ZEVALIN- ibritumomab tiuxetan kit
Zevalin by
Drug Labeling and Warnings
Zevalin by is a Prescription medication manufactured, distributed, or labeled by Acrotech Biopharma Inc, Ajinomoto Althea, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ZEVALIN safely and effectively. See full prescribing information for ZEVALIN.
ZEVALIN® (ibritumomab tiuxetan) injection, for intravenous use
Initial U.S. Approval: 2002WARNING: SERIOUS INFUSION REACTIONS, PROLONGED AND SEVERE CYTOPENIAS, and SEVERE CUTANEOUS AND MUCOCUTANEOUS REACTIONS
See full prescribing information for complete boxed warning
- Serious Infusion Reactions, some fatal, may occur within 24 hours of rituximab infusion. (5.1)
- Prolonged and Severe Cytopenias occur in most patients. (5.2)
- Severe Cutaneous and Mucocutaneous Reactions, some fatal, reported with Zevalin therapeutic regimen. (5.3, 6.2)
- Do not exceed 32 mCi (1184 MBq) of Y-90 Zevalin. (2.2)
INDICATIONS AND USAGE
Zevalin is a CD20-directed radiotherapeutic antibody administered as part of the Zevalin therapeutic regimen indicated for the treatment of adult patients with:
- relapsed or refractory, low-grade or follicular B-cell non-Hodgkin's lymphoma (NHL) (1.1).
- previously untreated follicular NHL who achieve a partial or complete response to first-line chemotherapy (1.2).
DOSAGE AND ADMINISTRATION
- Day 1: Administer rituximab 250 mg/m2 intravenous infusion. (2.2)
- Day 7, 8, or 9:
- Administer rituximab 250 mg/m2 intravenous infusion. (2.2)
- o If platelets at least 150,000/mm3: Within 4 hours after rituximab infusion, administer 0.4 mCi/kg (14.8 MBq per kg) Y-90 Zevalin intravenous infusion.
- o If platelets 100,000 to 149,000/mm3 in relapsed or refractory patients: Within 4 hours after rituximab infusion, administer 0.3 mCi/kg (11.1 MBq per kg) Y-90 Zevalin intravenous infusion.
DOSAGE FORMS AND STRENGTHS
- Injection: 3.2 mg per 2 mL in a single-dose vial. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Serious Infusion Reactions: Immediately discontinue rituximab and Y-90 Zevalin. (5.1, 6.1)
- Prolonged and Severe Cytopenias: Do not administer Zevalin to patients with ≥ 25% lymphoma marrow involvement or impaired bone marrow reserve. (5.2, 6.1)
- Severe Cutaneous and Mucocutaneous Reactions: Discontinue rituximab and Zevalin infusions if patients develop severe cutaneous or mucocutaneous reactions. (5.3, 6.2)
- Development of Leukemia and Myelodysplastic Syndrome: Monitor patients for hematological toxicity including secondary malignancies.(5.4, 6.1)
- Extravasation: Monitor for extravasation and terminate infusion if it occurs. Resume infusion in another limb. (5.5, 6.2)
- Immunization: Do not administer live viral vaccines to patients who recently received Zevalin. (5.6)
- Embryo-fetal Toxicity: May cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.8, 8.1, 8.3)
ADVERSE REACTIONS
Common adverse reactions (> 10%) in clinical trials were: cytopenias, fatigue, nasopharyngitis, nausea, abdominal pain, asthenia, cough, diarrhea, and pyrexia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Acrotech Biopharma LLC at 1-866-298-8433 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Monitor patients receiving medications that interfere with platelet function or coagulation more frequently for thrombocytopenia. (7)
USE IN SPECIFIC POPULATIONS
- Lactation: Advise women not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFUSION REACTIONS, PROLONGED AND SEVERE CYTOPENIAS, and SEVERE CUTANEOUS AND MUCOCUTANEOUS REACTIONS
1 INDICATIONS AND USAGE
1.1 Relapsed or Refractory, Low-grade or Follicular NHL
1.2 Previously Untreated Follicular NHL
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing Schedule
2.2 Zevalin Therapeutic Regimen Dosage and Administration
2.3 Directions for Preparation of Radiolabeled Y-90 Zevalin Doses
2.4 Procedure for Determining Radiochemical Purity
2.5 Radiation Dosimetry
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infusion Reactions
5.2 Prolonged and Severe Cytopenias
5.3 Severe Cutaneous and Mucocutaneous Reactions
5.4 Risk of Developing Myelodysplastic Syndrome, Leukemia, and Other Malignancies
5.5 Extravasation
5.6 Risks of Immunization
5.7 Radionuclide Precautions
5.8 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
6.3 Immunogenicity
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Relapsed or Refractory, Low-grade or Follicular Lymphoma
14.2 Follicular, B-Cell NHL Upon Completion of First-Line Chemotherapy
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFUSION REACTIONS, PROLONGED AND SEVERE CYTOPENIAS, and SEVERE CUTANEOUS AND MUCOCUTANEOUS REACTIONS
Serious Infusion Reactions: Deaths have occurred within 24 hours of rituximab infusion, an essential component of the Zevalin therapeutic regimen. These fatalities were associated with hypoxia, pulmonary infiltrates, acute respiratory distress syndrome, myocardial infarction, ventricular fibrillation, or cardiogenic shock. Most (80%) fatalities occurred with the first rituximab infusion [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)]. Discontinue rituximab and Y-90 Zevalin infusions in patients who develop severe infusion reactions.
Prolonged and Severe Cytopenias: Y-90 Zevalin administration results in severe and prolonged cytopenias in most patients. Do not administer Y-90 Zevalin to patients with ≥ 25% lymphoma marrow involvement and/or impaired bone marrow reserve [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Severe Cutaneous and Mucocutaneous Reactions: Severe cutaneous and mucocutaneous reactions, some fatal, can occur with the Zevalin therapeutic regimen. Discontinue rituximab and Y-90 Zevalin infusions in patients experiencing severe cutaneous or mucocutaneous reactions [see Warnings and Precautions (5.3) and Adverse Reactions (6.2)].
Dosing: The dose of Y-90 Zevalin should not exceed 32.0 mCi (1184 MBq) [see Dosage and Administration (2.2)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing Schedule
- Administer the Zevalin therapeutic regimen as outlined below.
- Initiate the Zevalin therapeutic regimen following recovery of platelet counts to 150,000/mm3 or more at least 6 weeks, but no more than 12 weeks, following the last dose of first-line chemotherapy.
- Only administer rituximab/Zevalin in facilities where immediate access to resuscitative measures is available.
Overview of Dosing Schedule
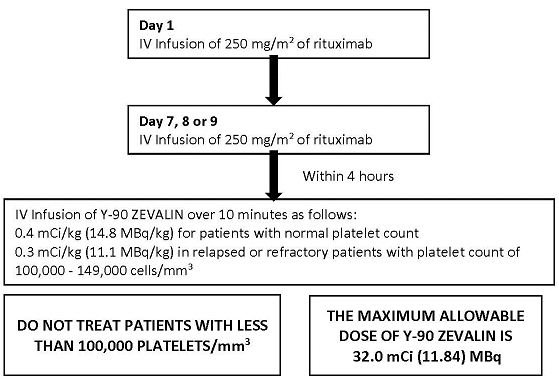
2.2 Zevalin Therapeutic Regimen Dosage and Administration
Day 1:
- Premedicate with acetaminophen 650 mg orally and diphenhydramine 50 mg orally prior to rituximab infusion.
- Administer rituximab 250 mg/m2 intravenously at an initial rate of 50 mg/hr. In the absence of infusion reactions, escalate the infusion rate in 50 mg/hr increments every 30 minutes to a maximum of 400 mg/hr. Do not mix or dilute rituximab with other drugs.
- Immediately stop the rituximab infusion for serious infusion reactions and discontinue the Zevalin therapeutic regimen [see Boxed Warning and Warnings and Precautions (5.1)].
- Temporarily slow or interrupt the rituximab infusion for less severe infusion reactions. If symptoms improve, continue the infusion at one-half the previous rate.
Day 7, 8 or 9:
- Premedicate with acetaminophen 650 mg orally and diphenhydramine 50 mg orally prior to rituximab infusion.
- Administer rituximab 250 mg/m2 intravenously at an initial rate of 100 mg/hr. Increase rate by 100 mg/hr increments at 30 minute intervals, to a maximum of 400 mg/hr, as tolerated. If infusion reactions occurred during rituximab infusion on Day 1 of treatment, administer rituximab at an initial rate of 50 mg/hr and escalate the infusion rate in 50 mg/hr increments every 30 minutes to a maximum of 400 mg/hr.
- Administer Y-90 Zevalin injection through a free flowing intravenous line within 4 hours following completion of rituximab infusion. Use a 0.22 micron low-protein-binding in-line filter between the syringe and the infusion port. After infusion, flush the line with at least 10 mL of normal saline.
- o If platelet count at least 150,000/mm3, administer Y-90 Zevalin over 10 minutes as an intravenous infusion at a dose of Y-90 0.4 mCi per kg (14.8 MBq per kg) actual body weight.
- o If platelet count 100,000 to 149,000/mm3, in relapsed or refractory patients, administer Y-90 Zevalin over 10 minutes as an intravenous infusion at a dose of Y-90 0.3 mCi per kg (11.1 MBq per kg) actual body weight.
- o Do not administer more than 32 mCi (1184 MBq) Y-90 Zevalin dose regardless of the patient’s body weight.
- Monitor patients closely for evidence of extravasation during the infusion of Y-90 Zevalin. Immediately stop infusion and restart in another limb if any signs or symptoms of extravasation occur [see Warnings and Precautions (5.5)].
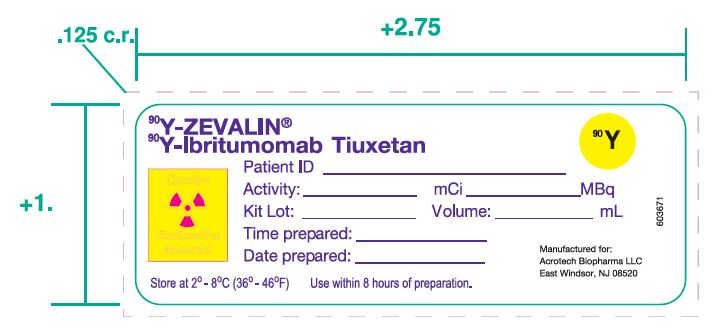
2.3 Directions for Preparation of Radiolabeled Y-90 Zevalin Doses
A clearly-labeled kit is required for preparation of Yttrium-90 (Y-90) Zevalin. Follow the detailed instructions for the preparation of radiolabeled Zevalin [see Dosage and Administration (2.4)].
Required materials not supplied in the kit:
- 1. Yttrium-90 Chloride Sterile Solution
- 2. Three sterile 1 mL plastic syringes
- 3. One sterile 3 mL plastic syringe
- 4. Two sterile 10 mL plastic syringes with 18-20 G needles
- 5. ITLC silica gel strips
- 6. 0.9% Sodium Chloride aqueous solution for the chromatography solvent
- 7. Developing chamber for chromatography
- 8. Suitable radioactivity counting apparatus
- 9. Filter, 0.22 micrometer, low-protein-binding
- 10. Appropriate acrylic shielding for reaction vial and syringe for Y-90
Method:
- 1. Allow contents of the refrigerated Y-90 Zevalin kit (Zevalin vial, 50 mM sodium acetate vial, and formulation buffer vial) to reach room temperature.
- 2. Place the empty reaction vial in an appropriate acrylic shield.
- 3. Determine the amount of each component needed:
- a. Calculate volume of Y-90 Chloride equivalent to 40 mCi based on the activity concentration of the Y-90 Chloride stock.
- b. The volume of 50 mM Sodium Acetate solution needed is 1.2 times the volume of Y-90 Chloride solution determined in step 3.a, above.
- c. Calculate the volume of formulation buffer needed to bring the reaction vial contents to a final volume of 10 mL.
- 4. Transfer the calculated volume of 50 mM Sodium Acetate to the empty reaction vial. Coat the entire inner surface of the reaction vial by gentle inversion or rolling.
- 5. Transfer 40 mCi of Y-90 Chloride to the reaction vial using an acrylic shielded syringe. Mix the two solutions by gentle inversion or rolling.
- 6. Transfer 1.3 mL of Zevalin (ibritumomab tiuxetan) to the reaction vial. Do not shake or agitate the vial contents.
- 7. Allow the labeling reaction to proceed at room temperature for 5 minutes. A shorter or longer reaction time may adversely alter the final labeled product.
- 8. Immediately after the 5-minute incubation period, transfer the calculated volume of formulation buffer from step 3.c. to the reaction vial. Gently add the formulation buffer down the side of the reaction vial. If necessary, withdraw an equal volume of air to normalize pressure.
- 9. Measure the final product for total activity using a radioactivity calibration system suitable for the measurement of Y-90.
- 10. Using the supplied labels, record the date and time of preparation, the total activity and volume, and the date and time of expiration, and affix these labels to the shielded reaction vial container.
- 11. Patient Dose: Calculate the volume required for a Y-90 Zevalin dose [see Dosage and Administration (2.2)]. Withdraw the required volume from the reaction vial. Assay the syringe in the dose calibrator suitable for the measurement of Y-90. The measured dose must be within 10% of the prescribed dose of Y-90 Zevalin and must not exceed 32 mCi (1184 MBq). Using the supplied labels, record the patient identifier, total activity and volume and the date and time of expiration, and affix these labels to the syringe and shielded unit dose container.
- 12. Determine Radiochemical Purity [see Dosage and Administration (2.4)].
- 13. Store Yttrium-90 Zevalin at 2-8°C (36-46°F) until use and administer within 8 hours of radiolabeling. Immediately prior to administration, assay the syringe and contents using a radioactivity calibration system suitable for the measurement of Y-90.
2.4 Procedure for Determining Radiochemical Purity
Use the following procedures for radiolabeling Y-90 Zevalin:
- 1. Place a small drop of Y-90 Zevalin at the origin of an ITLC silica gel strip.
- 2. Place the ITLC silica gel strip into a chromatography chamber with the origin at the bottom and the solvent front at the top. Allow the solvent (0.9% NaCl) to migrate at least 5 cm from the bottom of the strip. Remove the strip from the chamber and cut the strip in half. Count each half of the ITLC silica gel strip for one minute (CPM) with a suitable counting apparatus.
- 3. Calculate the percent RCP as follows:
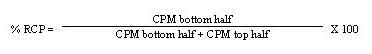
- 4. Repeat the ITLC procedure if the radiochemical purity is <95%. If repeat testing confirms that radiochemical purity is <95%, do not administer the Y-90 Zevalin dose.
2.5 Radiation Dosimetry
During clinical trials with Zevalin, estimations of radiation-absorbed doses for Y-90 Zevalin were performed using sequential whole body images and the MIRDOSE 3 software program. The estimated radiation absorbed doses to organs and marrow from a course of the Zevalin therapeutic regimen are summarized in Table 1. Absorbed dose estimates for the lower large intestine, upper large intestine, and small intestine have been modified from the standard MIRDOSE 3 output to account for the assumption that activity is within the intestine wall rather than the intestine contents.
Table 1. Estimated Radiation Absorbed Doses from Y-90 Zevalin - * Organ region of interest
- † Sacrum region of interest
- ‡ Whole body region of interest
Organ
Y-90 Zevalin cGy /mCi (mGy/MBq)
Median
Range
Spleen*
34.78 (9.4)
6.66 - 74.00 (1.8 - 20.0)
Liver*
17.76 (4.8)
10.73 - 29.97 (2.9 - 8.1)
Lower Large Intestinal Wall*
17.39 (4.7)
11.47 - 30.34 (3.1 - 8.2)
Upper Large Intestinal Wall*
13.32 (3.6)
7.40 - 24.79 (2.0 - 6.7)
Heart Wall*
10.73 (2.9)
5.55 - 11.84 (1.5 - 3.2)
Lungs*
7.4 (2)
4.44 - 12.58 (1.2 -3.4)
Testes*
5.55 (1.5)
3.70 - 15.91 (1.0 - 4.3)
Small Intestine*
5.18 (1.4)
2.96 - 7.77 (0.8 - 2.1)
Red Marrow†
4.81 (1.3)
2.22 - 6.66 (0.6 - 1.8)
Urinary Bladder Wall‡
3.33 (0.9)
2.59 - 4.81 (0.7 - 1.3)
Bone Surfaces†
3.33 (0.9)
1.85 - 4.44 (0.5 - 1.2)
Total Body‡
1.85 (0.5)
1.48 - 2.59 (0.4 - 0.7)
Ovaries‡
1.48 (0.4)
1.11 - 1.85 (0.3 - 0.5)
Uterus‡
1.48 (0.4)
1.11 - 1.85 (0.3 - 0.5)
Adrenals‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Brain‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Breasts‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Gallbladder Wall‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Muscle‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Pancreas‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Skin‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Stomach‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Thymus‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Thyroid‡
1.11 (0.3)
0.74 - 1.85 (0.2 - 0.5)
Kidneys*
0.37 (0.1)
0.00 - 1.11 (0.0 - 0.3)
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infusion Reactions
Rituximab, alone or as a component of the Zevalin therapeutic regimen, can cause severe, including fatal, infusion reactions. These reactions typically occur during the first rituximab infusion with time to onset of 30 to 120 minutes. Signs and symptoms of severe infusion reactions may include urticaria, hypotension, angioedema, hypoxia, bronchospasm, pulmonary infiltrates, acute respiratory distress syndrome, myocardial infarction, ventricular fibrillation, and cardiogenic shock. Temporarily slow or interrupt the rituximab infusion for less severe infusion reactions. Immediately discontinue rituximab and Y-90 Zevalin administration for severe infusion reactions. Only administer rituximab/Zevalin in facilities where immediate access to resuscitative measures is available [see Boxed Warning and Dosage and Administration (2.2)]. See also prescribing information for rituximab.
5.2 Prolonged and Severe Cytopenias
Cytopenias with delayed onset and prolonged duration, some complicated by hemorrhage and severe infection, are the most common severe adverse reactions of the Zevalin therapeutic regimen. When used according to recommended doses, the incidences of severe thrombocytopenia and neutropenia are greater in patients with mild baseline thrombocytopenia (≥ 100,000 but ≤ 149,000/mm3) compared to those with normal pretreatment platelet counts. Severe cytopenias persisting more than 12 weeks following administration can occur. Monitor complete blood counts (CBC) and platelet counts following the Zevalin therapeutic regimen weekly until levels recover or as clinically indicated [see Boxed Warning and Adverse Reactions (6.1)].
Do not administer the Zevalin therapeutic regimen to patients with ≥ 25% lymphoma marrow involvement and/or impaired bone marrow reserve. Monitor patients for cytopenias and their complications (e.g., febrile neutropenia, hemorrhage) for up to 3 months after use of the Zevalin therapeutic regimen. Avoid using drugs which interfere with platelet function or coagulation following the Zevalin therapeutic regimen.
5.3 Severe Cutaneous and Mucocutaneous Reactions
Erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, bullous dermatitis, and exfoliative dermatitis, some fatal, were reported in post-marketing experience. The time to onset of these reactions was variable, ranging from a few days to 4 months after administration of the Zevalin therapeutic regimen. Discontinue the Zevalin therapeutic regimen in patients experiencing a severe cutaneous or mucocutaneous reaction [see Boxed Warning and Adverse Reactions (6.2)].
5.4 Risk of Developing Myelodysplastic Syndrome, Leukemia, and Other Malignancies
The radiation dose resulting from therapeutic exposure to Y-90 radiolabeled Zevalin may result in secondary malignancies.
Myelodysplastic syndrome (MDS) and/or acute myelogenous leukemia (AML) were reported in 5.2% (11/211) of patients with relapsed or refractory NHL enrolled in clinical studies and 1.5% (8/535) of patients included in the expanded-access trial, with median follow-up of 6.5 and 4.4 years, respectively. Among the 19 reported cases, the median time to the diagnosis of MDS or AML was 1.9 years following treatment with the Zevalin therapeutic regimen; however, the cumulative incidence continues to increase [see Adverse Reactions (6.1)].
Among 204 patients receiving Y-90 Zevalin following first-line chemotherapy, 26 (12.7%) patients in the Zevalin arm developed a second primary malignancy compared to 14 (6.8%) of patients in the control arm. Seven patients (3.4%, 7/204) were diagnosed with MDS/AML after receiving Zevalin, compared to one patient (0.5%, 1/205) in the control arm, with a median follow-up of 7.3 years. Deaths due to second primary malignancy included 8 (3.9%) patients in the Zevalin arm compared to 3 (1.5%) patients in the control arm. Deaths due to MDS/AML included five (2.5%) patients in the Zevalin arm compared to no patients in the control arm.
Monitor patients for hematological toxicity including development of MDS or AML.
5.5 Extravasation
Monitor patients closely for evidence of extravasation during Zevalin infusion. Immediately terminate the infusion if signs or symptoms of extravasation occur and restart in another limb [see Dosage and Administration (2.2)].
5.6 Risks of Immunization
The safety of immunization with live viral vaccines following the Zevalin therapeutic regimen has not been studied. Do not administer live viral vaccines to patients who have recently received Zevalin. The ability to generate an immune response to any vaccine following the Zevalin therapeutic regimen has not been studied.
5.7 Radionuclide Precautions
During and after radiolabeling Zevalin with Y-90, minimize radiation exposure to patients and to medical personnel, consistent with institutional good radiation safety practices and patient management procedures.
5.8 Embryo-Fetal Toxicity
Based on its radioactivity, Y-90 Zevalin may cause fetal harm when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment and for a minimum of twelve months after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Serious Infusion Reactions [see Boxed Warning and Warnings and Precautions (5.1)].
- Prolonged and Severe Cytopenias [see Boxed Warning and Warnings and Precautions (5.2)].
- Severe Cutaneous and Mucocutaneous Reactions [see Boxed Warning and Warnings and Precautions (5.3)].
- Leukemia and Myelodysplastic Syndrome [see Warnings and Precautions (5.4)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The reported safety data reflects exposure to Zevalin in 349 patients with relapsed or refractory, low-grade, follicular or transformed NHL across 5 trials (4 single arm and 1 randomized) and in 206 patients with previously untreated follicular NHL in a randomized trial (FIT study) who received any portion of the Zevalin therapeutic regimen. The safety data reflect exposure to Zevalin in 270 patients with relapsed or refractory NHL with platelet counts ≥150,000/ mm3 who received 0.4 mCi/kg (14.8 MBq/kg) of Y-90 Zevalin (Group 1 in Table 4), 65 patients with relapsed or refractory NHL with platelet counts of ≥ 100,000 but ≤ 149,000 /mm3 who received 0.3 mCi/kg (11.1 MBq/kg) of Y-90 Zevalin (Group 2 in Table 4), and 204 patients with previously untreated NHL with platelet counts ≥150,000/ mm3 who received 0.4 mCi/kg (14.8 MBq/kg) of Y-90 Zevalin; all patients received a single course of Zevalin.
The most common adverse reactions of Zevalin are cytopenias, fatigue, nasopharyngitis, nausea, abdominal pain, asthenia, cough, diarrhea, and pyrexia.
The most serious adverse reactions of Zevalin are prolonged and severe cytopenias (thrombocytopenia, anemia, lymphopenia, neutropenia) and secondary malignancies.
Because the Zevalin therapeutic regimen includes the use of rituximab, see prescribing information for rituximab.
Table 2 displays selected adverse reaction incidence rates in patients who received any portion of the Zevalin therapeutic regimen (n=206) or no further therapy (n=203) following first-line chemotherapy (FIT study).
Table 2. Per-Patient Incidence (%) of Selected* Adverse Reactions Occurring in ≥ 5% of Patients with Previously Untreated Follicular NHL Treated with the Zevalin Therapeutic Regimen - * Between-group difference of ≥5%
- † NCI CTCAE version 2.0
Zevalin (n=206)
Observation (n=203)
All Grades†
Grade† 3-4
All Grades†
Grade† 3-4
%
%
%
%
Gastrointestinal Disorders
Abdominal pain
17
2
13
<1
Diarrhea
11
0
3
0
Nausea
18
0
2
0
Body as a Whole
Asthenia
15
1
8
<1
Fatigue
33
1
9
0
Influenza-like illness
8
0
3
0
Pyrexia
10
3
4
0
Musculoskeletal
Myalgia
9
0
3
0
Metabolism
Anorexia
8
0
2
0
Respiratory, Thoracic & Media
Cough
11
<1
5
0
Pharyngolaryngeal pain
7
0
2
0
Epistaxis
5
2
<1
0
Nervous System
Dizziness
7
0
2
0
Vascular
Hypertension
7
3
2
<1
Skin & Subcutaneous
Night sweats
8
0
2
0
Petechiae
8
2
0
0
Pruritus
7
0
1
0
Rash
7
0
<1
0
Infections & Infestations
Bronchitis
8
0
3
0
Nasopharyngitis
19
0
10
0
Rhinitis
8
0
2
0
Sinusitis
7
<1
<1
0
Urinary tract infection
7
<1
3
0
Blood and Lymphatic System
Thrombocytopenia
62
51
1
0
Neutropenia
45
41
3
2
Anemia
22
5
4
0
Leukopenia
43
36
4
1
Lymphopenia
26
18
9
5
Table 3 shows hematologic toxicities in 349 Zevalin-treated patients with relapsed or refractory, low-grade, follicular or transformed B-cell NHL. Grade 2-4 hematologic toxicity occurred in 86% of Zevalin-treated patients.
Table 3. Per-Patient Incidence (%) of Hematologic Adverse Reactions in Patients with Relapsed or Refractory Low-grade, Follicular or Transformed B-cell NHL* (N = 349 - * Occurring within the 12 weeks following the first rituximab infusion of the Zevalin therapeutic regimen
All Grades
%
Grade 3-4
%
Thrombocytopenia
95
63
Neutropenia
77
60
Anemia
61
17
Ecchymosis
7
<1
Prolonged and Severe Cytopenias
Patients in clinical studies were not permitted to receive hematopoietic growth factors beginning 2 weeks prior to administration of the Zevalin therapeutic regimen.
The incidence and duration of severe hematologic toxicity in previously treated NHL patients (N=335) and in previously untreated patients (FIT study) receiving Y-90 Zevalin are shown in Table 4.
Table 4. Severe Hematologic Toxicity in Patients Receiving Zevalin - * Day from last ANC ≥1000/mm3 to first ANC ≥1000/mm3 following nadir, censored at next treatment or death
- † Day from nadir to first count at level of Grade 1 toxicity or baseline
- ‡ Day from last platelet count ≥50,000/mm3 to day of first platelet count ≥50,000/mm3 following nadir, censored at next treatment or death
Baseline Platelet Count
Group 1
(n=270)
≥ 150,000/mm3
Group 2
(n=65 )
≥ 100,000 but ≤ 149,000/mm3
FIT study
(n=204)
≥ 150,000/mm3
Y-90 Zevalin Dose
0.4 mCi/kg
(14.8 MBq/kg)
0.3 mCi/kg
(11.1 MBq/kg)
0.4 mCi/kg
(14.8 MBq/kg)
ANC
Median nadir ( per mm3)
800
600
721
Per Patient Incidence
ANC <1000/mm3
57%
74%
65%
Per Patient Incidence
ANC <500/mm3
30%
35%
26%
Median Duration (Days)* ANC <1000/mm3 22
29
29
Median Time to Recovery† 12
13
15
Platelets
Median nadir (per mm3)
41,000
24,000
42,000
Per Patient Incidence
Platelets <50,000/mm3
61%
78%
61%
Per Patient Incidence
Platelets <10,000/mm3
10%
14%
4%
Median Duration (Days)‡Platelets <50,000/mm3 24
35
26
Median Time to Recovery†
13
14
14
Cytopenias were more severe and more prolonged among eleven (5%) patients who received Zevalin after first-line fludarabine or a fludarabine-containing chemotherapy regimen as compared to patients receiving non-fludarabine-containing regimens. Among these eleven patients, the median platelet nadir was 13,000/mm3 with a median duration of platelets below 50,000/mm3 of 56 days and the median time for platelet recovery from nadir to Grade 1 toxicity or baseline was 35 days. The median ANC was 355/mm3, with a median duration of ANC below 1,000/mm3 of 37 days and the median time for ANC recovery from nadir to Grade 1 toxicity or baseline was 20 days.
The median time to cytopenia was similar across patients with relapsed/refractory NHL and those completing first-line chemotherapy, with median ANC nadir at 61-62 days, platelet nadir at 49-53 days, and hemoglobin nadir at 68-69 days after Y-90-Zevalin administration.
Information on hematopoietic growth factor use and platelet transfusions is based on 211 patients with relapsed/refractory NHL and 206 patients following first-line chemotherapy. Filgrastim was given to 13% of patients and erythropoietin to 8% with relapsed or refractory disease; 14% of patients receiving Zevalin following first-line chemotherapy received granulocyte-colony stimulating factors and 5% received erythopoiesis-stimulating agents. Platelet transfusions were given to approximately 22% of all Zevalin-treated patients. Red blood cell transfusions were given to 20% of patients with relapsed or refractory NHL and 2% of patients receiving Zevalin following first-line chemotherapy.
Infections
In relapsed or refractory NHL patients, infections occurred in 29% of 349 patients during the first 3 months after initiating the Zevalin therapeutic regimen and 3% developed serious infections (urinary tract infection, febrile neutropenia, sepsis, pneumonia, cellulitis, colitis, diarrhea, osteomyelitis, and upper respiratory tract infection). Life-threatening infections were reported in 2% (sepsis, empyema, pneumonia, febrile neutropenia, fever, and biliary stent-associated cholangitis). From 3 months to 4 years after Zevalin treatment, 6% of patients developed infections; 2% were serious (urinary tract infection, bacterial or viral pneumonia, febrile neutropenia, perihilar infiltrate, pericarditis, and intravenous drug-associated viral hepatitis) and 1% were life-threatening infections (bacterial pneumonia, respiratory disease, and sepsis).
When administered following first-line chemotherapy (Table 2), Grade 3-4 infections occurred in 8% of Zevalin treated patients and in 2% of controls and included neutropenic sepsis (1%), bronchitis, catheter sepsis, diverticulitis, herpes zoster, influenza, lower respiratory tract infection, sinusitis, and upper respiratory tract infection.
Leukemia and Myelodysplastic Syndrome
Among 746 patients with relapsed/refractory NHL, 19 (2.6%) patients developed MDS/AML with a median follow-up of 4.4 years. The overall incidence of MDS/AML among the 211 patients included in the clinical studies was 5.2% (11/211), with a median follow-up of 6.5 years and median time to development of MDS/AML of 2.9 years. The cumulative Kaplan-Meier estimated incidence of MDS/secondary leukemia in this patient population was 2.2% at 2 years and 5.9% at 5 years. The incidence of MDS/AML among the 535 patients in the expanded access programs was 1.5% (8/535) with a median follow-up of 4.4 years and median time to development of MDS/AML of 1.5 years. Multiple cytogenetic abnormalities were described, most commonly involving chromosomes 5 and/or 7. The risk of MDS/AML was not associated with the number of prior treatments (0-1 versus 2-10).
Among 204 patients receiving Y-90-Zevalin following first-line treatment, 7 (3%) patients developed MDS/AML between approximately 2 to 7 years after Zevalin administration [see Warnings and Precautions (5.4)].
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of the Zevalin therapeutic regimen in hematologic malignancies. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors: (1) seriousness of the reaction, (2) frequency of reporting, or (3) strength of causal connection to the Zevalin therapeutic regimen.
- Cutaneous and mucocutaneous reactions: erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis, bullous dermatitis, and exfoliative dermatitis [see Boxed Warning and Warnings and Precautions (5.3)].
- Infusion site erythema and ulceration following extravasation [see Warnings and Precautions (5.5)].
- Radiation injury in tissues near areas of lymphomatous involvement within a month of Zevalin administration.
6.3 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparisons of the incidence of antibodies to the Zevalin therapeutic regimen in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
HAMA and HACA response data on 446 patients from 8 clinical studies conducted over a 10-year time period are available. Overall, 11/446 (2.5%) had evidence of either HAMA formation (N=8) or HACA formation (N=4). Six of these patients developed HAMA/HACA after treatment with Zevalin and 5 were HAMA/HACA positive at baseline. Of the 6 who were HAMA/HACA positive, only one was positive for both. Furthermore, in 6 of the 11 patients, the HAMA/HACA reverted to negative within 2 weeks to 3 months. No patients had increasing levels of HAMA/HACA at the end of the studies.
Only 6/446 patients (1.3%) had developed evidence of antibody formation after treatment with Zevalin, and of these, many either reverted to negative or decreased over time. This data demonstrates that HAMA/HACA develop infrequently, are typically transient, and do not increase with time.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its radioactivity, Y-90 Zevalin may cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Immunoglobulins are known to cross the placenta. There are no available data on Zevalin use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Advise women of childbearing potential to use adequate contraception for a minimum of twelve months. Inform women who become pregnant while receiving Zevalin of the potential fetal risks.
The estimated background risk of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. However, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.
8.2 Lactation
Risk Summary
There are no data on the presence of Zevalin or its metabolites in human milk, the effects of Zevalin on the breastfed child, or its effects on milk production. Because human IgG is excreted in human milk, it is expected that Zevalin would be present in human milk. Due to the potential for serious adverse reactions in a breastfeeding child from Zevalin, advise lactating women to avoid breastfeeding during treatment with the Zevalin therapeutic regimen and for 6 months after the last dose.
8.3 Females and Males of Reproductive Potential
Zevalin may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Conduct pregnancy testing in females of reproductive potential prior to treatment with Zevalin.
Contraception
Females
Based on its radioactivity, Y-90 Zevalin may cause fetal harm. Advise females of reproductive potential to use effective contraceptive methods during treatment and for 12 months after the last dose of the Zevalin therapeutic regimen [see Clinical Pharmacology (12.1)].
Males
Based on its radioactivity, Y-90 Zevalin may cause fetal harm. Advise males with female partners of reproductive potential to use effective contraceptive methods during treatment and for 12 months after the final dose of the Zevalin therapeutic regimen [see Clinical Pharmacology (12.1)].
Infertility
Based on its radioactivity, there is a potential risk that the Zevalin therapeutic regimen could cause toxic effects on the male and female gonads [see Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of Zevalin have not been established in pediatric patients.
8.5 Geriatric Use
Of 349 patients with relapsed/refractory NHL treated with the Zevalin therapeutic regimen in clinical studies, 38% (132 patients) were age 65 years and over, while 12% (41 patients) were age 75 years and over.
Of 414 patients enrolled in the FIT study (Zevalin following first-line chemotherapy) 206 patients received Zevalin. Of these patients 14% (29 patients) were 65 years and over, while 2% (4 patients) were 75 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, but greater sensitivity of some older individuals cannot be ruled out.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Zevalin (ibritumomab tiuxetan) is the immunoconjugate resulting from a stable thiourea covalent bond between the monoclonal antibody ibritumomab and the linker-chelator tiuxetan [N-[2-bis(carboxymethyl)amino]-3-(p-isothiocyanatophenyl)-propyl]-[N-[2-bis(carboxymethyl)amino]-2-(methyl)-ethyl]glycine. This linker-chelator provides a high affinity, conformationally restricted chelation site for Yttrium-90. The approximate molecular weight of ibritumomab tiuxetan is 148 kD. The antibody moiety of Zevalin is ibritumomab, a murine IgG1 kappa monoclonal antibody directed against the CD20 antigen.
Ibritumomab tiuxetan is a clear, colorless, sterile, pyrogen-free, preservative-free solution that may contain translucent particles. Each single-use vial includes 3.2 mg of ibritumomab tiuxetan in 2 mL of 0.9% Sodium Chloride.
Physical/Radiochemical Characteristics of Y-90
Yttrium-90 decays by emission of beta particles, with a physical half-life of 64.1 hours (2.67 days). The product of radioactive decay is non-radioactive Zirconium-90. The range of beta particles in soft tissue (χ90) is 5 mm. Radiation emission data for Y-90 are summarized in Table 5.
Table 5. Principal Y-90 Radiation Emission Data Radiation
Mean % per
Disintegration
Mean
Energy (keV)
Beta minus
100
750-935
External Radiation
The exposure rate for 1 mCi (37 MBq) of Y-90 is 8.3 x 10-3 C/kg/hr (32 R/hr) at the mouth of an open Y-90 vial.
To allow correction for physical decay of Y-90, the fractions that remain at selected intervals before and after the time of calibration are shown in Table 6.
Table 6. Physical Decay Chart: Y-90 Half-life 2.67 Days (64.1 Hours) Calibration Time (Hrs.)
Fraction Remaining
Calibration Time (Hrs.)
Fraction Remaining
-36
1.48
0
1.00
-24
1.30
1
0.99
-12
1.14
2
0.98
-8
1.09
3
0.97
-7
1.08
4
0.96
-6
1.07
5
0.95
-5
1.06
6
0.94
-4
1.04
7
0.93
-3
1.03
8
0.92
-2
1.02
12
0.88
-1
1.01
24
0.77
0
1.00
36
0.68
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ibritumomab tiuxetan binds specifically to the CD20 antigen (human B-lymphocyte-restricted differentiation antigen, Bp35). The apparent affinity (KD) of ibritumomab tiuxetan for the CD20 antigen ranges between approximately 14 to 18 nM. The CD20 antigen is expressed on pre-B and mature B lymphocytes and on > 90% of B-cell non-Hodgkin’s lymphomas (NHL). The CD20 antigen is not shed from the cell surface and does not internalize upon antibody binding.
The chelate tiuxetan, which tightly binds Y-90, is covalently linked to ibritumomab. The beta emission from Y-90 induces cellular damage by the formation of free radicals in the target and neighboring cells.
Ibritumomab tiuxetan binding was observed in vitro on lymphoid cells of the bone marrow, lymph node, thymus, red and white pulp of the spleen, and lymphoid follicles of the tonsil, as well as lymphoid nodules of other organs such as the large and small intestines.
12.2 Pharmacodynamics
In clinical studies, administration of the Zevalin therapeutic regimen resulted in sustained depletion of circulating B cells. At four weeks, the median number of circulating B cells was zero (range, 0-1084/mm3). B-cell recovery began at approximately 12 weeks following treatment, and the median level of B cells was within the normal range (32 to 341/mm3) by 9 months after treatment. Median serum levels of IgG and IgA remained within the normal range throughout the period of B-cell depletion. Median IgM serum levels dropped below normal (median 49 mg/dL, range 13-3990 mg/dL) after treatment and recovered to normal values by 6-months post therapy.
12.3 Pharmacokinetics
Pharmacokinetic and biodistribution studies were performed using In-111 Zevalin (5 mCi [185 MBq] In-111, 1.6 mg ibritumomab tiuxetan). In an early study designed to assess the need for pre-administration of unlabeled antibody, only 18% of known sites of disease were imaged when In-111 Zevalin was administered without unlabeled ibritumomab. When preceded by unlabeled ibritumomab (1.0 mg/kg or 2.5 mg/kg), In-111 Zevalin detected 56% and 92% of known disease sites, respectively. These studies were conducted with a Zevalin therapeutic regimen that included unlabeled ibritumomab.
In pharmacokinetic studies of patients receiving the Zevalin therapeutic regimen, the mean effective half-life for Y-90 activity in blood was 30 hours, and the mean area under the fraction of injected activity (FIA) vs. time curve in blood was 39 hours. Over 7 days, a median of 7.2% of the injected activity was excreted in urine.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity and mutogenicity studies have not been conducted. However, radiation is a potential carcinogen and mutagen.
No animal studies have been performed to determine the effects of Zevalin on fertility in males or females. In clinical studies, the Zevalin therapeutic regimen results in a significant radiation dose to the testes: the radiation dose to the ovaries has not been established [see Dosage and Administration (2.5)]. There is a potential risk that the Zevalin therapeutic regimen could cause toxic effects on the male and female gonads. Effective contraceptive methods should be used during treatment and for up to 12 months following the Zevalin therapeutic regimen.
-
14 CLINICAL STUDIES
14.1 Relapsed or Refractory, Low-grade or Follicular Lymphoma
A study to evaluate the efficacy and safety of Zevalin (referred to as Study 106-06) was a single arm study of 54 patients with relapsed follicular lymphoma, who were refractory to rituximab treatment. Patients had a World Health Organization (WHO) Performance Status (PS) 0-2, <25% bone marrow involvement by NHL, no prior bone marrow transplantation, and acceptable hematologic, renal, and hepatic function. Refractoriness to rituximab was defined as failure to achieve a complete or partial response or time-to-disease-progression (TTP) of < 6 months. The main efficacy outcome measure of the study was the overall response rate (ORR) using the International Workshop Response Criteria (IWRC). Other efficacy outcome measures included time-to-disease-progression (TTP) and duration of response (DR). Table 7 summarizes efficacy data from Study 106-06.
Another study to evaluate the efficacy and safety of Zevalin (referred to as Study 106-04) was a randomized (1:1), open-label, multicenter study comparing the Zevalin therapeutic regimen with rituximab. The trial was conducted in 130 patients with relapsed or refractory low-grade or follicular non-Hodgkin's lymphoma (NHL); no patient had received prior rituximab. Patients had histologically confirmed NHL requiring therapy, a WHO PS 0-2, <25% bone marrow involvement by NHL, no prior bone marrow transplantation, and acceptable hematologic function. Sixty-four patients received the Zevalin therapeutic regimen, and 66 patients received rituximab given as an IV infusion at 375 mg per m2 weekly times 4 doses. The main efficacy outcome measure of the study was ORR using the IWRC. The ORR was significantly higher for patients receiving the Zevalin therapeutic regimen (83% vs. 55%, p<0.001). Time-to-disease-progression was not significantly different between study arms. Table 7 summarizes efficacy data from Study 106-04.
Table 7. Summary of Efficacy Data* - * IWRC: International Workshop Response Criteria
- † CRu and CR: Unconfirmed and confirm complete response
- ‡ Estimated with observed range
- § Duration of response: interval from the onset of response to disease progression
- ¶ “+” indicates an ongoing response
- # Time to Disease Progression: interval from the first infusion to disease progression
Study 106-06
Study 106-04
Zevalin
therapeutic regimen
N = 54
Zevalin
therapeutic regimen
N = 64
Rituximab
N = 66
Overall Response Rate (%)
74
83
55
Complete Response Rate† (%)
15
38
18
Median DR‡§
(Months)
[Range¶]
6.4
[0.5-49.9+]
14.3
[1.8-47.6+]
11.5
[1.2-49.7+]
Median TTP‡# (Months)
[[Range¶]
6.8
[1.1-50.9+]
12.1
[2.1-49.0+]
10.1
[0.7-51.3+]
A single-arm study to evaluate the efficacy and safety of Zevalin (referred to as Study 106-05) was conducted in 30 patients, of whom 27 had relapsed or refractory low-grade, follicular NHL and a platelet count 100,000 to 149,000/mm3. Patients with ≥ 25% lymphomatous marrow involvement, prior myeloablative therapy with stem cell support, prior external beam radiation to > 25% of active marrow or neutrophil count <1,500/mm3 were ineligible for Study 106-05. All patients received Y-90 Zevalin [0.3 mCi per kg (11.1 MBq per kg)]. Objective, durable clinical responses were observed [89% ORR (95% CI: 70-97%) with a median duration of response of 11.6 months (range: 1.0-42.4+ months)].
14.2 Follicular, B-Cell NHL Upon Completion of First-Line Chemotherapy
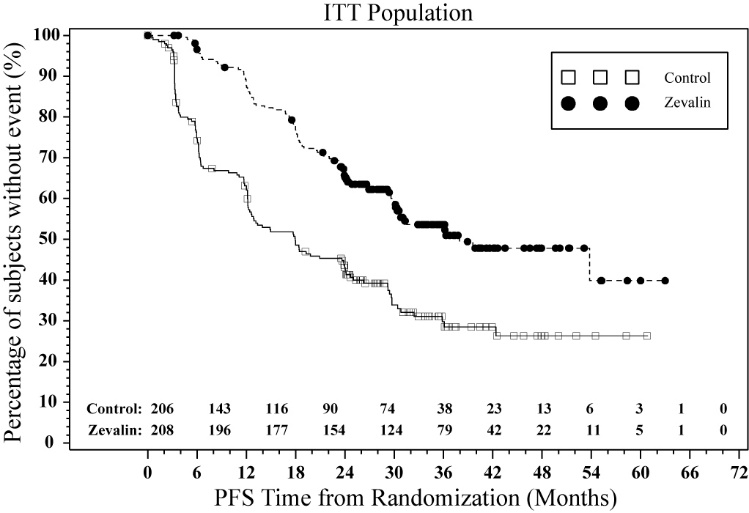
The FIT (First-line Indolent Trial) study (NCT00185393) was a multi-center, randomized, open-label study conducted in patients with follicular NHL with a partial (PR) or complete response (CR/CRu) upon completion of first-line chemotherapy. Randomization was stratified by center and response to first-line therapy (CR or PR). Key eligibility criteria were <25% bone marrow involvement, no prior external beam radiation or myeloablative therapy, and recovery of platelets to normal levels. Patients were randomized to receive Zevalin (n=208) or no further therapy (n=206). Y-90 Zevalin was administered at least 6 weeks but no more than 12 weeks following the last dose of chemotherapy. The main efficacy outcome measure was progression-free survival (PFS) assessed by study investigators using the International Workshop to Standardize Response Criteria for non-Hodgkin’s Lymphoma (1999).
Among the 414 patients, 49% were male, 99% were Caucasian, 12% were ≥65 years old, 83% had a WHO performance status of 0, and 65% had Stage IV disease. Thirty-nine (9.5%) patients received single agent chlorambucil, 22 (5%) patients received fludarabine or a fludarabine-containing regimen, 294 (71%) patients received cyclophosphamide-containing combination chemotherapy [CHOP (31%); CHOP-like (15%); CVP/COP (26%)] and 59 (14%) patients received rituximab-containing combination chemotherapy as first-line treatment.
Progression-free survival was significantly prolonged among Zevalin-treated patients compared to those receiving no further treatment [median PFS 38 months vs. 18 months; HR 0.46 (95% CI: 0.35, 0.60) p<0.0001 Cox model stratified by response to first-line therapy and initial treatment strategy (immediate vs. watch-and-wait)]. The number of patients who died was too small to permit a reliable comparison on survival.
The results for PFS are presented in Figure 1 .
-
16 HOW SUPPLIED/STORAGE AND HANDLING
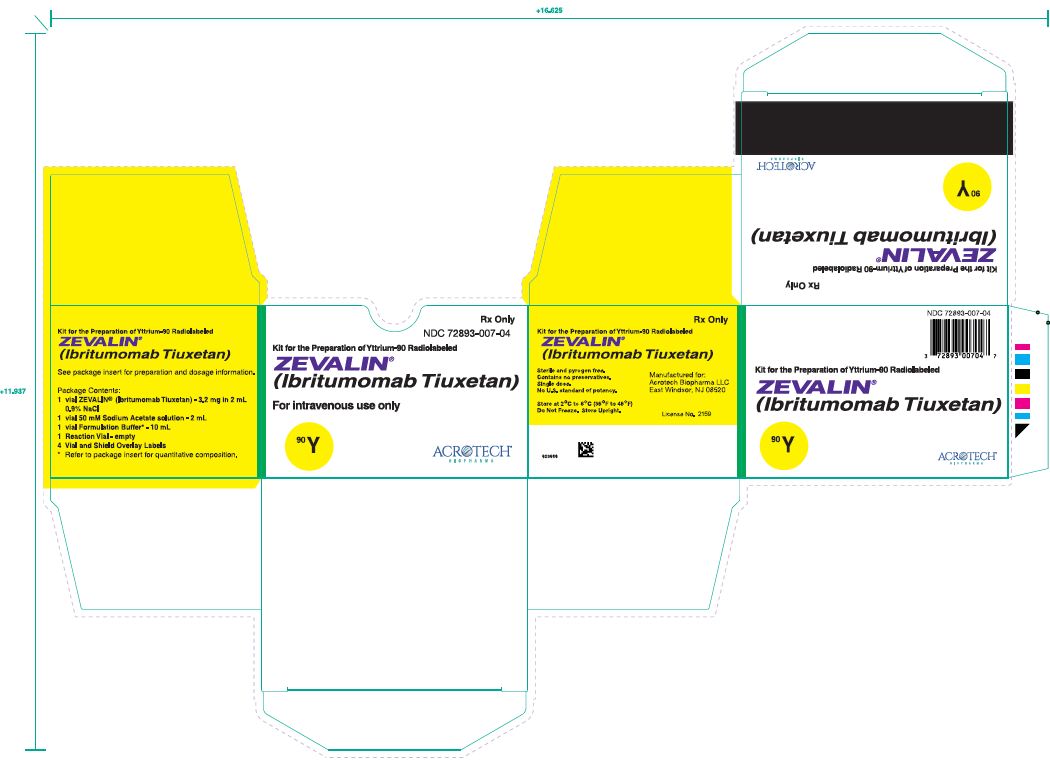
A kit is used for preparing Y-90 radiolabeled Zevalin (NDC: 72893-007-04). The contents of all vials are sterile, pyrogen-free, contain no preservatives, and are not radioactive. The kit contains four identification labels and the following four vials:
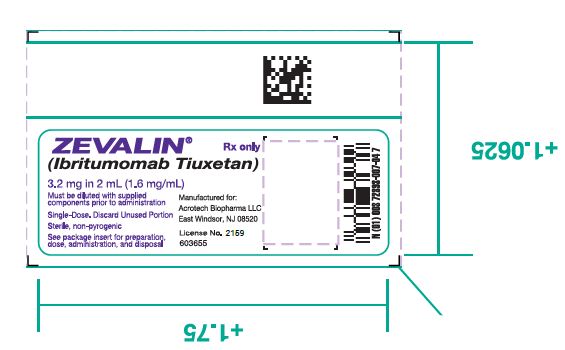
- 1. One (1) Zevalin vial containing 3.2 mg ibritumomab tiuxetan in 2 mL 0.9% Sodium Chloride as a clear, colorless solution.
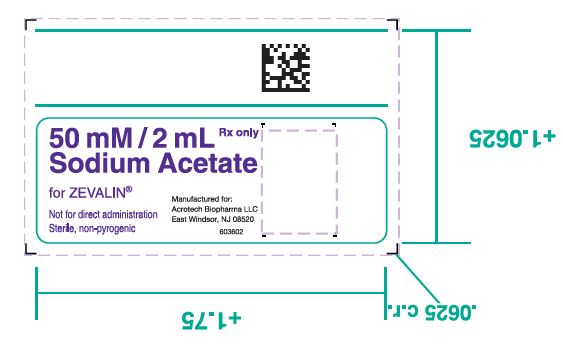
- 2. One (1) 50 mM Sodium Acetate Vial containing 13.6 mg Sodium Acetate trihydrate in 2 mL Water for Injection, USP as a clear, colorless solution.
- 3. One (1) Formulation Buffer Vial containing 750 mg Albumin (Human), 76 mg Sodium Chloride, 28 mg Sodium Phosphate Dibasic Dodecahydrate, 4 mg Pentetic Acid, 2 mg Potassium Phosphate Monobasic and 2 mg Potassium Chloride in 10 mL Water for Injection, pH 7.1 as a clear yellow to amber colored solution.
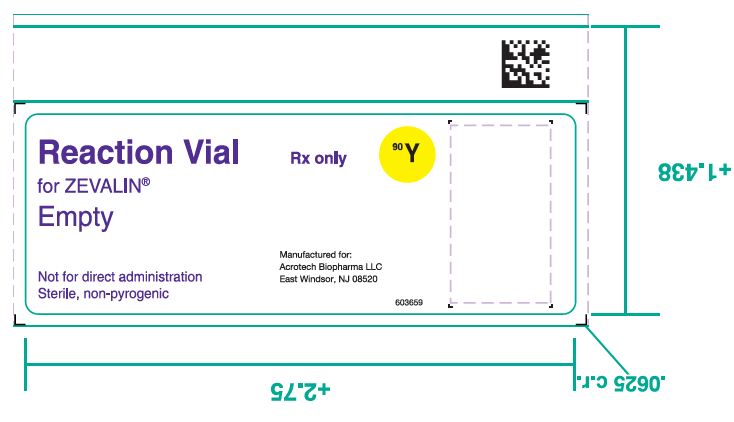
- 4. One (1) empty Reaction Vial.
Yttrium-90 Chloride Sterile Solution is shipped directly from the supplier upon placement of an order for the Y-90 Zevalin kit.
Rituximab must be ordered separately.
Storage
Store the kit at 2-8°C (36-46°F). Do not freeze.
-
17 PATIENT COUNSELING INFORMATION
Advise patients:
- To contact a healthcare professional for severe signs and symptoms of infusion reactions.
- To take premedications as prescribed [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- To report any signs or symptoms of cytopenias (bleeding, easy bruising, petechiae or purpura, pallor, weakness or fatigue) [see Warnings and Precautions (5.2)].
- To avoid medications that interfere with platelet function, except as directed by a healthcare professional [see Warnings and Precautions (5.2)].
- To seek prompt medical evaluation for diffuse rash, bullae, or desquamation of the skin or oral mucosa [see Warnings and Precautions (5.3)].
- To immediately report symptoms of infection (e.g. pyrexia) [see Adverse Reactions (6.2)].
- That immunization with live viral vaccines is not recommended for 12 months following the Zevalin therapeutic regimen [see Warnings and Precautions (5.6)].
- To use effective contraceptive methods during treatment and for a minimum of 12 months following Zevalin therapy [see Warnings and Precautions (5.8)], Use in Specific Populations (8.1, 8.3) and Nonclinical Toxicology (13.1)].
- To discontinue breastfeeding during and for 6 months after the last dose of Zevalin treatment [see Use In Specific Populations (8.2)].
Zevalin® (ibritumomab tiuxetan)
Manufactured for:
Acrotech Biopharma LLC
East Windsor, NJ 08520U.S. License No. 2159
Zevalin® is a registered trademark of Acrotech Biopharma LLC and its subsidiaries.
Protected by U.S. Patent Nos. 5,736,137, 5,776,456, 5,843,439, 6,207,858, 6,399,061, 6,682,734, 6,994,840, 7,229,620, 7,381,560, 7,422,739 and other patents and patents pending
- 18. PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
ZEVALIN
ibritumomab tiuxetan kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72893-007 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72893-007-04 1 in 1 CARTON; Type 0: Not a Combination Product 02/19/2002 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL 2 mL Part 2 1 VIAL 10 mL Part 3 1 VIAL 2 mL Part 1 of 3 ZEVALIN
ibritumomab tiuxetan injectionProduct Information Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IBRITUMOMAB TIUXETAN (UNII: 4Q52C550XK) (IBRITUMOMAB TIUXETAN - UNII:4Q52C550XK) IBRITUMOMAB TIUXETAN 1.6 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.8 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125019 02/19/2002 Part 2 of 3 FORMULATION BUFFER
ibritumomab tiuxetan injectionProduct Information Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 75 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 7.6 mg in 1 mL SODIUM PHOSPHATE, DIBASIC, DODECAHYDRATE (UNII: E1W4N241FO) 2.8 mg in 1 mL PENTETIC ACID (UNII: 7A314HQM0I) 0.4 mg in 1 mL POTASSIUM PHOSPHATE, MONOBASIC (UNII: 4J9FJ0HL51) 0.2 mg in 1 mL POTASSIUM CHLORIDE (UNII: 660YQ98I10) 0.2 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125019 02/19/2002 Part 3 of 3 SODIUM ACETATE
ibritumomab tiuxetan injectionProduct Information Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength SODIUM ACETATE (UNII: 4550K0SC9B) 6.8 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125019 02/19/2002 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125019 02/19/2002 Labeler - Acrotech Biopharma LLC (116965616) Registrant - Acrotech Biopharma LLC (116965616) Establishment Name Address ID/FEI Business Operations Ajinomoto Bio-Pharma Services 023050730 MANUFACTURE(72893-007)
Trademark Results [Zevalin]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ZEVALIN 75637474 2643264 Live/Registered |
RIT ONCOLOGY, LLC 1999-02-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.