RUZURGI- amifampridine tablet
RUZURGI by
Drug Labeling and Warnings
RUZURGI by is a Prescription medication manufactured, distributed, or labeled by Jacobus Pharmaceutical Company, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use RUZURGI safely and effectively. See full prescribing information for RUZURGI.
RUZURGI (amifampridine) tablets for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
RUZURGI is a potassium channel blocker indicated for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in patients 6 to less than 17 years of age. (1)
DOSAGE AND ADMINISTRATION
- Patients 6 to less than 17 years of age weighing 45 kg or more:
- Initial dosage is 15 mg to 30 mg daily, in divided doses
- Increase daily in 5 mg to 10 mg increments, divided in up to 5 doses daily
- Maximum single dose is 30 mg; maximum daily dosage is 100 mg (2.1)
- Patients 6 to less than 17 years of age weighing less than 45 kg:
- Initial dosage is 7.5 mg to 15 mg daily, in divided doses
- Increase daily in 2.5 mg to 5 mg increments, divided in up to 5 doses daily
- Maximum single dose is 15 mg; maximum daily dosage is 50 mg (2.1)
- When patients require a dosage in less than 5 mg increments, have difficulty swallowing, or require feeding tubes, a 1 mg/mL suspension can be prepared. (2.2)
- For patients with renal or hepatic impairment or who are known N-acetyltransferase 2 poor metabolizers, use the lowest recommended initial dosage. (2.3, 2.4, 2.5)
DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg, functionally scored (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
The most common adverse reactions (incidence at least 10% and at least 2% greater than placebo) are paresthesia/dysesthesia, abdominal pain, dyspepsia, dizziness, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Jacobus at 609-921-7447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Drugs that lower seizure threshold: The concomitant use of RUZURGI and drugs that lower seizure threshold may lead to an increased risk of seizures. (7.1)
- Drugs with cholinergic effects: The concomitant use of RUZURGI and drugs with cholinergic effects (e.g., direct or indirect cholinesterase inhibitors) may increase the cholinergic effects of RUZURGI and of those drugs, and increase the risk of adverse reactions. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2019
- Patients 6 to less than 17 years of age weighing 45 kg or more:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
2.3 Patients with Renal Impairment
2.4 Patients with Hepatic Impairment
2.5 Known N-acetyltransferase 2 (NAT2) Poor Metabolizers
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Seizures
5.2 Hypersensitivity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Drugs that Lower Seizure Threshold
7.2 Drugs with Cholinergic Effects
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 NAT2 Poor Metabolizers
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended oral dosage for pediatric patients 6 to less than 17 years of age is dependent on body weight and is included in Table 1. Dosage should be increased based on clinical response and tolerability.
If a dose is missed, patients should not take double or extra doses.
Table 1: Recommended Dosage for Pediatric Patients 6 to Less Than 17 Years of Age *see Dosage and Administration (2.2) for method to achieve these doses
Age and Body Weight Initial Dosage Titration Regimen Maximum
Single DoseMaximum
Total Daily
Maintenance DosagePediatric patients 6 to less than 17 years of age weighing 45 kg or more 15 mg to 30 mg daily, in divided doses (2 to 3 times per day) Increase daily in 5 mg to 10 mg increments, divided in up to 5 doses per day 30 mg 100 mg Pediatric patients 6 to less than 17 years of age weighing less than 45 kg 7.5 mg* to 15 mg daily, in divided doses (2 to 3 times per day) Increase daily in 2.5 mg* to 5 mg increments, divided in up to 5 doses per day 15 mg 50 mg 2.2 Administration Instructions
RUZURGI can be taken without regard to food.
Preparation of 1 mg/mL Suspension
When patients require a dosage in less than 5 mg increments, have difficulty swallowing tablets, or require feeding tubes, a 1 mg/mL suspension can be prepared (e.g., by placing three 10 mg tablets in a 30 mL container, adding 30 mL of sterile water, and shaking well for 30 seconds).
Crushing the tablets prior to making the suspension is not necessary. After preparation of the suspension, an oral syringe can be used to draw up and administer the correct dose by mouth or by feeding tube. Refrigerate the suspension between doses and shake well before drawing up each dose. The suspension can be stored under refrigeration for up to 24 hours. Discard any unused portion of the suspension after 24 hours.
2.3 Patients with Renal Impairment
The recommended starting dosage of RUZURGI in pediatric patients weighing 45 kg or more with renal impairment (creatinine clearance 15 to 90 mL/min) is 15 mg daily taken orally in divided doses. The recommended starting dosage for pediatric patients weighing less than 45 kg with renal impairment is 7.5 mg daily taken orally in divided doses [see Dosage and Administration (2.1) and Use in Specific Populations (8.6)]. No dosage recommendations for RUZURGI can be made for patients with end-stage renal disease.
2.4 Patients with Hepatic Impairment
The recommended starting dosage of RUZURGI in pediatric patients weighing 45 kg or more with any degree of hepatic impairment is 15 mg daily taken orally in divided doses. The recommended starting dosage for pediatric patients weighing less than 45 kg with any degree of hepatic impairment is 7.5 mg daily taken orally in divided doses [see Dosage and Administration (2.1) and Use in Specific Populations (8.7)].
2.5 Known N-acetyltransferase 2 (NAT2) Poor Metabolizers
The recommended starting dosage of RUZURGI in pediatric patients weighing 45 kg or more who are known N-acetyltransferase 2 (NAT2) poor metabolizers is 15 mg daily taken orally in divided doses. The recommended starting dosage in pediatric patients weighing less than 45 kg who are known NAT2 poor metabolizers is 7.5 mg daily taken orally in divided doses [see Dosage and Administration (2.1), Use in Specific Populations (8.8), and Clinical Pharmacology (12.5)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Seizures
RUZURGI can cause seizures. Seizures have been observed in patients with and without a history of seizures taking RUZURGI at the recommended doses, and at various times after initiation of treatment. Many of the patients were taking medications or had comorbid medical conditions that may have lowered the seizure threshold [see Drug Interactions (7.1)]. Seizures may be dose-dependent. Because seizure events were captured retrospectively from expanded access programs, it is not possible to reliably estimate their frequency with use of RUZURGI. Consider discontinuation or dose-reduction of RUZURGI in patients who have a seizure while on treatment. RUZURGI is contraindicated in patients with a history of seizures.
5.2 Hypersensitivity
In clinical trials, hypersensitivity reactions and anaphylaxis associated with RUZURGI administration have not been reported. Anaphylaxis has been reported in patients taking another aminopyridine; therefore, it may occur with RUZURGI. If anaphylaxis occurs, administration of RUZURGI should be discontinued and appropriate therapy initiated.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
In a double-blind, 3-way crossover, pharmacology study to assess the effects of RUZURGI on QTc interval prolongation, RUZURGI was administered at doses greater than the maximum recommended dose (120 mg administered as 4 equal doses of 30 mg at 4-hour intervals) to 52 healthy adult volunteers [see Clinical Pharmacology (12.2)]. Adverse reactions that occurred in at least 5% of subjects during RUZURGI treatment and with incidence at least 2% greater than during placebo treatment are displayed in Table 2.
Table 2: Adverse Reactions Occurring in at Least 5% of Subjects During RUZURGI Treatment and With at Least 2% Greater Incidence Than Placebo * Includes paresthesia, dysesthesia, and oral dysesthesias.
** Includes abdominal pain and upper abdominal pain.
RUZURGI
(N=52)
%Placebo Adverse Reaction (N=49)
%Paresthesia/Dysesthesia* 69
252 Abdominal pain** 0 Dyspepsia 17 2 Dizziness 12 0 Nausea 10 2 Back pain 8 2 Hypoesthesia 6 0 Muscle spasms 6 2 Subjects classified as poor metabolizers based on rate of metabolism were more likely to experience adverse reactions during RUZURGI treatment than intermediate or normal metabolizers [see Clinical Pharmacology (12.5)].
Expanded Access Experience
In expanded access programs, 162 patients with LEMS (54% female) were treated with RUZURGI. Among patients with available exposure data, the median duration of treatment was 1.7 years (range 1 day to 27.6 years) for a total of 766.4 person years. Patient age at the time RUZURGI was initiated ranged from 21 to 84 years (mean 58.7 years). The median of the maximum total daily dosage was 75 mg/day.
In general, the most frequent adverse reactions observed in the expanded access programs were similar to those observed in the QT study. Additionally, the following adverse reactions were reported in ≥5% of patients: falls, diarrhea, pneumonia, dyspnea, arthralgia, asthenia, depression, dysphagia, headache, insomnia, vision blurred, anemia, anxiety, constipation, feeling cold, gastroesophageal reflux disease, and pain. Because these reactions were captured retrospectively from expanded access programs, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Pediatric Patients (6 to Less than 17 Years of Age)
Safety of RUZURGI was evaluated in pediatric LEMS and non-LEMS patients 6 to less than 17 years of age who were treated in expanded access programs. There were 15 patients ages 6 to less than 17 years who received RUZURGI, of whom 9 received RUZURGI for at least 1 year. Adverse reactions reported in pediatric patients 6 to less than 17 years of age were similar to those seen in adult patients.
-
7 DRUG INTERACTIONS
7.1 Drugs that Lower Seizure Threshold
The concomitant use of RUZURGI and drugs that lower seizure threshold may lead to an increased risk of seizures [see Warnings and Precautions (5.1)]. The decision to administer RUZURGI concomitantly with drugs that lower the seizure threshold should be carefully considered in light of the severity of the associated risks.
7.2 Drugs with Cholinergic Effects
The concomitant use of RUZURGI and drugs with cholinergic effects (e.g., direct or indirect cholinesterase inhibitors) may increase the cholinergic effects of RUZURGI and of those drugs and increase the risk of adverse reactions [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no data on the developmental risk associated with the use of RUZURGI in pregnant women.
Animal studies to assess the potential adverse effects of amifampridine on embryofetal development have not been conducted.
In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
8.2 Lactation
Risk Summary
There are no data on the presence of amifampridine or the 3-N-acetyl-amifampridine metabolite in human milk, the effects on the breastfed infant, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for RUZURGI and any potential adverse effects on the breastfed infant from RUZURGI or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of RUZURGI have been established in patients 6 to less than 17 years of age. Use of RUZURGI in patients 6 to less than 17 years of age is supported by evidence from adequate and well-controlled studies of RUZURGI in adults with LEMS, pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients, and safety data from pediatric patients 6 to less than 17 years of age [see Adverse Reactions (6.1) and Clinical Pharmacology (12.3)].
Safety and effectiveness in pediatric patients below the age of 6 years have not been established.
8.6 Renal Impairment
Renal clearance is an elimination pathway for amifampridine and the inactive metabolite, 3-N-acetyl amifampridine [see Clinical Pharmacology (12.3)]. Therefore, in patients with renal impairment, RUZURGI should be initiated at the lowest recommended starting dosage and patients should be closely monitored for adverse reactions [see Dosage and Administration (2.3)]. Consider dosage modification or discontinuation of RUZURGI for patients with renal impairment as needed based on clinical effect and tolerability. No dosage recommendations for RUZURGI can be made for patients with end-stage renal disease (CLcr < 15 mL/min or patients requiring dialysis).
8.7 Hepatic Impairment
The effects of RUZURGI have not been studied in patients with hepatic impairment. RUZURGI is extensively metabolized by N-acetyltransferase 2 (NAT2), and hepatic impairment may cause an increase in exposure. Therefore, initiate RUZURGI in patients with any degree of hepatic impairment at the lowest recommended starting dosage and monitor for adverse reactions [see Dosage and Administration (2.4)]. Consider dosage modification or discontinuation of RUZURGI for patients with hepatic impairment as needed based on clinical effect and tolerability.
8.8 NAT2 Poor Metabolizers
Exposure of RUZURGI is increased in patients who are N-acetyltransferase 2 (NAT2) poor metabolizers [see Clinical Pharmacology (12.5)]. Therefore, initiate RUZURGI in patients who are known NAT2 poor metabolizers at the lowest recommended starting dosage and monitor for adverse reactions [see Dosage and Administration (2.5)]. Consider dosage modification of RUZURGI for patients who are known NAT2 poor metabolizers as needed based on clinical effect and tolerability.
-
10 OVERDOSAGE
In case reports, events reported after intake of RUZURGI at doses of 300 mg per day or greater (more than three times the maximum recommended daily dosage) include vomiting, nystagmus, seizures and status epilepticus, rhabdomyolysis, chest pain, diaphoresis, palpitations, paroxysmal supraventricular tachycardia, transient QTc prolongation, aspiration with acute respiratory failure, and cardiac arrest.
Patients with suspected overdose with RUZURGI should be monitored for signs or symptoms of exaggerated RUZURGI adverse reactions or effects, and appropriate symptomatic treatment instituted immediately.
-
11 DESCRIPTION
RUZURGI (amifampridine) is a potassium channel blocker available as functionally scored tablets for oral administration.
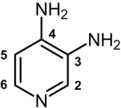
The chemical name of amifampridine is 3,4-diaminopyridine (CAS 54-96-6).
It is a white to off-white, crystalline solid with a molecular formula of C5H7N3 and a molecular weight of 109.13 g/mol. It is sparingly soluble in water. A 1% aqueous solution of amifampridine has a pH of 10.8 at 25°C.
The structural formula is
Each RUZURGI tablet contains 10 mg of amifampridine. Inactive ingredients consist of colloidal silicon dioxide, dibasic calcium phosphate dihydrate, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which amifampridine exerts its therapeutic effect in LEMS patients has not been fully elucidated. Amifampridine is a broad spectrum potassium channel blocker.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of RUZURGI on QTc interval prolongation was studied in a double-blind, randomized, placebo- and positive-controlled study in 52 healthy volunteers (including 23 subjects with poor metabolizer phenotype). Study participants were administered 120 mg RUZURGI in 4 equal doses of 30 mg at 4-hour intervals (Dose 1, 2, 3, and 4) [see Clinical Pharmacology (12.5)]. RUZURGI did not prolong the QTc interval to any clinically relevant extent. In vitro, RUZURGI did not inhibit the human ether-à-go-go-related gene ion channel.
12.3 Pharmacokinetics
The pharmacokinetics of amifampridine form RUZURGI is approximately dose proportional. Steady state was generally reached within 1 day of dosing. Multiple dosing resulted in no accumulation of amifampridine and only moderate accumulation of the 3-N-acetyl amifampridine metabolite [see Clinical Pharmacology (12.5)].
Absorption
The absolute bioavailability of RUZURGI has not been assessed. RUZURGI is absorbed in an approximately dose-proportional manner with a median time to maximum concentration (tmax) of 0.5 hours post administration.
Effect of Food
Compared to administration of RUZURGI in the fasting state, administration of the 20 and 30 mg dose levels of RUZURGI with a standard high fat meal resulted in significant decrease in Cmax (41% and 52%, respectively) and an increase in median tmax to 1.0 hour; AUC0-last was only significantly reduced for the 30 mg dose (23%) [see Dosage and Adminstration (2.2)].
Distribution
In healthy volunteers, the volume of distribution for plasma amifampridine indicated that RUZURGI is a drug with a moderate to high volume of distribution.
In vitro human plasma protein binding of amifampridine and 3-N-acetyl amifampridine was 25.3% and 43.3%, respectively.
Elimination
Metabolism
In vitro studies with complimentary DNA expressed human N-acetyltransferase (NAT) enzyme preparations indicate that amifampridine is rapidly metabolized by the N-acetyltransferase 2 (NAT2) enzyme to the 3-N-acetyl amifampridine metabolite. Metabolism of amifampridine by N-acetyltransferase 1 (NAT1) may also occur but at a much slower rate.
Amifampridine does not undergo glucuronidation or sulfonation.
Excretion
Following oral administration of a single 20 or 30 mg dose of RUZURGI to healthy volunteers, amifampridine apparent oral clearance (CL/F) was 149 to 214 L/h, the average elimination half-life (t1/2) was 3.6 to 4.2 hours. The average t1/2 of the 3-N-acetyl amifampridine metabolite was 4.1 to 4.8 hours.
The majority (>65%) of RUZURGI administered to healthy volunteers was recovered in urine as either the parent compound or the 3-N-acetyl amifampridine metabolite.
Specific Populations
Pediatric Patients (6 to Less than 17 Years of Age)
A population pharmacokinetic analysis showed that body weight significantly correlates with the clearance of amifampridine; clearance increased with an increase in body weight. A weight-based dosing regimen is necessary to achieve amifampridine exposures in pediatric patients 6 to less than 17 years of age similar to those observed in adults at effective doses of RUZURGI [see Indications and Usage (1) and Clinical Studies (14)].
Drug Interaction Studies
In vitro studies
Amifampridine is not metabolized by cytochrome P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4.
In vitro studies with human liver microsomes indicated that amifampridine and 3-N-acetyl amifampridine were not direct or time-dependent inhibitors of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4.
In vitro studies in cryopreserved human hepatocytes indicated that amifampridine did not induce CYP isoforms CYP1A2, CYP2B6, or CYP3A4.
Based on in vitro studies with Caco-2 cells amifampridine is unlikely to act as a substrate or inhibitor of the P glycoprotein transporter. Amifampridine is not an inhibitor of the BCRP transporter.
In vitro studies with Chinese hamster ovary cells expressing human OATP1B1, OATP1B3, OAT1, and OCT2 and Madin-Darby canine kidney cells expressing human OAT3 indicated that amifampridine is a weak inhibitor of OCT2, but is not an inhibitor of OAT1, OAT3, OATP1B1, or OATP1B3. The studies also indicated that amifampridine is not a substrate for OAT1, OAT3, or OCT2 transporters.
In vivo studies
Controlled clinical drug interaction studies have not been performed with RUZURGI.
Co-administration of intravenous amifampridine and intravenous pyridostigmine led to a 21% elevation in maximum pyridostigmine serum concentrations, but did not significantly affect the pharmacokinetics of amifampridine [see Drug Interactions (7.2)].
12.5 Pharmacogenomics
Genetic variants in the N-acetyltransferase gene 2 (NAT2) affect the rate and extent of RUZURGI metabolism. In normal healthy volunteers, poor metabolizers, also referred to as “slow acetylators” (i.e., carriers of two reduced function alleles) had higher average plasma amifampridine concentrations than intermediate metabolizers, also referred to as “intermediate acetylators” (i.e., carriers of one reduced and one normal function alleles), and normal metabolizers, also referred to as “fast/rapid acetylators” (i.e., carriers of two normal function alleles).
In the TQT study [see Clinical Pharmacology (12.2)], poor metabolizers (N=19) had 1.1 to 3.7 times higher AUC0-4h and 1.3 to 3.7 times higher Cmax than intermediate metabolizers (N=21), following the first dose. Poor metabolizers had 6.0 to 8.5 times higher AUC0-4h and 6.1 to 7.6 times higher Cmax than normal metabolizers (N=3), following the first dose.
In the general population, the NAT2 poor metabolizer phenotype prevalence is 40–60% in the White and African American populations, and in 10–30% in Asian ethnic populations (individuals of Japanese, Chinese, or Korean descent).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Mutagenesis
Amifampridine was negative for mutagenicity in an in vitro bacterial reverse mutation (Ames) assay and for clastogenicity in in vivo mouse micronucleus and chromosomal aberration assay. Amifampridine was positive for clastogenicity in an in vitro mouse lymphoma assay in the absence of metabolic activation.
-
14 CLINICAL STUDIES
The efficacy of RUZURGI for the treatment of LEMS was established by Study 1, a randomized, double-blind, placebo-controlled, withdrawal study (NCT: 01511978). Study 1 enrolled patients with an established diagnosis of LEMS, confirmed by documentation and an independent neurologist review. Patients were required to be on an adequate and stable dosage (30 mg to 100 mg daily for at least 3 months) of RUZURGI prior to entering the study.
The primary measure of efficacy was the categorization of the degree of change (e.g., greater than 30% deterioration) in the Triple Timed Up and Go test (3TUG) upon withdrawal of active medication, when compared with the time-matched average of the 3TUG assessments at baseline.
The 3TUG is a measure of the time it takes a person to rise from a chair, walk 3 meters, and return to the chair for 3 consecutive laps without pause. Higher 3TUG scores represent greater impairment.
The secondary efficacy endpoint was the self-assessment scale for LEMS-related weakness (W-SAS), a scale from -3 to 3 assessing a person's feeling of weakening or strengthening from baseline. A higher positive W-SAS score indicates a perceived greater improvement of strength. A more negative score indicates perceived greater weakening.
After an initial open-label run-in phase, 32 patients were randomized in a double-blind fashion to either continue treatment with RUZURGI (n = 14) or switch to placebo over a 3-day downward titration (n = 18) period. Following the downward titration period, patients remained on blinded RUZURGI or placebo for 16 more hours. Efficacy was assessed 2 hours after the last dose of the downward titration period. Patients were allowed to use stable dosages of peripherally-acting cholinesterase inhibitors or oral immunosuppressants. Seventy-nine percent of patients randomized to RUZURGI were receiving cholinesterase inhibitors, versus 83% in the placebo group, and 29% of patients randomized to RUZURGI were receiving an immunosuppressant therapy, versus 39% in the placebo group.
Randomized patients had a median age of 56 years (range: 23 to 83 years), 66% were female, and 91% were White. Ninety-seven percent of patients had a diagnosis of autoimmune LEMS, and 3% of patients had a diagnosis of paraneoplastic LEMS.
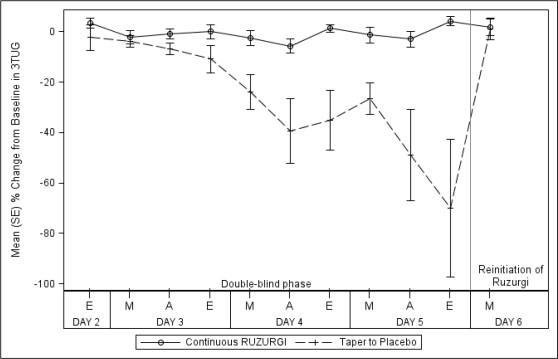
All 32 patients completed the study. None of the patients randomized to continue RUZURGI experienced a greater than 30% deterioration in the final post-dose 3TUG test. In contrast, 72% (13/18) of those randomized to placebo experienced a greater than 30% deterioration in the final 3TUG test (p < 0.0001). Patients who were randomized to placebo returned to baseline after restarting RUZURGI. Figure 1 shows the time course of the mean percent change from baseline on the 3TUG during the double-blind phase and with reinitiation of RUZURGI.
Figure 1: Mean Percent Change From Baseline in Post-dose 3TUG Time During the Double-blind Phase of the Study and Return to Baseline Upon Reinitiation of RUZURGI
3TUG=Triple Timed Up and Go; A=afternoon; E=evening; M=morning.
The W-SAS score showed a significantly greater decrease in patients randomized to placebo (-2.4) than in those who continued treatment with RUZURGI ( -0.2; p < 0.0001), indicating that patients who were randomized to placebo perceived a worsening of weakness compared to those who remained on RUZURGI.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
RUZURGI (amifampridine) 10 mg functionally scored tablets are oval, white to off-white, and debossed “10 │ 110” on one side and “JACOBUS” on the other side. RUZURGI is supplied as follows:
- Bottles of 100 tablets: NDC: 49938-110-01
-
17 PATIENT COUNSELING INFORMATION
Advise the patient and/or caregiver to read the Food and Drug Administration-approved patient labeling (Medication Guide and Instructions for Use).
Risk of Seizures
Inform patients and/or caregivers that RUZURGI can cause seizures, and to notify their healthcare provider if they experience a seizure [see Warnings and Precautions (5.1)].
RUZURGI Dosing
Instruct patients to take RUZURGI exactly as prescribed. Patients should carefully follow the dose escalation schedule provided by their healthcare provider to safely achieve the therapeutic dosage [see Dosage and Administration (2.1)]. Inform patients that the tablets may be divided in half at the score, if needed. Refer patients and/or caregivers to the Instructions for Use if they require a dosage in less than 5 mg increments, have difficulty swallowing tablets, or require feeding tubes [see Dosage and Administration (2.2)]. Instruct patients not to take a double dose after they miss a dose of RUZURGI, as this may increase their risk of seizure.
Hypersensitivity
Instruct patients and/or caregivers to inform their healthcare provider if they have signs or symptoms of hypersensitivity, and to seek emergency help if signs and symptoms of anaphylaxis occur [see Warnings and Precautions (5.2)].
Drug Interactions
Instruct patients to notify their healthcare provider prior to starting any new medication, including over-the-counter drugs [see Drug Interactions (7)].
Storage
Advise patients and/or caregivers to store the tablets in the pharmacy dispensed container at controlled room temperature, for a period not to exceed 3 months [see How Supplied/Storage and Handling (16.2)].
Instruct patients and/or caregivers who prepare the 1 mg/mL suspension of RUZURGI that it should be prepared daily and refrigerated between doses. The suspension can be stored under refrigeration for up to 24 hours. Instruct the patient to discard any unused portion of the suspension after 24 hours.
Distributed by Jacobus Pharmaceutical Company, Inc.
Princeton, NJ 08540
Manufactured by Jacobus Pharmaceutical Company, Inc. Plainsboro, NJ
RUZURGI® is a registered trademark of Jacobus Pharmaceutical Company, Inc.
-
MEDICATION GUIDE
MEDICATION GUIDE
RUZURGI® (rew-ZUR-jee)
(amifampridine) tablets, for oral useRead this Medication Guide before you start taking RUZURGI and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. What is the most important information I should know about RUZURGI? RUZURGI can cause seizures.
- You could have a seizure even if you never had a seizure before.
-
Do not take RUZURGI if you have ever had a seizure.
Stop taking RUZURGI and call your doctor right away if you have a seizure while taking RUZURGI. What is RUZURGI?
RUZURGI is a prescription medicine used to treat Lambert-Eaton myasthenic syndrome (LEMS) in children 6 to less than 17 years of age.
It is not known if RUZURGI is safe or effective in children less than 6 years of age.Do not take RUZURGI if you:
- have ever had a seizure.
- are allergic to amifampridine or another aminopyridine. Talk to your doctor if you are not sure.
Before you take RUZURGI, tell your doctor about all of your medical conditions including if you:
- are taking another aminopyridine, such as as compounded 3,4-diaminopyridine (3,4-DAP), 4-aminopyridine, or pyridostigmine.
- have had a seizure.
- have kidney problems.
- have liver problems.
- are pregnant or planning to become pregnant. It is not known if RUZURGI can harm your unborn baby. You and your doctor should decide if you will take RUZURGI while you are pregnant.
- are breastfeeding or plan to breastfeed. It is not known if RUZURGI passes into your breastmilk. Talk to your doctor about the best way to feed your baby while taking RUZURGI.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. How should I take RUZURGI?
- See the detailed “Instructions for Use” on how to take and prepare a suspension of RUZURGI if your dose is less than 5mg, you have trouble swallowing tablets, or you need a feeding tube.
- Take RUZURGI exactly as your doctor tells you to take it.
- Do not change your dose of RUZURGI.
- Do not stop taking RUZURGI without first talking to your doctor.
- RUZURGI tablets are scored and can be cut if less than a full tablet is needed for you to get the right dose.
- RUZURGI can be taken with or without food.
- If you miss a dose of RUZURGI, skip that dose and take your next dose at your next scheduled dose time.
-
Do not take RUZURGI together with other medicines known to increase the risk of seizures such as aminopyridine medicines, including:
- compounded 3,4-diaminopyridine (amifampridine)
- amifampridine phosphate
- 4-aminopyridine
- If you take too much RUZURGI, call your doctor or go to the nearest hospital emergency room right away.
What are the possible side effects of RUZURGI?
RUZURGI may cause serious side effects, including:
- Seizures. See “What is the most important information I should know about RUZURGI?”
-
Serious allergic reactions, such as anaphylaxis. RUZURGI can cause serious allergic reactions. Stop taking RUZURGI and call your doctor right away or get emergency medical help if you have:
- shortness of breath or trouble breathing
- swelling of your throat or tongue
- hives
The most common side effects of RUZURGI include:
- tingling around the mouth, tongue, face, fingers, toes, and other body parts
- stomach pain
- indigestion
- dizziness
- nausea
Tell your doctor if you have any side effect that bothers you or does not go away. These are not all the possible side effects of RUZURGI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store RUZURGI?
- Store RUZURGI tablets in the container from the pharmacy at room temperature between 68°F to 77°F (20°C to 25°C) for up to 3 months.
- Refrigerate prepared RUZURGI oral suspension between doses for up to 24 hours.
- Safely throw away medicine that is no longer needed or out of date. Keep RUZURGI and all medicines out of the reach of children.
General Information about the safe and effective use of RUZURGI
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use RUZURGI for a condition for which it was not prescribed. Do not give RUZURGI to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk with your doctor or pharmacist. You can ask your pharmacist or doctor for information about RUZURGI that is written for health professionals.What are the ingredients in RUZURGI?
Active ingredient: amifampridine (also called 3,4-diaminopyridine)
Inactive ingredients: colloidal silicon dioxide, dibasic calcium phosphate dihydrate, magnesium stearate, microcrystalline cellulose, and sodium starch glycolateDistributed by Jacobus Pharmaceutical Company, Inc.
Princeton, NJ 08540
RUZURGI® is a registered trademark of Jacobus Pharmaceutical Company, Inc.
Manufactured by Jacobus Pharmaceutical Company, Inc. Plainsboro, NJ For more information, go to www.RUZURGI.com or call 609-921-7447.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Issued: 5/2019 -
INSTRUCTIONS FOR USE
How should I store RUZURGI?
- Store RUZURGI tablets in the container from the pharmacy at room temperature between 68°F to 77°F (20°C to 25°C) for up to 3 months.
- Refrigerate prepared RUZURGI oral suspension between doses.
- Safely throw away unused RUZURGI oral suspension after 24 hours.
- Keep RUZURGI and all medicines out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration
Issued: 5/2019
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Carton Label
NDC: 49938-110-01
Ruzurgi®
(amifampridine)
Tablets10 mg
Rx Only
100 Tablets
Refrigerate prior to dispensing
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - Bottle Label
NDC: 49938-110-01
Ruzurgi®
(amifampridine)
Tablets10 mg
Rx Only
100 Tablets
Refrigerate prior to dispensing
-
INGREDIENTS AND APPEARANCE
RUZURGI
amifampridine tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 49938-110 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Amifampridine (UNII: RU4S6E2G0J) (Amifampridine - UNII:RU4S6E2G0J) Amifampridine 10 mg Inactive Ingredients Ingredient Name Strength Silicon dioxide (UNII: ETJ7Z6XBU4) dibasic calcium phosphate dihydrate (UNII: O7TSZ97GEP) magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose 101 (UNII: 7T9FYH5QMK) sodium starch glycolate type A potato (UNII: 5856J3G2A2) Product Characteristics Color white (white) Score 2 pieces Shape OVAL (OVAL) Size 11mm Flavor Imprint Code JACOBUS;10;110 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 49938-110-01 1 in 1 CARTON 07/02/2019 1 100 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209321 07/02/2019 Labeler - Jacobus Pharmaceutical Company,Inc (088805734)
 Empty 30 mL Bottle
Empty 30 mL Bottle
 Sterile Water
Sterile Water
 10 mL Oral Syringe with a Catheter Tip
10 mL Oral Syringe with a Catheter Tip
 Place one (1) 10 mg Ruzurgi (amifampridine) tablet in a 30 mL bottle.
Place one (1) 10 mg Ruzurgi (amifampridine) tablet in a 30 mL bottle. Fill an oral syringe with 10 mL of sterile water and inject the contents into the 30 mL bottle.
Fill an oral syringe with 10 mL of sterile water and inject the contents into the 30 mL bottle. Secure the cap onto the bottle and shake well for 30 seconds.
Secure the cap onto the bottle and shake well for 30 seconds. Place three (3) 10 mg Ruzurgi (amilampridine) tablets in a 30 mL bottle.
Place three (3) 10 mg Ruzurgi (amilampridine) tablets in a 30 mL bottle. Fill on oral syringe with 10 mL of sterile water and inject the contents into the 30 mL bottle. This step must be performed for a total of three (3) times to create a volume of 30 mL which is equal to a 30 mg dose.
Fill on oral syringe with 10 mL of sterile water and inject the contents into the 30 mL bottle. This step must be performed for a total of three (3) times to create a volume of 30 mL which is equal to a 30 mg dose. Secure the cap onto the bottle and shake well for 30 seconds.
Secure the cap onto the bottle and shake well for 30 seconds. Remove the bottle cap and use an oral syringe with a catheter tip to measure the prescribed dose.
Remove the bottle cap and use an oral syringe with a catheter tip to measure the prescribed dose. Administer by mouth. For patients requiring more than 10 mg for each dose, repeat steps 4 and 5 until the prescribed dose is given.
Administer by mouth. For patients requiring more than 10 mg for each dose, repeat steps 4 and 5 until the prescribed dose is given. Remove the bottle cap and use an oral syringe with a catheter tip to measure the prescribed dose.
Remove the bottle cap and use an oral syringe with a catheter tip to measure the prescribed dose. Inject the medicine using the oral syringe with a catheter tip into the feeding tube right away. For patients requiring more than 10 mg for each dose, repeat steps 4 and 5 until the prescribed dose is given.
Inject the medicine using the oral syringe with a catheter tip into the feeding tube right away. For patients requiring more than 10 mg for each dose, repeat steps 4 and 5 until the prescribed dose is given. To flush the feeding tube, refill the syringe with 10 mL of sterile water.
To flush the feeding tube, refill the syringe with 10 mL of sterile water. Shake the syringe, insert the catheter tip into the feeding tube to flush any remaining medicine from the feeding tube into the stomach.
Shake the syringe, insert the catheter tip into the feeding tube to flush any remaining medicine from the feeding tube into the stomach.

Trademark Results [RUZURGI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 RUZURGI 87638007 5950658 Live/Registered |
Jacobus Pharmaceutical Co., Inc. 2017-10-09 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.