reactions (≥25%) for BRAFTOVI, in combination with binimetinib, are fatigue, nausea, vomiting, abdominal pain, and arthralgia. (6.1)
CRC: Most common adverse reactions (≥25%) for BRAFTOVI, in combination with cetuximab, are fatigue, nausea, diarrhea, dermatitis acneiform, abdominal pain, decreased appetite, arthralgia, and rash. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Array BioPharma at 1-844-792-7729 or FDA at 1-800-FDA-1088 or www.fda.gaov/medwatch.
DRUG INTERACTIONS
- Strong or moderate CYP3A4 inhibitors: Avoid coadministration. If unavoidable, reduce BRAFTOVI dosage. (2.6, 7.1)
- Strong or moderate CYP3A4 inducers: Avoid coadministration. (7.1)
- Sensitive CYP3A4 substrates: Coadministration with BRAFTOVI may increase toxicity or decrease efficacy of these agents. Avoid coadministration of BRAFTOVI with hormonal contraceptives. (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
1.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
1.3 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage for BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
2.3 Recommended Dosage for BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
2.4 Administration
2.5 Dosage Modifications for Adverse Reactions
2.6 Dose Modifications for Coadministration with Strong or Moderate CYP3A4 Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
5.2 Tumor Promotion in BRAF Wild-Type Tumors
5.3 Hemorrhage
5.4 Uveitis
5.5 QT Prolongation
5.6 Embryo-Fetal Toxicity
5.7 Risks Associated with BRAFTOVI as a Single Agent
5.8 Risks Associated with Combination Treatment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BRAFTOVI
7.2 Effect of BRAFTOVI on Other Drugs
7.3 Drugs That Prolong the QT Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
14.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
BRAFTOVI® is indicated, in combination with binimetinib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test [see Dosage and Administration (2.1)].
1.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
BRAFTOVI is indicated, in combination with cetuximab, for the treatment of adult patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation, as detected by an FDA-approved test, after prior therapy [see Dosage and Administration (2.1)].
1.3 Limitations of Use
BRAFTOVI is not indicated for treatment of patients with wild-type BRAF melanoma or wild-type BRAF CRC [see Warnings and Precautions (5.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
Confirm the presence of a BRAF V600E or V600K mutation in tumor specimens prior to initiating BRAFTOVI [see Warnings and Precautions (5.2), Clinical Studies (14.1)]. Information on FDA-approved tests for the detection of BRAF V600E and V600K mutations in melanoma is available at: http://www.fda.gov/CompanionDiagnostics.
BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
Confirm the presence of a BRAF V600E mutation in tumor specimens prior to initiating BRAFTOVI [see Warnings and Precautions (5.2), Clinical Studies 14.2)]. Information on FDA-approved tests for the detection of BRAF V600E mutations in CRC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage for BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
The recommended dosage of BRAFTOVI is 450 mg (six 75 mg capsules) orally once daily in combination with binimetinib until disease progression or unacceptable toxicity. Refer to the binimetinib prescribing information for recommended binimetinib dosing information.
2.3 Recommended Dosage for BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
The recommended dosage of BRAFTOVI is 300 mg (four 75 mg capsules) orally once daily in combination with cetuximab until disease progression or unacceptable toxicity. Refer to the cetuximab prescribing information for recommended cetuximab dosing information.
2.4 Administration
BRAFTOVI may be taken with or without food [see Clinical Pharmacology (12.3)]. Do not take a missed dose of BRAFTOVI within 12 hours of the next dose of BRAFTOVI.
Do not take an additional dose if vomiting occurs after BRAFTOVI administration but continue with the next scheduled dose.
2.5 Dosage Modifications for Adverse Reactions
BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
If binimetinib is withheld, reduce BRAFTOVI to a maximum dose of 300 mg (four 75 mg capsules) once daily until binimetinib is resumed [see Warnings and Precautions (5.7)].
Dose reductions for adverse reactions associated with BRAFTOVI are presented in Table 1.
Table 1: Recommended Dose Reductions for BRAFTOVI for Adverse Reactions – Melanoma Action Recommended Dose First Dose Reduction 300 mg (four 75 mg capsules) orally once daily Second Dose Reduction 225 mg (three 75 mg capsules) orally once daily Subsequent Modification Permanently discontinue if unable to tolerate BRAFTOVI 225 mg (three 75 mg capsules) once daily BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
If cetuximab is discontinued, discontinue BRAFTOVI.
Dose reductions for adverse reactions associated with BRAFTOVI are presented in Table 2.
Table 2: Recommended Dose Reductions for BRAFTOVI for Adverse Reactions – CRC Action Recommended Dose First Dose Reduction 225 mg (three 75 mg capsules) orally once daily Second Dose Reduction 150 mg (two 75 mg capsules) orally once daily Subsequent Modification Permanently discontinue if unable to tolerate BRAFTOVI 150 mg (two 75 mg capsules) once daily BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma and BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
Dosage modifications for adverse reactions associated with BRAFTOVI are presented in Table 3.
Table 3: Recommended Dosage Modifications for BRAFTOVI for Adverse Reactions Severity of Adverse Reaction* Dose Modification for BRAFTOVI - * National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
- † Dose modification of BRAFTOVI when administered with binimetinib or with cetuximab is not recommended for new primary cutaneous malignancies; ocular events other than uveitis, iritis, and iridocyclitis; interstitial lung disease/pneumonitis; cardiac dysfunction; creatine phosphokinase (CPK) elevation; rhabdomyolysis; and venous thromboembolism.
New Primary Malignancies [see Warnings and Precautions (5.1)] Non-Cutaneous RAS Mutation-positive Malignancies Permanently discontinue BRAFTOVI. Uveitis [see Warnings and Precautions (5.4)] - Grade 1–3
If Grade 1 or 2 does not respond to specific ocular therapy, or for Grade 3 uveitis, withhold BRAFTOVI for up to 6 weeks. - If improved, resume at same or reduced dose.
- If not improved, permanently discontinue BRAFTOVI.
- Grade 4
Permanently discontinue BRAFTOVI. QTc Prolongation [see Warnings and Precautions (5.5)] - QTcF greater than 500 ms and less than or equal to 60 ms increase from baseline
Withhold BRAFTOVI until QTcF less than or equal to 500 ms. Resume at reduced dose. - If more than one recurrence, permanently discontinue BRAFTOVI.
- QTcF greater than 500 ms and greater than 60 ms increase from baseline
Permanently discontinue BRAFTOVI. Hepatotoxicity - Grade 2 AST or ALT increased
Maintain BRAFTOVI dose. - If no improvement within 4 weeks, withhold BRAFTOVI until improves to Grade 0–1 or to pretreatment/baseline levels and then resume at same dose.
- Grade 3 or 4 AST or ALT increased
See Other Adverse Reactions. Dermatologic (other than Hand-foot Skin Reaction [HFSR]) - Grade 2
If no improvement within 2 weeks, withhold BRAFTOVI until Grade 0–1. Resume at same dose. - Grade 3
Withhold BRAFTOVI until Grade 0–1. Resume at same dose if first occurrence or reduce dose if recurrent. - Grade 4
Permanently discontinue BRAFTOVI. Other Adverse Reactions (including Hemorrhage [see Warnings and Precautions (5.3)] and HFSR)† - Recurrent Grade 2 or
- First occurrence of any Grade 3
Withhold BRAFTOVI for up to 4 weeks. - If improves to Grade 0–1 or to pretreatment/baseline level, resume at reduced dose.
- If no improvement, permanently discontinue BRAFTOVI.
- First occurrence of any Grade 4
Permanently discontinue BRAFTOVI or
Withhold BRAFTOVI for up to 4 weeks.- If improves to Grade 0–1 or to pretreatment/baseline level, then resume at reduced dose.
- If no improvement, permanently discontinue BRAFTOVI.
- Recurrent Grade 3
Consider permanently discontinuing BRAFTOVI. - Recurrent Grade 4
Permanently discontinue BRAFTOVI. Refer to the binimetinib or cetuximab prescribing information for dose modifications for adverse reactions associated with each product, as appropriate.
2.6 Dose Modifications for Coadministration with Strong or Moderate CYP3A4 Inhibitors
Avoid coadministration of BRAFTOVI with strong or moderate CYP3A4 inhibitors. If coadministration is unavoidable, reduce the BRAFTOVI dose according to the recommendations in Table 4. After the inhibitor has been discontinued for 3 to 5 elimination half-lives, resume the BRAFTOVI dose that was taken prior to initiating the CYP3A4 inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Table 4: Recommended Dose Reductions for BRAFTOVI for Coadministration with Strong or Moderate CYP3A4 Inhibitors Current Daily Dose* Dose for Coadministration with Moderate CYP3A4 Inhibitor Dose for Coadministration with Strong CYP3A4 Inhibitor - * Current daily dose refers to recommended dose of BRAFTOVI based on indication or reductions for adverse reactions based on dosing recommendations in Table 1 (Melanoma) and Table 2 (CRC).
- † Encorafenib exposure at the 75 mg QD BRAFTOVI dosage when coadministered with a strong CYP3A4 inhibitor is expected to be higher than at the 150 mg QD dosage in the absence of a CYP3A4 inhibitor and similar to exposure at the 225 mg QD dosage in the absence of a CYP3A4 inhibitor. Monitor patients closely for adverse reactions and use clinical judgement when using BRAFTOVI with strong CYP3A4 inhibitors at the 150 mg dose level.
450 mg 225 mg (three 75 mg capsules) 150 mg (two 75 mg capsules) 300 mg 150 mg (two 75 mg capsules) 75 mg 225 mg 75 mg 75 mg 150 mg 75 mg 75 mg† - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
New primary malignancies, cutaneous and non-cutaneous, have been observed in patients treated with BRAF inhibitors and can occur with BRAFTOVI.
Cutaneous Malignancies
In COLUMBUS, cutaneous squamous cell carcinoma (cuSCC), including keratoacanthoma (KA), occurred in 2.6%, and basal cell carcinoma occurred in 1.6% of patients who received BRAFTOVI in combination with binimetinib. Median time to first occurrence of cuSCC/KA was 5.8 months (range 1 to 9 months) [see Adverse Reactions (6.1)].
For patients who received BRAFTOVI as a single agent, cuSCC/KA was reported in 8%, basal cell carcinoma in 1%, and a new primary melanoma in 5% of patients.
In BEACON CRC, cuSCC/KA occurred in 1.4% of patients with CRC, and a new primary melanoma occurred in 1.4% of patients who received BRAFTOVI in combination with cetuximab.
Perform dermatologic evaluations prior to initiating treatment, every 2 months during treatment, and for up to 6 months following discontinuation of treatment. Manage suspicious skin lesions with excision and dermatopathologic evaluation. Dose modification is not recommended for new primary cutaneous malignancies.
Non-Cutaneous Malignancies
Based on its mechanism of action, BRAFTOVI may promote malignancies associated with activation of RAS through mutation or other mechanisms [see Warnings and Precautions (5.2)]. Monitor patients receiving BRAFTOVI for signs and symptoms of non-cutaneous malignancies. Discontinue BRAFTOVI for RAS mutation-positive non-cutaneous malignancies [see Dosage and Administration (2.5)].
5.2 Tumor Promotion in BRAF Wild-Type Tumors
In vitro experiments have demonstrated paradoxical activation of MAP-kinase signaling and increased cell proliferation in BRAF wild-type cells, which are exposed to BRAF inhibitors. Confirm evidence of BRAF V600E or V600K mutation prior to initiating BRAFTOVI [see Indications and Usage (1), Dosage and Administration (2.1)].
5.3 Hemorrhage
In COLUMBUS, hemorrhage occurred in 19% of patients receiving BRAFTOVI in combination with binimetinib; Grade 3 or greater hemorrhage occurred in 3.2% of patients. The most frequent hemorrhagic events were gastrointestinal, including rectal hemorrhage (4.2%), hematochezia (3.1%), and hemorrhoidal hemorrhage (1%). Fatal intracranial hemorrhage in the setting of new or progressive brain metastases occurred in 1.6% of patients.
In BEACON CRC, hemorrhage occurred in 19% of patients receiving BRAFTOVI in combination with cetuximab; Grade 3 or higher hemorrhage occurred in 1.9% of patients, including fatal gastrointestinal hemorrhage in 0.5% of patients. The most frequent hemorrhagic events were epistaxis (6.9%), hematochezia (2.3%) and rectal hemorrhage (2.3%).
Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.5), Adverse Reactions (6.1)].
5.4 Uveitis
Uveitis, including iritis and iridocyclitis, has been reported in patients treated with BRAFTOVI in combination with binimetinib. In COLUMBUS, the incidence of uveitis among patients treated with BRAFTOVI in combination with binimetinib was 4%.
Assess for visual symptoms at each visit. Perform an ophthalmologic evaluation at regular intervals and for new or worsening visual disturbances, and to follow new or persistent ophthalmologic findings. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.5), Adverse Reactions (6.1)].
5.5 QT Prolongation
BRAFTOVI is associated with dose-dependent QTc interval prolongation in some patients [see Clinical Pharmacology (12.2)]. In COLUMBUS, an increase in QTcF to > 500 ms was measured in 0.5% (1/192) of patients who received BRAFTOVI in combination with binimetinib.
Monitor patients who already have or who are at significant risk of developing QTc prolongation, including patients with known long QT syndromes, clinically significant bradyarrhythmias, severe or uncontrolled heart failure and those taking other medicinal products associated with QT prolongation. Correct hypokalemia and hypomagnesemia prior to and during BRAFTOVI administration. Withhold, reduce dose, or permanently discontinue for QTc > 500 ms [see Dosage and Administration (2.5), Adverse Reactions (6.1)].
5.6 Embryo-Fetal Toxicity
Based on its mechanism of action, BRAFTOVI can cause fetal harm when administered to a pregnant woman. Encorafenib produced embryo-fetal developmental changes in rats and rabbits and was an abortifacient in rabbits at doses greater than or equal to those resulting in exposures approximately 26 (in the rat) and 178 (in the rabbit) times the human exposure at the recommended dose of 450 mg, with no clear findings at lower doses.
Advise women of the potential risk to a fetus. Advise females of reproductive potential to use an effective, non-hormonal method of contraception since BRAFTOVI can render hormonal contraceptives ineffective, during treatment and for 2 weeks after the final dose of BRAFTOVI [see Use in Specific Populations (8.1, 8.3)].
5.7 Risks Associated with BRAFTOVI as a Single Agent
BRAFTOVI when used as a single agent is associated with an increased risk of certain adverse reactions compared to when BRAFTOVI is used in combination with binimetinib. In COLUMBUS, Grades 3 or 4 dermatologic reactions occurred in 21% of patients treated with BRAFTOVI single agent compared to 2% of patients treated with BRAFTOVI in combination with binimetinib [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
If binimetinib is temporarily interrupted or permanently discontinued, reduce the dose of BRAFTOVI as recommended [see Dosage and Administration (2.5)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- Hemorrhage [see Warnings and Precautions (5.3)]
- Uveitis [see Warnings and Precautions (5.4)]
- QT Prolongation [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
The safety of BRAFTOVI in combination with binimetinib is described in 192 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma who received BRAFTOVI (450 mg once daily) in combination with binimetinib (45 mg twice daily) in a randomized open-label, active-controlled trial (COLUMBUS).
The COLUMBUS trial [see Clinical Studies (14.1)] excluded patients with a history of Gilbert's syndrome, abnormal left ventricular ejection fraction, prolonged QTc (>480 ms), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of exposure was 11.8 months for patients treated with BRAFTOVI in combination with binimetinib and 6.2 months for patients treated with vemurafenib.
The most common (≥ 25%) adverse reactions in patients receiving BRAFTOVI in combination with binimetinib were fatigue, nausea, vomiting, abdominal pain, and arthralgia.
Adverse reactions leading to dose interruptions of BRAFTOVI occurred in 30% of patients receiving BRAFTOVI in combination with binimetinib; the most common were nausea (7%), vomiting (7%), and pyrexia (4%). Adverse reactions leading to dose reductions of BRAFTOVI occurred in 14% of patients receiving BRAFTOVI in combination with binimetinib; the most common were arthralgia (2%), fatigue (2%), and nausea (2%). Five percent (5%) of patients receiving BRAFTOVI in combination with binimetinib experienced an adverse reaction that resulted in permanent discontinuation of BRAFTOVI; the most common were hemorrhage in 2% and headache in 1% of patients.
Table 5 and Table 6 present adverse drug reactions and laboratory abnormalities, respectively, identified in COLUMBUS. The COLUMBUS trial was not designed to demonstrate a statistically significant difference in adverse reaction rates for BRAFTOVI in combination with binimetinib, as compared to vemurafenib, for any specific adverse reaction listed in Table 5.
Table 5: Adverse Reactions Occurring in ≥10% of Patients Receiving BRAFTOVI in Combination with Binimetinib in COLUMBUS* Adverse Reaction BRAFTOVI with binimetinib
N=192Vemurafenib
N=186All Grades
(%)Grades 3 and 4†
(%)All Grades
(%)Grades 3 and 4
(%)- * Grades per National Cancer Institute CTCAE v4.03.
- † Grade 4 adverse reactions limited to fatigue (n=1), pruritus (n=1) and rash (n=1) in the BRAFTOVI with binimetinib arm.
- ‡ Represents a composite of multiple, related preferred terms.
General Disorders and Administration Site Conditions Fatigue‡ 43 3 46 6 Pyrexia‡ 18 4 30 0 Gastrointestinal Disorders Nausea 41 2 34 2 Vomiting‡ 30 2 16 1 Abdominal pain‡ 28 4 16 1 Constipation 22 0 6 1 Musculoskeletal and Connective Tissue Disorders Arthralgia‡ 26 1 46 6 Myopathy‡ 23 0 22 1 Pain in extremity 11 1 13 1 Skin and Subcutaneous Tissue Disorders Hyperkeratosis‡ 23 1 49 1 Rash‡ 22 1 53 13 Dry skin‡ 16 0 26 0 Alopecia‡ 14 0 38 0 Pruritus‡ 13 1 21 1 Nervous System Disorders Headache‡ 22 2 20 1 Dizziness‡ 15 3 4 0 Peripheral neuropathy‡ 12 1 13 2 Vascular Disorders Hemorrhage‡ 19 3 9 2 BRAFTOVI when used as a single agent increases the risk of certain adverse reactions compared to BRAFTOVI in combination with binimetinib. In patients receiving BRAFTOVI 300 mg orally once daily as a single agent, the following adverse reactions were observed at a higher rate (≥ 5%) compared to patients receiving BRAFTOVI in combination with binimetinib: palmar-plantar erythrodysesthesia syndrome (51% vs. 7%), hyperkeratosis (57% vs. 23%), dry skin (38% vs. 16%), erythema (16% vs. 7%), rash (41% vs. 22%), alopecia (56% vs. 14%), pruritus (31% vs. 13%), arthralgia (44% vs. 26%), myopathy (33% vs. 23%), back pain (15% vs. 9%), dysgeusia (13% vs. 6%), and acneiform dermatitis (8% vs. 3%).
Other clinically important adverse reactions occurring in <10% of patients who received BRAFTOVI in combination with binimetinib were:
Nervous system disorders: Facial paresis
Gastrointestinal disorders: Pancreatitis
Skin and subcutaneous tissue disorders: Panniculitis
Immune system disorders: Drug hypersensitivity
Table 6: Laboratory Abnormalities Occurring in ≥10% (All Grades) of Patients Receiving BRAFTOVI in Combination with Binimetinib in COLUMBUS* Laboratory Abnormality BRAFTOVI with binimetinib*
N=192Vemurafenib*
N=186All Grades
(%)Grades 3 and 4
(%)All Grades
(%)Grades 3 and 4
(%)- * Grades per National Cancer Institute CTCAE v4.03.
Hematology Anemia 36 3.6 34 2.2 Leukopenia 13 0 10 0.5 Lymphopenia 13 2.1 30 7 Neutropenia 13 3.1 4.8 0.5 Chemistry Increased Creatinine 93 3.6 92 1.1 Increased Gamma Glutamyl Transferase 45 11 34 4.8 Increased ALT 29 6 27 2.2 Increased AST 27 2.6 24 1.6 Hyperglycemia 28 5 20 2.7 Increased Alkaline Phosphatase 21 0.5 35 2.2 Hyponatremia 18 3.6 15 0.5 Hypermagnesemia 10 1.0 26 0.5 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
The safety of BRAFTOVI 300 mg once daily in combination with cetuximab (400 mg/m2 initial dose, followed by 250 mg/m2 weekly) was evaluated in 216 patients with BRAF V600E mutation-positive metastatic CRC in a randomized, open-label, active-controlled trial (BEACON CRC). The BEACON CRC trial [see Clinical Studies (14.2)] excluded patients with a history of Gilbert's syndrome, abnormal left ventricular ejection fraction, prolonged QTc (> 480 ms), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of exposure was 4.4 months for patients treated with BRAFTOVI in combination with cetuximab and 1.6 months for patients treated with either irinotecan or infusional 5-fluorouracil (5-FU)/folinic acid (FA)/irinotecan (FOLFIRI) in combination with cetuximab.
The most common (≥ 25%) adverse reactions in patients receiving BRAFTOVI in combination with cetuximab were fatigue, nausea, diarrhea, dermatitis acneiform, abdominal pain, decreased appetite, arthralgia, and rash.
Adverse reactions leading to dose interruptions of BRAFTOVI occurred in 33% of patients receiving BRAFTOVI in combination with cetuximab; the most common were vomiting (4%), fatigue (4%), nausea (4%), pyrexia (3%), and diarrhea (3%). Adverse reactions leading to dose reductions of BRAFTOVI occurred in 9% of patients receiving BRAFTOVI in combination with cetuximab; the most common were fatigue (2%), arthralgia (2%), and peripheral neuropathy (2%). Ten percent (10%) of patients receiving BRAFTOVI in combination with cetuximab experienced an adverse reaction that resulted in permanent discontinuation of BRAFTOVI. None of the adverse reactions leading to permanent discontinuation of BRAFTOVI occurred in more than one patient (>0.5%).
Table 7 and Table 8 present adverse drug reactions and laboratory abnormalities, respectively, identified in BEACON CRC.
Table 7: Adverse Reactions Occurring in ≥ 10% of Patients Receiving BRAFTOVI in Combination with Cetuximab in BEACON CRC* Adverse Reaction BRAFTOVI with cetuximab
N=216Irinotecan with cetuximab or FOLFIRI with cetuximab
N=193All Grades
(%)≥ Grade 3†
(%)All Grades
(%)≥ Grade 3
(%)- * Grades per National Cancer Institute CTCAE v4.03.
- † Grade 4–5 adverse reactions in the BRAFTOVI with cetuximab arm were limited to Grade 5 hemorrhage (n=1).
- ‡ Represents a composite of multiple, related preferred terms.
General Disorders and Administration Site Conditions Fatigue‡ 51 7 50 8 Pyrexia‡ 17 1 15 1 Gastrointestinal Disorders Nausea 34 1 41 1 Diarrhea‡ 33 2 48 10 Abdominal pain‡ 30 4 32 5 Vomiting 21 1 29 3 Constipation 15 0 18 1 Metabolism and Nutrition Disorders Decreased appetite 27 1 27 3 Musculoskeletal and Connective Tissue Disorders Arthralgia‡ 27 1 3 0 Myopathy‡ 15 1 4 0 Pain in extremity 10 0 1 0 Skin and Subcutaneous Tissue Disorders Dermatitis acneiform‡ 32 1 43 3 Rash‡ 26 0 26 2 Pruritus‡ 14 0 6 0 Melanocytic nevus 14 0 0 0 Dry skin‡ 13 0 12 1 Nervous System Disorders Headache‡ 20 0 3 0 Peripheral neuropathy‡ 12 1 6 0 Vascular Disorders Hemorrhage‡ 19 2 9 0 Psychiatric Disorders Insomnia‡ 13 0 6 0 Other clinically important adverse reactions occurring in <10% of patients who received BRAFTOVI in combination with cetuximab were:
Gastrointestinal disorders: Pancreatitis
Table 8: Laboratory Abnormalities Occurring in ≥10% (All Grades) of Patients Receiving BRAFTOVI in Combination with Cetuximab in BEACON CRC* Laboratory Abnormality† BRAFTOVI with cetuximab Irinotecan with cetuximab or FOLFIRI with cetuximab All Grades
(%)Grades 3 and 4
(%)All Grades
(%)Grades 3 and 4
(%)- * Grades per National Cancer Institute CTCAE v4.03.
- † Based on the number of patients with available baseline and at least one on-treatment laboratory test.
Hematology Anemia 34 4 48 5 Lymphopenia 24 7 35 5 Increased Activated Partial Thromboplastin Time 13 1 7 1 Chemistry Hypomagnesemia 19 0 22 1 Increased Alkaline Phosphatase 18 4 30 7 Increased ALT 17 0 29 3 Increased AST 15 1 22 2 Hypokalemia 12 3 32 5 Hyponatremia 11 2 13 2 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on BRAFTOVI
Strong or Moderate CYP3A4 Inhibitors
Coadministration of BRAFTOVI with a strong or moderate CYP3A4 inhibitor increases encorafenib plasma concentrations [see Clinical Pharmacology (12.3)] and may increase encorafenib adverse reactions. Avoid coadministration of BRAFTOVI with strong or moderate CYP3A4 inhibitors, including grapefruit juice. If coadministration is unavoidable, reduce the BRAFTOVI dose [see Dosage and Administration (2.6)].
Strong or Moderate CYP3A4 Inducers
Coadministration of BRAFTOVI with a strong or moderate CYP3A4 inducer may decrease encorafenib plasma concentrations [see Clinical Pharmacology (12.3)] and may decrease encorafenib efficacy. Avoid coadministration of BRAFTOVI with strong or moderate CYP3A4 inducers.
7.2 Effect of BRAFTOVI on Other Drugs
Sensitive CYP3A4 Substrates
Coadministration of BRAFTOVI with sensitive CYP3A4 substrates may increase adverse reactions or decrease efficacy of these agents.
Coadministration of BRAFTOVI with hormonal contraceptives (CYP3A4 substrates) can result in decreased concentrations and loss of hormonal contraceptive efficacy. Avoid coadministration of BRAFTOVI with hormonal contraceptives [see Use in Specific Populations (8.3)].
7.3 Drugs That Prolong the QT Interval
BRAFTOVI is associated with dose-dependent QTc interval prolongation [see Warnings and Precautions (5.5), Clinical Pharmacology (12.2)]. Avoid coadministration of BRAFTOVI with drugs known to prolong the QT/QTc interval.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, BRAFTOVI can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available clinical data on the use of BRAFTOVI during pregnancy. In animal reproduction studies, encorafenib produced embryo-fetal developmental changes in rats and rabbits and was an abortifacient in rabbits at doses greater than or equal to those resulting in exposures approximately 26 (in the rat) and 178 (in the rabbit) times the human exposure at the clinical dose of 450 mg, with no clear findings at lower doses (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In reproductive toxicity studies, administration of encorafenib to rats during the period of organogenesis resulted in maternal toxicity, decreased fetal weights, and increased incidence of total skeletal variations at a dose of 20 mg/kg/day (approximately 26 times the human exposure based on area under the concentration-time curve [AUC] at the recommended clinical dose of 450 mg once daily). In pregnant rabbits, administration of encorafenib during the period of organogenesis resulted in maternal toxicity, decreased fetal body weights, increased incidence of total skeletal variations and increased post-implantation loss, including total loss of pregnancy at a dose of 75 mg/kg/day (approximately 178 times the human exposure based on AUC at the recommended clinical dose of 450 mg once daily). While formal placental transfer studies have not been performed, encorafenib exposure in the fetal plasma of both rats and rabbits was up to 1.7% and 0.8%, respectively, of maternal exposure.
8.2 Lactation
Risk Summary
There are no data on the presence of encorafenib or its metabolites in human milk or the effects of encorafenib on the breastfed infant, or on milk production. Because of the potential for serious adverse reactions from BRAFTOVI in breastfed infants, advise women not to breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating BRAFTOVI [see Use in Specific Populations (8.1)].
Contraception
BRAFTOVI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise females of reproductive potential to use effective contraception during treatment with BRAFTOVI and for 2 weeks after the final dose. Counsel patients to use a non-hormonal method of contraception since BRAFTOVI has the potential to render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Infertility
Males
Based on findings in male rats at doses approximately 13 times the human exposure at the 450 mg clinical dose, use of BRAFTOVI may impact fertility in males [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of BRAFTOVI have not been established in pediatric patients.
8.5 Geriatric Use
Of the 690 patients with BRAF mutation-positive melanoma who received BRAFTOVI at doses between 300 mg and 600 mg once daily in combination with binimetinib (45 mg twice daily) across multiple clinical trials, 20% were aged 65 to 74 years and 8% were aged 75 years and older [see Clinical Studies (14.1)].
Of the 216 patients with BRAF V600E mutation positive metastatic CRC who received BRAFTOVI 300 mg QD in combination with cetuximab, 62 (29%) were 65 years of age to up to 75 years of age, while 20 (9%) were 75 years of age and over [see Clinical Studies (14.2)].
No overall differences in the safety or effectiveness of BRAFTOVI plus binimetinib or BRAFTOVI plus cetuximab were observed in elderly patients as compared to younger patients.
8.6 Hepatic Impairment
No BRAFTOVI dosage adjustment is recommended in patients with mild hepatic impairment (Child-Pugh Class A) [see Clinical Pharmacology (12.3)]. A recommended dosage has not been established in patients with moderate (Child-Pugh Class B) or severe (Child-Pugh Class C) hepatic impairment.
8.7 Renal Impairment
No BRAFTOVI dosage adjustment is recommended in patients with mild to moderate renal impairment (CLcr 30 to < 90 mL/min) [see Clinical Pharmacology (12.3)]. A recommended dosage has not been established in patients with severe renal impairment (CLcr <30 mL/min).
- 10 OVERDOSAGE
-
11 DESCRIPTION
Encorafenib is a kinase inhibitor. The chemical name is methyl N-{(2S)-1-[(4-{3-[5-chloro-2-fluoro-3-(methanesulfonamido)phenyl]-1-(propan-2-yl)-1H-pyrazol-4-yl}pyrimidin-2-yl)amino]propan-2-yl}carbamate. The molecular formula is C22H27ClFN7O4S and the molecular weight is 540 daltons. The chemical structure of encorafenib is shown below:
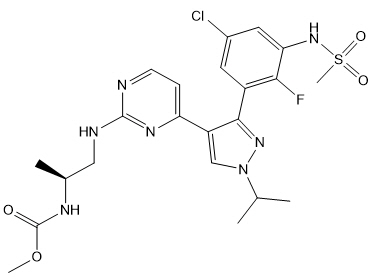
Encorafenib is a white to almost white powder. In aqueous media, encorafenib is slightly soluble at pH 1, very slightly soluble at pH 2, and insoluble at pH 3 and higher.
BRAFTOVI (encorafenib) capsules for oral use contain 75 mg of encorafenib with the following inactive ingredients: copovidone, poloxamer 188, microcrystalline cellulose, succinic acid, crospovidone, colloidal silicon dioxide, magnesium stearate (vegetable origin). The capsule shell contains gelatin, titanium dioxide, iron oxide red, iron oxide yellow, ferrosoferric oxide, monogramming ink (pharmaceutical glaze, ferrosoferric oxide, propylene glycol).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Encorafenib is a kinase inhibitor that targets BRAF V600E, as well as wild-type BRAF and CRAF in in vitro cell-free assays with IC50 values of 0.35, 0.47, and 0.3 nM, respectively. Mutations in the BRAF gene, such as BRAF V600E, can result in constitutively activated BRAF kinases that may stimulate tumor cell growth. Encorafenib was also able to bind to other kinases in vitro including JNK1, JNK2, JNK3, LIMK1, LIMK2, MEK4, and STK36 and reduce ligand binding to these kinases at clinically achievable concentrations (≤0.9 µM).
Encorafenib inhibited in vitro growth of tumor cell lines expressing BRAF V600 E, D, and K mutations. In mice implanted with tumor cells expressing BRAF V600E, encorafenib induced tumor regressions associated with RAF/MEK/ERK pathway suppression.
Encorafenib and binimetinib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared with either drug alone, coadministration of encorafenib and binimetinib resulted in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. Additionally, the combination of encorafenib and binimetinib delayed the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared to either drug alone.
In the setting of BRAF-mutant CRC, induction of EGFR-mediated MAPK pathway activation has been identified as a mechanism of resistance to BRAF inhibitors. Combinations of a BRAF inhibitor and agents targeting EGFR have been shown to overcome this resistance mechanism in nonclinical models. Coadministration of encorafenib and cetuximab had an anti-tumor effect greater than either drug alone, in a mouse model of colorectal cancer with mutated BRAF V600E.
12.2 Pharmacodynamics
Cardiac Electrophysiology
A dedicated study to evaluate the QT prolongation potential of BRAFTOVI has not been conducted. BRAFTOVI is associated with dose-dependent QTc interval prolongation. Based on a central tendency analysis of QTc in a study of adult patients with melanoma who received the recommended dose of BRAFTOVI in combination with binimetinib, the largest mean (90% CI) QTcF change from baseline (ΔQTcF) was 18 (14 to 22) ms [see Warnings and Precautions (5.5)].
12.3 Pharmacokinetics
The pharmacokinetics of encorafenib were studied in healthy subjects and patients with solid tumors, including advanced and unresectable or metastatic cutaneous melanoma harboring a BRAF V600E or V600K mutation and BRAF V600E mutation-positive metastatic CRC. After a single dose, systemic exposure of encorafenib was dose proportional over the dose range of 50 mg to 700 mg (0.1 to 1.6 times the maximum recommended dose of 450 mg). After once-daily dosing, systemic exposure of encorafenib was less than dose proportional over the dose range of 50 mg to 800 mg (0.1 to 1.8 times the maximum recommended dose of 450 mg). Steady-state was reached within 15 days, with exposure being 50% lower compared to Day 1; intersubject variability (CV%) of AUC ranged from 12% to 69%.
Absorption
The median Tmax of encorafenib is 2 hours. At least 86% of the dose is absorbed.
Effect of Food
Following administration of a single dose of BRAFTOVI 100 mg (0.2 times the maximum recommended dose of 450 mg) with a high-fat, high-calorie meal (consisting of approximately 150 calories from protein, 350 calories from carbohydrates, and 500 calories from fat) the mean maximum encorafenib concentration (Cmax) decreased by 36% and there was no effect on AUC.
Distribution
The geometric mean (CV%) of apparent volume of distribution is 164 L (70%). The protein binding of encorafenib is 86% in vitro. The blood-to-plasma concentration ratio is 0.58.
Elimination
The mean (CV%) terminal half-life (t1/2) of encorafenib is 3.5 hours (17%), and the apparent clearance is 14 L/h (54%) at day 1, increasing to 32 L/h (59%) at steady-state at the maximum recommended dose of 450 mg.
Specific Populations
No clinically significant differences in the pharmacokinetics of encorafenib were observed based on age (19 to 94 years), sex, body weight (34 to 168 kg), mild hepatic impairment (Child-Pugh Class A), and mild or moderate renal impairment (CLcr 30 to < 90 mL/min). The effect of race or ethnicity, moderate or severe hepatic impairment (Child-Pugh Class B or C), and severe renal impairment (CLcr <30 mL/min) on encorafenib pharmacokinetics have not been studied.
Drug Interaction Studies
Clinical Studies
CYP3A4 Inhibitors: Coadministration of posaconazole (strong CYP3A4 inhibitor) or diltiazem (moderate CYP3A4 inhibitor) increased AUC of encorafenib by 3- and 2-fold, respectively, and increased Cmax by 68% and 45%, respectively, after a single dose of 50 mg BRAFTOVI (0.1 times the maximum recommended dose of 450 mg).
CYP3A4 Inducers: The effect of a CYP3A4 inducer on encorafenib exposure has not been studied. However, encorafenib (CYP3A4 inducer in vitro) exposures were lower at steady-state compared to the first dose in clinical studies, suggesting CYP3A4 auto-induction.
Proton Pump Inhibitors: No clinically significant differences in encorafenib pharmacokinetics were observed when coadministered with rabeprazole.
In Vitro Studies
CYP/UGT Enzymes: Encorafenib is a reversible inhibitor of UGT1A1, CYP1A2, CYP2B6, CYP2C8/9, CYP2D6, and CYP3A, and a time-dependent inhibitor of CYP3A4 at clinically relevant plasma concentrations. Encorafenib is an inducer of CYP2B6, CYP2C9, and CYP3A4 at clinically relevant plasma concentrations.
Transporters Systems: Encorafenib is a substrate of P-glycoprotein (P-gp) but not of breast cancer resistance protein (BCRP), multidrug resistance-associated protein 2 (MRP2), organic anion transporting polypeptide (OATP1B1, OATP1B3) or organic cation transporter (OCT1) at clinically relevant plasma concentrations.
Encorafenib is an inhibitor of P-gp, BCRP, OCT2, organic anion transporter (OAT1, OAT3), OATP1B1, and OATP1B3, but not of OCT1 or MRP2 at clinically relevant plasma concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with encorafenib have not been conducted. Encorafenib was not genotoxic in studies evaluating reverse mutations in bacteria, chromosomal aberrations in mammalian cells, or micronuclei in bone marrow of rats.
No dedicated fertility studies were performed with encorafenib in animals. In a general toxicology study in rats, decreased testes and epididymis weights, tubular degeneration in testes, and oligospermia in epididymides were observed at doses approximately 13 times the human exposure at the 450 mg clinical dose based on AUC. No effects on reproductive organs were observed in either sex in any of the non-human primate toxicity studies.
13.2 Animal Toxicology and/or Pharmacology
Adverse histopathology findings of hyperplasia and hyperkeratosis occurred in the stomach of rats at encorafenib doses of 20 mg/kg/day (approximately 14 times the human exposure at the 450 mg clinical dose based on AUC) or greater, in both 4 and 13-week studies.
-
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
BRAFTOVI in combination with binimetinib was evaluated in a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID™BRAF assay. Patients were permitted to have received immunotherapy in the adjuvant setting and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior use of BRAF inhibitors or MEK inhibitors was prohibited. Randomization was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), and prior immunotherapy for unresectable or metastatic disease (yes versus no).
Patients were randomized (1:1:1) to receive BRAFTOVI 450 mg once daily in combination with binimetinib 45 mg twice daily (BRAFTOVI in combination with binimetinib), BRAFTOVI 300 mg once daily, or vemurafenib 960 mg twice daily. Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved dosing (BRAFTOVI 450 mg in combination with binimetinib 45 mg) are described below.
The major efficacy outcome measure was progression-free survival (PFS), as assessed by a blinded independent central review, to compare BRAFTOVI in combination with binimetinib with vemurafenib. Additional efficacy outcome measures included overall survival (OS), as well as objective response rate (ORR) and duration of response (DoR) which were assessed by central review.
A total of 577 patients were randomized, 192 to the BRAFTOVI in combination with binimetinib arm, 194 to the BRAFTOVI arm, and 191 to the vemurafenib arm. Of the 383 patients randomized to either the BRAFTOVI in combination with binimetinib or the vemurafenib arms, the median age was 56 years (20 to 89 years), 59% were male, 91% were White, and 72% had baseline ECOG performance status of 0. Ninety-five percent (95%) had metastatic disease, 65% were Stage IVM1c, and 4% received prior CTLA-4, PD-1, or PD-L1 directed antibodies. Twenty-eight percent (28%) had elevated baseline serum lactate dehydrogenase (LDH), 45% had ≥ 3 organs with tumor involvement at baseline, and 3% had brain metastases. Based on centralized testing, 100% of patients' tumors tested positive for BRAF mutations; BRAF V600E (88%), BRAF V600K (11%), or both (<1%).
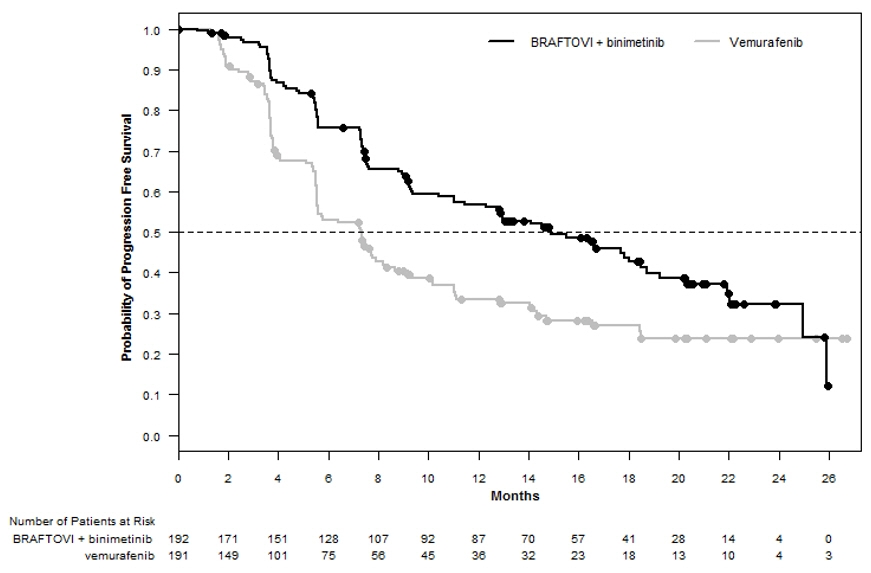
BRAFTOVI in combination with binimetinib demonstrated a statistically significant improvement in PFS compared to vemurafenib. Efficacy results are summarized in Table 9 and Figure 1.
Table 9: Efficacy Results for COLUMBUS BRAFTOVI with binimetinib
N=192Vemurafenib
N=191CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NE = Not estimable; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. - * Estimated with Cox proportional hazard model adjusted by the following stratification factors: American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c) and Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1).
- † Log-rank test adjusted by the same stratification factors.
- ‡ Based on a cutoff date of 17.6 months after the date of the PFS analysis.
Progression-Free Survival Number of events (%) 98 (51) 106 (55) Progressive disease 88 (46) 104 (54) Death 10 (5) 2 (1) Median PFS, months (95% CI) 14.9 (11, 18.5) 7.3 (5.6, 8.2) HR (95% CI)* 0.54 (0.41, 0.71) P-value† <0.0001 Overall Survival‡ Number of events (%) 105 (55) 127 (67) Median OS, months (95% CI) 33.6 (24.4, 39.2) 16.9 (14.0, 24.5) HR (95% CI)* 0.61 (0.47, 0.79) Overall Response Rate ORR (95% CI) 63% (56%, 70%) 40% (33%, 48%) CR 8% 6% PR 55% 35% Duration of Response Median DoR, months (95% CI) 16.6 (12.2, 20.4) 12.3 (6.9, 16.9) Figure 1: Kaplan-Meier Curves for Progression-Free Survival in COLUMBUS
14.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
BRAFTOVI in combination with cetuximab was evaluated in a randomized, active-controlled, open-label, multicenter trial (BEACON CRC; NCT02928224). Eligible patients were required to have BRAF V600E mutation-positive metastatic colorectal cancer (CRC), as detected using the Qiagen therascreen BRAF V600E RGQ polymerase chain reaction (PCR) Kit, with disease progression after 1 or 2 prior regimens. Other key eligibility criteria included absence of prior treatment with a RAF, MEK, or EGFR inhibitor, eligibility to receive cetuximab per local labeling with respect to tumor RAS status, and ECOG performance status (PS) 0–1. Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), prior use of irinotecan (yes versus no), and cetuximab product used (US-licensed versus EU-approved).
Patients were randomized 1:1:1 to one of the following treatment arms:
- BRAFTOVI 300 mg orally once daily in combination with cetuximab (BRAFTOVI/cetuximab arm)
- BRAFTOVI 300 mg orally once daily in combination with binimetinib and cetuximab
- Irinotecan with cetuximab or FOLFIRI with cetuximab (control arm)
The dosage of cetuximab in all patients was 400 mg/m2 intravenously for the first dose followed by 250 mg/m2 weekly.
Patients in the control arm received cetuximab with either irinotecan 180 mg/m2 intravenously on Days 1 and 15 of each 28-day cycle or FOLFIRI intravenously (irinotecan 180 mg/m2 on Days 1 and 15; folinic acid 400 mg/m2 on Days 1 and 15; then fluorouracil 400 mg/m2 bolus on Days 1 and 15 followed by fluorouracil 2400 mg/m2/day by continuous infusion over 2 days).
Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved regimen (BRAFTOVI in combination with cetuximab) are described below.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures included progression-free survival (PFS), overall response rate (ORR), and duration of response (DoR) as assessed by blinded independent central review (BICR). OS and PFS were assessed in all randomized patients. ORR and DoR were assessed in the subset of the first 220 patients included in the randomized portion of the BRAFTOVI/cetuximab and control arm of the study.
A total of 220 patients were randomized to the BRAFTOVI/cetuximab arm and 221 to the control arm. Of these 441 patients, the median age was 61 years; 53% were female; 80% were White and 15% were Asian. Fifty percent (50%) had baseline ECOG performance status of 0; 66% received 1 prior therapy and 34% received 2; 93% received prior oxaliplatin and 52% received prior irinotecan.
BRAFTOVI in combination with cetuximab demonstrated a statistically significant improvement in OS, ORR, and PFS compared to the active comparator. Efficacy results are summarized in Table 10 and Figure 2.
Table 10: Efficacy Results from BEACON CRC BRAFTOVI with cetuximab
N = 220Irinotecan with cetuximab or FOLFIRI with cetuximab
N = 221CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NR = Not reached; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. - * Stratified by ECOG PS, source of cetuximab (US-licensed versus EU-approved) and prior irinotecan use at randomization.
- † Stratified Cox proportional hazard model.
- ‡ Stratified log-rank test, tested at alpha level of 0.0084.
- § BRAFTOVI/cetuximab arm (n=113) and control arm (n=107).
- ¶ Cochran-Mantel-Haenszel test; tested at alpha level of 0.05.
- # Stratified log-rank test, tested at alpha level of 0.0234.
Overall Survival Number of Events (%) 93 (42) 114 (52) Median OS, months (95% CI) 8.4 (7.5, 11.0) 5.4 (4.8, 6.6) HR (95% CI)*,† 0.60 (0.45, 0.79) P-value*,‡ 0.0003 Overall Response Rate (per BICR) ORR (95% CI)§ 20% (13%, 29%) 2% (0%, 7%) CR 5% 0% PR 15% 2% P-value*,¶ <0.0001 Median DoR, months (95% CI) 6.1 (4.1, 8.3) NR (2.6, NR) Progression Free Survival (per BICR) Number of events (%) 133 (60) 128 (58) Progressive disease 110 (50) 101 (46) Death 23 (10) 27 (12) Median PFS, months (95% CI) 4.2 (3.7, 5.4) 1.5 (1.4, 1.7) HR (95% CI)*,† 0.40 (0.31, 0.52) P-value*,# < 0.0001 Figure 2: Kaplan-Meier Curves for Overall Survival in BEACON CRC
-
16 HOW SUPPLIED/STORAGE AND HANDLING
BRAFTOVI (encorafenib) is supplied as 75 mg hard gelatin capsules.
75 mg: stylized "A" on beige cap and "LGX 75mg" on white body, available in cartons (NDC: 70255-025-01) containing two bottles of 90 capsules each (NDC: 70255-025-02) and cartons (NDC: 70255-025-03) containing two bottles of 60 capsules each (NDC: 70255-025-04).
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Do not use if safety seal under cap is broken or missing. Dispense in original bottle. Do not remove desiccant. Protect from moisture. Keep container tightly closed.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the following:
New Primary Cutaneous Malignancies
Advise patients to contact their healthcare provider immediately for change in or development of new skin lesions [see Warnings and Precautions (5.1)].
Hemorrhage
Advise patients to notify their healthcare provider immediately with any symptoms suggestive of hemorrhage, such as unusual bleeding [see Warnings and Precautions (5.3)].
Uveitis
Advise patients to contact their healthcare provider if they experience any changes in their vision [see Warnings and Precautions (5.4)].
QT Prolongation
Advise patients that BRAFTOVI can cause QTc interval prolongation and to inform their physician if they have any QTc interval prolongation symptoms, such as syncope [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
- Advise females with reproductive potential of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with BRAFTOVI [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective non-hormonal contraception during treatment with BRAFTOVI and for 2 weeks after the final dose [Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential that BRAFTOVI may impair fertility [see Use in Specific Populations (8.3)].
Strong or Moderate CYP3A Inducers or Inhibitors
Coadministration of BRAFTOVI with a strong or moderate CYP3A inhibitor may increase encorafenib concentrations; while coadministration of BRAFTOVI with a strong or moderate CYP3A inducer may decrease encorafenib concentrations. Advise patients that they need to avoid certain medications while taking BRAFTOVI and to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Advise patients to avoid grapefruit or grapefruit juice while taking BRAFTOVI [see Drug Interactions (7.1)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 04/2020 MEDICATION GUIDE
BRAFTOVI® (braf-TOE-vee)
(encorafenib)
capsulesImportant information: BRAFTOVI is used with other medicines, either binimetinib or cetuximab. Read the Patient Information leaflet that comes with binimetinib if used with binimetinib, and talk to your healthcare provider about cetuximab if used with cetuximab. What is the most important information I should know about BRAFTOVI?
BRAFTOVI may cause serious side effects, including:-
Risk of new skin cancers. BRAFTOVI when used alone, or with binimetinib or cetuximab, may cause skin cancers called cutaneous squamous cell carcinoma or basal cell carcinoma.
Talk to your healthcare provider about your risk for these cancers.
Check your skin and tell your healthcare provider right away about any skin changes, including a:- new wart
- skin sore or reddish bump that bleeds or does not heal
- change in size or color of a mole
Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that develop during treatment with BRAFTOVI.
See "What are the possible side effects of BRAFTOVI?" for more information about side effects.
What is BRAFTOVI?
BRAFTOVI is a prescription medicine used:- in combination with a medicine called binimetinib to treat people with a type of skin cancer called melanoma:
- that has spread to other parts of the body or cannot be removed by surgery, and
- that has a certain type of abnormal "BRAF" gene
- in combination with a medicine called cetuximab, for the treatment of adults with cancer of your colon or rectum (colorectal cancer):
- that has been previously treated, and
- that has spread to other parts of the body, and
- that has a certain type of abnormal "BRAF" gene
It is not known if BRAFTOVI is safe and effective in children.Before taking BRAFTOVI, tell your healthcare provider about all of your medical conditions, including if you: - have had bleeding problems
- have eye problems
- have heart problems, including a condition called long QT syndrome
- have been told that you have low blood levels of potassium, calcium, or magnesium
- have liver or kidney problems
- are pregnant or plan to become pregnant. BRAFTOVI can harm your unborn baby.
- Females who are able to become pregnant should use effective non-hormonal birth control (contraception) during treatment with BRAFTOVI and for 2 weeks after the final dose of BRAFTOVI. Birth control methods that contain hormones (such as birth control pills, injections or transdermal systems) may not work as well during treatment with BRAFTOVI.
- Talk to your healthcare provider about birth control methods that may be right for you during this time.
- Your healthcare provider will do a pregnancy test before you start taking BRAFTOVI. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with BRAFTOVI.
- are breastfeeding or plan to breastfeed. It is not known if BRAFTOVI passes into your breast milk. Do not breastfeed during treatment with BRAFTOVI and for 2 weeks after the final dose of BRAFTOVI. Talk to your healthcare provider about the best way to feed your baby during this time.
BRAFTOVI and certain other medicines can affect each other, causing side effects or affecting how BRAFTOVI or the other medicines work.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take BRAFTOVI? - Take BRAFTOVI exactly as your healthcare provider tells you. Do not change your dose or stop taking BRAFTOVI unless your healthcare provider tells you to.
- Your healthcare provider may change your dose of BRAFTOVI, temporarily stop, or completely stop your treatment with BRAFTOVI if you develop certain side effects.
- For melanoma, take BRAFTOVI in combination with binimetinib by mouth one time each day.
- For colorectal cancer, take BRAFTOVI by mouth one time each day. You will also receive cetuximab through a vein in your arm (intravenously) given by your healthcare provider.
- BRAFTOVI may be taken with or without food.
- Avoid grapefruit during treatment with BRAFTOVI. Grapefruit products may increase the amount of BRAFTOVI in your body.
- If you miss a dose of BRAFTOVI, take it as soon as you remember. If it is within 12 hours of your next scheduled dose, take your next dose at your regular time. Do not make up for the missed dose.
- Do not take an extra dose if you vomit after taking your scheduled dose. Take your next dose at your regular time.
- If you stop treatment with binimetinib or cetuximab, talk to your healthcare provider about your BRAFTOVI treatment. Your BRAFTOVI dose may need to be changed or stopped.
What are the possible side effects of BRAFTOVI?
BRAFTOVI may cause serious side effects, including:
See "What is the most important information I should know about BRAFTOVI?"-
Bleeding problems. BRAFTOVI, when taken with binimetinib or cetuximab, can cause serious bleeding problems, including in your stomach or brain, that can lead to death. Call your healthcare provider and get medical help right away if you have any signs of bleeding, including:
- headaches, dizziness, or feeling weak
- cough up blood or blood clots
- vomit blood or your vomit looks like "coffee grounds"
- red or black stools that look like tar
-
Eye problems. Tell your healthcare provider right away if you develop any of these symptoms of eye problems:
- blurred vision, loss of vision, or other vision changes
- see colored dots
- see halos (blurred outline around objects)
- eye pain, swelling, or redness
- Changes in the electrical activity of your heart called QT prolongation. QT prolongation can cause irregular heartbeats that can be life threatening. Your healthcare provider should do tests before you start taking BRAFTOVI with binimetinib or cetuximab and during your treatment to check your body salts (electrolytes). Tell your healthcare provider right away if you feel faint, lightheaded, dizzy or if you feel your heart beating irregularly or fast while taking BRAFTOVI with binimetinib or cetuximab. These symptoms may be related to QT prolongation.
- fatigue
- nausea
- vomiting
- abdominal pain
- pain or swelling of your joints (arthralgia)
- fatigue
- nausea
- diarrhea
- acne-like rash (dermatitis acneiform)
- abdominal pain
- decreased appetite
- pain or swelling of your joints (arthralgia)
- rash
These are not all the possible side effects of BRAFTOVI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Array BioPharma Inc. at 1-844-792-7729.How should I store BRAFTOVI? - Store BRAFTOVI at room temperature between 68°F to 77°F (20°C to 25°C).
- Store BRAFTOVI in the original bottle.
- Keep the BRAFTOVI bottle tightly closed and protect it from moisture.
- BRAFTOVI comes with a desiccant packet in the bottle to help protect your medicine from moisture. Do not remove the desiccant packet from the bottle.
General information about the safe and effective use of BRAFTOVI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BRAFTOVI for a condition for which it was not prescribed. Do not give BRAFTOVI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about BRAFTOVI that is written for health professionals.What are the ingredients in BRAFTOVI?
Active ingredient: encorafenib
Inactive ingredients: copovidone, poloxamer 188, microcrystalline cellulose, succinic acid, crospovidone, colloidal silicon dioxide, and magnesium stearate of vegetable origin
Capsule shell: gelatin, titanium dioxide, iron oxide red, iron oxide yellow, ferrosoferric oxide, monogramming ink (pharmaceutical glaze, ferrosoferric oxide, propylene glycol)
Distributed by: Array BioPharma Inc., a wholly owned subsidiary of Pfizer Inc. Boulder, Colorado 80301.
BRAFTOVI® is a registered trademark of Array BioPharma Inc. in the United States and various other countries.
For more information, go to www.BRAFTOVIMEKTOVI.com or call 1-844-792-7729.
© 2020 Array BioPharma Inc. All rights reserved. -
Risk of new skin cancers. BRAFTOVI when used alone, or with binimetinib or cetuximab, may cause skin cancers called cutaneous squamous cell carcinoma or basal cell carcinoma.
- PRINCIPAL DISPLAY PANEL - 75 mg Capsule Bottle Carton - 025-01
-
INGREDIENTS AND APPEARANCE
BRAFTOVI
encorafenib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70255-025 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ENCORAFENIB (UNII: 8L7891MRB6) (ENCORAFENIB - UNII:8L7891MRB6) ENCORAFENIB 75 mg Inactive Ingredients Ingredient Name Strength COPOVIDONE K25-31 (UNII: D9C330MD8B) POLOXAMER 188 (UNII: LQA7B6G8JG) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SUCCINIC ACID (UNII: AB6MNQ6J6L) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) SHELLAC (UNII: 46N107B71O) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) Product Characteristics Color PINK (beige) , WHITE Score no score Shape CAPSULE Size 23mm Flavor Imprint Code A;LGX;75mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70255-025-01 2 in 1 CARTON 06/27/2018 1 NDC: 70255-025-02 90 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 70255-025-03 2 in 1 CARTON 06/27/2018 2 NDC: 70255-025-04 60 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210496 06/27/2018 Labeler - Array BioPharma Inc. (004047838)
Trademark Results [BRAFTOVI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 BRAFTOVI 90021001 not registered Live/Pending |
Array BioPharma Inc. 2020-06-25 |
 BRAFTOVI 87077386 5561579 Live/Registered |
Array Biopharma Inc. 2016-06-20 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.