ARCALYST- rilonacept injection, powder, lyophilized, for solution
Arcalyst by
Drug Labeling and Warnings
Arcalyst by is a Prescription medication manufactured, distributed, or labeled by Regeneron Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ARCALYST safely and effectively. See full prescribing information for ARCALYST.
ARCALYST® (rilonacept)
Injection for Subcutaneous Use
Initial U.S. Approval: 2008INDICATIONS AND USAGE
ARCALYST (rilonacept) is an interleukin-1 blocker indicated for the treatment of Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle-Wells Syndrome (MWS) in adults and children 12 and older. (1)
DOSAGE AND ADMINISTRATION
- Adult patients 18 yrs and older: Initiate treatment with a loading dose of 320 mg delivered as two, 2-mL, subcutaneous injections of 160 mg on the same day at two different sites. Continue dosing with a once-weekly injection of 160 mg administered as a single, 2-mL, subcutaneous injection. Do not administer ARCALYST more often than once weekly. (2)
- Pediatric patients aged 12 to 17 years: Initiate treatment with a loading dose of 4.4 mg/kg, up to a maximum of 320 mg, delivered as one or two subcutaneous injections with a maximum single-injection volume of 2 mL. Continue dosing with a once-weekly injection of 2.2 mg/kg, up to a maximum of 160 mg, administered as a single subcutaneous injection, up to 2 mL. If the initial dose is given as two injections, they should be given on the same day at two different sites. Do not administer ARCALYST more often than once weekly. (2)
DOSAGE FORMS AND STRENGTHS
Sterile, single-use 20-mL, glass vial containing 220 mg of rilonacept as a lyophilized powder for reconstitution. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interleukin-1 blockade may interfere with immune response to infections. Serious, life-threatening infections have been reported in patients taking ARCALYST. Discontinue treatment with ARCALYST if a patient develops a serious infection. Do not initiate treatment with ARCALYST in patients with active or chronic infections. (5.1)
- Hypersensitivity reactions associated with ARCALYST administration have been rare. If a hypersensitivity reaction occurs, discontinue administration of ARCALYST and initiate appropriate therapy. (5.5)
- Live vaccines should not be given concurrently with ARCALYST. Prior to initiation of therapy with ARCALYST, patients should receive all recommended vaccinations. (5.3)
ADVERSE REACTIONS
The most common adverse reactions reported by patients with CAPS treated with ARCALYST are injection-site reactions and upper respiratory tract infections. (6.2, 6.3)
To report SUSPECTED ADVERSE REACTIONS, contact Regeneron at 1-877-REGN-777 (1-877-734-6777) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
No formal drug interaction studies have been conducted with ARCALYST. (7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Dosing
2.3 Preparation for Administration
2.4 Administration
2.5 Stability and Storage
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Immunosuppression
5.3 Immunizations
5.4 Lipid Profile Changes
5.5 Hypersensitivity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Injection-Site Reactions
6.3 Infections
6.4 Malignancies
6.5 Hematologic Events
6.6 Immunogenicity
6.7 Lipid Profiles
7 DRUG INTERACTIONS
7.1 TNF-Blocking Agent and IL-1 Blocking Agent
7.2 Cytochrome P450 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
8.7 Patients with Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.2 Dosing
Adult patients 18 years and older: Treatment should be initiated with a loading dose of 320 mg delivered as two, 2-mL, subcutaneous injections of 160 mg each given on the same day at two different sites. Dosing should be continued with a once-weekly injection of 160 mg administered as a single, 2-mL, subcutaneous injection. ARCALYST should not be given more often than once weekly. Dosage modification is not required based on advanced age or gender.
Pediatric patients aged 12 to 17 years: Treatment should be initiated with a loading dose of 4.4 mg/kg, up to a maximum of 320 mg, delivered as one or two subcutaneous injections with a maximum single-injection volume of 2 mL. Dosing should be continued with a once-weekly injection of 2.2 mg/kg, up to a maximum of 160 mg, administered as a single subcutaneous injection, up to 2 mL. If the initial dose is given as two injections, they should be given on the same day at two different sites. ARCALYST should not be given more often than once weekly.
2.3 Preparation for Administration
Each single-use vial of ARCALYST contains a sterile, white to off-white, preservative-free, lyophilized powder. Reconstitution with 2.3 mL of preservative-free Sterile Water for Injection (supplied separately) is required prior to subcutaneous administration of the drug.
2.4 Administration
Using aseptic technique, withdraw 2.3 mL of preservative-free Sterile Water for Injection through a 27-gauge, ½-inch needle attached to a 3-mL syringe and inject the preservative-free Sterile Water for Injection into the drug product vial for reconstitution. The needle and syringe used for reconstitution with preservative-free Sterile Water for Injection should then be discarded and should not be used for subcutaneous injections. After the addition of preservative-free Sterile Water for Injection, the vial contents should be reconstituted by shaking the vial for approximately one minute and then allowing it to sit for one minute. The resulting 80-mg/mL solution is sufficient to allow a withdrawal volume of up to 2 mL for subcutaneous administration. The reconstituted solution is viscous, clear, colorless to pale yellow, and essentially free from particulates. Prior to injection, the reconstituted solution should be carefully inspected for any discoloration or particulate matter. If there is discoloration or particulate matter in the solution, the product in that vial should not be used.
Using aseptic technique, withdraw the recommended dose volume, up to 2 mL (160 mg), of the solution with a new 27-gauge, ½-inch needle attached to a new 3-mL syringe for subcutaneous injection. EACH VIAL SHOULD BE USED FOR A SINGLE DOSE ONLY. Discard the vial after withdrawal of drug.
Sites for subcutaneous injection, such as the abdomen, thigh, or upper arm, should be rotated. Injections should never be made at sites that are bruised, red, tender, or hard.
2.5 Stability and Storage
The lyophilized ARCALYST product is to be stored refrigerated at 2° to 8°C (36° to 46°F) inside the original carton to protect it from light. Do not use beyond the date stamped on the label. After reconstitution, ARCALYST may be kept at room temperature, should be protected from light, and should be used within three hours of reconstitution. ARCALYST does not contain preservatives; therefore, unused portions of ARCALYST should be discarded.
-
3 DOSAGE FORMS AND STRENGTHS
ARCALYST is supplied in sterile, single-use, 20-mL, glass vials. Each vial contains 220 mg of rilonacept as a white to off-white, preservative-free, lyophilized powder. Reconstitution with 2.3 mL of preservative-free Sterile Water for Injection is required prior to subcutaneous administration of the drug. The reconstituted ARCALYST is a viscous, clear, colorless to pale yellow, essentially free from particulates, 80-mg/mL solution.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
Interleukin-1 (IL-1) blockade may interfere with the immune response to infections. Treatment with another medication that works through inhibition of IL-1 has been associated with an increased risk of serious infections, and serious infections have been reported in patients taking ARCALYST [see Clinical Studies (14)]. There was a greater incidence of infections in patients on ARCALYST compared with placebo. In the controlled portion of the study, one infection was reported as severe, which was bronchitis in a patient on ARCALYST.
In an open-label extension study, one patient developed bacterial meningitis and died [see Adverse Reactions (6.3)]. ARCALYST should be discontinued if a patient develops a serious infection. Treatment with ARCALYST should not be initiated in patients with an active or chronic infection.
In clinical studies, ARCALYST has not been administered concomitantly with tumor necrosis factor (TNF) inhibitors. An increased incidence of serious infections has been associated with administration of an IL-1 blocker in combination with TNF inhibitors. Taking ARCALYST with TNF inhibitors is not recommended because this may increase the risk of serious infections.
Drugs that affect the immune system by blocking TNF have been associated with an increased risk of reactivation of latent tuberculosis (TB). It is possible that taking drugs such as ARCALYST that block IL-1 increases the risk of TB or other atypical or opportunistic infections. Healthcare providers should follow current CDC guidelines both to evaluate for and to treat possible latent tuberculosis infections before initiating therapy with ARCALYST.
5.2 Immunosuppression
The impact of treatment with ARCALYST on active and/or chronic infections and the development of malignancies is not known [see Adverse Reactions (6.3)]. However, treatment with immunosuppressants, including ARCALYST, may result in an increase in the risk of malignancies.
5.3 Immunizations
Since no data are available on either the efficacy of live vaccines or on the risks of secondary transmission of infection by live vaccines in patients receiving ARCALYST, live vaccines should not be given concurrently with ARCALYST. In addition, because ARCALYST may interfere with normal immune response to new antigens, vaccinations may not be effective in patients receiving ARCALYST. No data are available on the effectiveness of vaccination with inactivated (killed) antigens in patients receiving ARCALYST.
Because IL-1 blockade may interfere with immune response to infections, it is recommended that prior to initiation of therapy with ARCALYST adult and pediatric patients receive all recommended vaccinations, as appropriate, including pneumococcal vaccine and inactivated influenza vaccine. (See current Recommended Immunizations schedules at the website of the Centers for Disease Control and Prevention. http://www.cdc.gov/vaccines/schedules/index.html).
5.4 Lipid Profile Changes
Patients should be monitored for changes in their lipid profiles and provided with medical treatment if warranted [see Adverse Reactions (6.7)].
-
6 ADVERSE REACTIONS
Six serious adverse reactions were reported by four patients during the clinical program. These serious adverse reactions were Mycobacterium intracellulare infection; gastrointestinal bleeding and colitis; sinusitis and bronchitis; and Streptococcus pneumoniae meningitis [see Adverse Reactions (6.3)].
The most commonly reported adverse reaction associated with ARCALYST was injection-site reaction (ISR) [see Adverse Reactions (6.2)]. The next most commonly reported adverse reaction was upper respiratory infection [see Adverse Reactions (6.3)].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described herein reflect exposure to ARCALYST in 600 patients, including 85 exposed for at least 6 months and 65 exposed for at least one year. These included patients with CAPS, patients with other diseases, and healthy volunteers. Approximately 60 patients with CAPS have been treated weekly with 160 mg of ARCALYST. The pivotal trial population included 47 patients with CAPS. These patients were between the ages of 22 and 78 years (average 51 years). Thirty-one patients were female and 16 were male. All of the patients were White/Caucasian. Six pediatric patients (12-17 years) were enrolled directly into the open-label extension phase.
6.1 Clinical Trial Experience
Part A of the clinical trial was conducted in patients with CAPS who were naïve to treatment with ARCALYST. Part A of the study was a randomized, double-blind, placebo-controlled, six-week study comparing ARCALYST to placebo [see Clinical Studies (14)]. Table 1 reflects the frequency of adverse events reported by at least two patients during Part A.
Table 1: Most Frequent Adverse Reactions (Part A, Reported by at Least Two Patients) Adverse Event ARCALYST 160 mg
(n = 23)Placebo
(n= 24)Any AE 17 (74%) 13 (54%) Injection-site reactions 11 (48%) 3 (13%) Upper respiratory tract infection 6 (26%) 1 (4%) Nausea 1 (4%) 3 (13%) Diarrhea 1 (4%) 3 (13%) Sinusitis 2 (9%) 1 (4%) Abdominal pain upper 0 2 (8%) Cough 2 (9%) 0 Hypoesthesia 2 (9%) 0 Stomach discomfort 1 (4%) 1 (4%) Urinary tract infection 1 (4%) 1 (4%) 6.2 Injection-Site Reactions
In patients with CAPS, the most common and consistently reported adverse event associated with ARCALYST was injection-site reaction (ISR). The ISRs included erythema, swelling, pruritus, mass, bruising, inflammation, pain, edema, dermatitis, discomfort, urticaria, vesicles, warmth and hemorrhage. Most injection-site reactions lasted for one to two days. No ISRs were assessed as severe, and no patient discontinued study participation due to an ISR.
6.3 Infections
During Part A, the incidence of patients reporting infections was greater with ARCALYST (48%) than with placebo (17%). In Part B, randomized withdrawal, the incidence of infections were similar in the ARCALYST (18%) and the placebo patients (22%). Part A of the trial was initiated in the winter months, while Part B was predominantly performed in the summer months.
In placebo-controlled studies across a variety of patient populations encompassing 360 patients treated with rilonacept and 179 treated with placebo, the incidence of infections was 34% and 27% (2.15 per patient-exposure year and 1.81 per patient-exposure year), respectively, for rilonacept and placebo.
Serious Infections: One patient receiving ARCALYST for an unapproved indication in another study developed an infection in his olecranon bursa with Mycobacterium intracellulare. The patient was on chronic glucocorticoid treatment. The infection occurred after an intraarticular glucocorticoid injection into the bursa with subsequent local exposure to a suspected source of mycobacteria. The patient recovered after the administration of the appropriate antimicrobial therapy. One patient treated for another unapproved indication developed bronchitis/sinusitis, which resulted in hospitalization. One patient died in an open-label study of CAPS from Streptococcus pneumoniae meningitis.
6.5 Hematologic Events
One patient in a study in an unapproved indication developed transient neutropenia (ANC < 1 × 109/L) after receiving a large dose (2000 mg intravenously) of ARCALYST. The patient did not experience any infection associated with the neutropenia.
6.6 Immunogenicity
Antibodies directed against the receptor domains of rilonacept were detected by an ELISA assay in patients with CAPS after treatment with ARCALYST. Nineteen of 55 patients (35%) who had received ARCALYST for at least 6 weeks tested positive for treatment-emergent binding antibodies on at least one occasion. Of the 19, seven tested positive at the last assessment (Week 18 or 24 of the open-label extension period), and five patients tested positive for neutralizing antibodies on at least one occasion. There was no correlation of antibody activity and either clinical effectiveness or safety.
The data reflect the percentage of patients whose test results were positive for antibodies to the rilonacept receptor domains in specific assays, and are highly dependent on the sensitivity and specificity of the assays. The observed incidence of antibody (including neutralizing antibody) positivity in an assay is highly dependent on several factors including assay sensitivity and specificity, assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to rilonacept with the incidence of antibodies to other products may be misleading.
6.7 Lipid Profiles
Cholesterol and lipid levels may be reduced in patients with chronic inflammation. Patients with CAPS treated with ARCALYST experienced increases in their mean total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides. The mean increases from baseline for total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides were 19 mg/dL, 2 mg/dL, 10 mg/dL, and 57 mg/dL respectively after 6 weeks of open-label therapy. Physicians should monitor the lipid profiles of their patients (for example after 2-3 months) and consider lipid-lowering therapies as needed based upon cardiovascular risk factors and current guidelines.
-
7 DRUG INTERACTIONS
7.1 TNF-Blocking Agent and IL-1 Blocking Agent
Specific drug interaction studies have not been conducted with ARCALYST. Concomitant administration of another drug that blocks IL-1 with a TNF-blocking agent in another patient population has been associated with an increased risk of serious infections and an increased risk of neutropenia. The concomitant administration of ARCALYST with TNF-blocking agents may also result in similar toxicities and is not recommended [see Warnings and Precautions (5.1)]. The concomitant administration of ARCALYST with other drugs that block IL-1 has not been studied. Based upon the potential for pharmacologic interactions between rilonacept and a recombinant IL-1ra, concomitant administration of ARCALYST and other agents that block IL-1 or its receptors is not recommended.
7.2 Cytochrome P450 Substrates
The formation of CYP450 enzymes is suppressed by increased levels of cytokines (e.g., IL-1) during chronic inflammation. Thus it is expected that for a molecule that binds to IL-1, such as rilonacept, the formation of CYP450 enzymes could be normalized. This is clinically relevant for CYP450 substrates with a narrow therapeutic index, where the dose is individually adjusted (e.g., warfarin). Upon initiation of ARCALYST, in patients being treated with these types of medicinal products, therapeutic monitoring of the effect or drug concentration should be performed and the individual dose of the medicinal product may need to be adjusted as needed.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Rare pregnancy outcomes reported postmarketing and from clinical trials, with very limited use of ARCALYST in pregnant women, are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There may be risks to the mother and fetus associated with Cryopyrin Associated Periodic Syndromes (CAPS) (see Clinical Considerations). In an animal reproduction study, subcutaneous administration of rilonacept to pregnant monkeys during the period of organogenesis was complicated by losses of drug exposure as the study progressed, due to anti-drug antibody formation at all doses, but a dose-related increase in exposure was still evident. There were no treatment-related effects on fetal survival or development of malformations with doses up to 11 times the maximum recommended human dose (MRHD). Increased incidences of lumbar ribs, a skeletal variation, were observed in fetuses at doses approximately 2 times the MRHD and higher that slightly exceeded incidences in both control animals and the historical control database (see Data). There were findings of multiple fusion and absence of the ribs and thoracic vertebral bodies and arches in one fetus of the only pregnant monkey with exposure to rilonacept during the later period of gestation associated with a dose approximately 6 times the MRHD (see Data). The relationship of these findings in a single fetus to drug treatment was unclear, as these findings were not evident in fetuses from pregnant monkeys that had higher exposures to rilonacept during the period of organogenesis associated with a dose approximately 11 times the MRHD.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and of miscarriage is 15–20%, respectively.
Data
Animal Data
In an embryo-fetal development study, pregnant cynomolgus monkeys received rilonacept at subcutaneous doses of 0, 5, 15 or 30 mg/kg given twice a week from gestation days 20 to 48. The study was complicated by losses of drug exposure as the study progressed, due to anti-drug antibody formation at all doses, but a dose-related increase in exposure was still evident. There were no treatment-related effects on fetal survival or development of malformations with doses up to 11 times the MRHD (on a mg/kg basis with maternal subcutaneous doses up to 30 mg/kg). Increased incidences of lumbar ribs, a skeletal variation, were observed in fetuses at doses approximately 2 times the MRHD and higher (on a mg/kg basis with maternal subcutaneous doses of 5 mg/kg and higher) that slightly exceeded incidences in both control animals and the historical control database. There were findings of multiple fusion and absence of the ribs and thoracic vertebral bodies and arches in one fetus of the only pregnant monkey with exposure to rilonacept during the later period of gestation associated with a dose 6 times the MRHD (on a mg/kg basis with a maternal subcutaneous dose of 15 mg/kg). The relationship of these findings in a single fetus to drug treatment was unclear, as these findings were not evident in fetuses from pregnant monkeys that had higher exposures to rilonacept during the period of organogenesis associated with a dose approximately 11 times the MRHD (on a mg/kg basis with a maternal subcutaneous dose of 30 mg/kg). All doses of rilonacept reduced maternal serum levels of estradiol up to 64% compared to controls. In pre- and postnatal development studies in the mouse model using a murine analog of rilonacept (subcutaneous doses of 0, 20, 100 or 200 mg/kg), there was a small increase in the number of stillbirths in dams treated with 200 mg/kg three times per week.
8.2 Lactation
Risk Summary
There is no information on the presence of rilonacept in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ARCALYST and any potential adverse effects on the breastfed child from ARCALYST or from the underlying maternal condition.
8.4 Pediatric Use
Six pediatric patients with CAPS between the ages of 12 and 16 were treated with ARCALYST at a weekly, subcutaneous dose of 2.2 mg/kg (up to a maximum of 160 mg) for 24-weeks during the open-label extension phase. These patients showed improvement from baseline in their symptom scores and in objective markers of inflammation (e.g. Serum Amyloid A and C-Reactive Protein). The adverse events included injection site reactions and upper respiratory symptoms as were commonly seen in the adult patients.
The trough drug levels for four pediatric patients measured at the end of the weekly dose interval (mean 20 mcg/mL, range 3.6 to 33 mcg/mL) were similar to those observed in adult patients with CAPS (mean 24 mcg/mL, range 7 to 56 mcg/mL).
Safety and effectiveness in pediatric patients below the age of 12 have not been established.
When administered to pregnant primates, rilonacept treatment may have contributed to alterations in bone ossification in the fetus. It is not known if ARCALYST will alter bone development in pediatric patients. Pediatric patients treated with ARCALYST should undergo appropriate monitoring for growth and development. [see Use in Specific Populations (8.1)]
8.5 Geriatric Use
In the placebo-controlled clinical studies in patients with CAPS and other indications, 70 patients randomized to treatment with ARCALYST were ≥ 65 years of age, and 6 were ≥ 75 years of age. In the CAPS clinical trial, efficacy, safety and tolerability were generally similar in elderly patients as compared to younger adults; however, only ten patients ≥ 65 years old participated in the trial. In an open-label extension study of CAPS, a 71 year old woman developed bacterial meningitis and died [see Adverse Reactions (6.3)]. Age did not appear to have a significant effect on steady-state trough concentrations in the clinical study.
-
10 OVERDOSAGE
There have been no reports of overdose with ARCALYST. Maximum weekly doses of up to 320 mg have been administered subcutaneously for up to approximately 18 months in a small number of patients with CAPS and up to 6 months in patients with an unapproved indication in clinical trials without evidence of dose-limiting toxicities. In addition, ARCALYST given intravenously at doses up to 2000 mg monthly in another patient population for up to six months were tolerated without dose-limiting toxicities. The maximum amount of ARCALYST that can be safely administered has not been determined.
In case of overdose, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects, and appropriate symptomatic treatment instituted immediately.
-
11 DESCRIPTION
Rilonacept is a dimeric fusion protein consisting of the ligand-binding domains of the extracellular portions of the human interleukin-1 receptor component (IL-1RI) and IL-1 receptor accessory protein (IL-1RAcP) linked in-line to the Fc portion of human IgG1. Rilonacept has a molecular weight of approximately 251 kDa. Rilonacept is expressed in recombinant Chinese hamster ovary (CHO) cells.
ARCALYST is supplied in single-use, 20-mL glass vials containing a sterile, white to off-white, lyophilized powder. Each vial of ARCALYST is to be reconstituted with 2.3 mL of Sterile Water for Injection. A volume of up to 2 mL can be withdrawn, which is designed to deliver 160 mg for subcutaneous administration only. The resulting solution is viscous, clear, colorless to pale yellow, and essentially free from particulates. Each vial contains 220 mg rilonacept. After reconstitution, each vial contains 80 mg/mL rilonacept, 46 mM histidine, 50 mM arginine, 3.0% (w/v) polyethylene glycol 3350, 2.0% (w/v) sucrose, and 1.0% (w/v) glycine at a pH of 6.5 ± 0.3. No preservatives are present.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CAPS refer to rare genetic syndromes generally caused by mutations in the NLRP-3 [Nucleotide-binding domain, leucine rich family (NLR), pyrin domain containing 3] gene (also known as Cold-Induced Auto-inflammatory Syndrome-1 [CIAS1]). CAPS disorders are inherited in an autosomal dominant pattern with male and female offspring equally affected. Features common to all disorders include fever, urticaria-like rash, arthralgia, myalgia, fatigue, and conjunctivitis.
In most cases, inflammation in CAPS is associated with mutations in the NLRP-3 gene which encodes the protein cryopyrin, an important component of the inflammasome. Cryopyrin regulates the protease caspase-1 and controls the activation of interleukin-1 beta (IL-1β). Mutations in NLRP-3 result in an overactive inflammasome resulting in excessive release of activated IL-1β that drives inflammation.
Rilonacept blocks IL-1β signaling by acting as a soluble decoy receptor that binds IL-1β and prevents its interaction with cell surface receptors. Rilonacept also binds IL-1α and IL-1 receptor antagonist (IL-1ra) with reduced affinity. The equilibrium dissociation constants for rilonacept binding to IL-1β, IL-1α and IL-1ra were 0.5 pM, 1.4 pM and 6.1 pM, respectively.
12.2 Pharmacodynamics
C-Reactive Protein (CRP) and Serum Amyloid A (SAA) are indicators of inflammatory disease activity that are elevated in patients with CAPS. Elevated SAA has been associated with the development of systemic amyloidosis in patients with CAPS. Compared to placebo, treatment with ARCALYST resulted in sustained reductions from baseline in mean serum CRP and SAA to normal levels during the clinical trial. ARCALYST also normalized mean SAA from elevated levels.
12.3 Pharmacokinetics
The average trough levels of rilonacept were approximately 24 mcg/mL at steady-state following weekly subcutaneous doses of 160 mg for up to 48 weeks in patients with CAPS. The steady-state appeared to be reached by 6 weeks.
No pharmacokinetic data are available in patients with hepatic or renal impairment.
No study was conducted to evaluate the effect of age, gender, or body weight on rilonacept exposure. Based on limited data obtained from the clinical study, steady state trough concentrations were similar between male and female patients. Age (26-78 years old) and body weight (50-120 kg) did not appear to have a significant effect on trough rilonacept concentrations. The effect of race could not be assessed because only Caucasian patients participated in the clinical study, reflecting the epidemiology of the disease.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies have not been performed to evaluate the carcinogenic potential of rilonacept.
A murine analog of rilonacept had no effects on fertility and reproductive performance in male and female mice at subcutaneous doses up to 200 mg/kg three times per week.
-
14 CLINICAL STUDIES
The safety and efficacy of ARCALYST for the treatment of CAPS was demonstrated in a randomized, double-blind, placebo-controlled study with two parts (A and B) conducted sequentially in the same patients with FCAS and MWS.
Part A was a 6-week, randomized, double-blind, parallel-group period comparing ARCALYST at a dose of 160 mg weekly after an initial loading dose of 320 mg to placebo. Part B followed immediately after Part A and consisted of a 9-week, patient-blind period during which all patients received ARCALYST 160 mg weekly, followed by a 9-week, double-blind, randomized withdrawal period in which patients were randomly assigned to either remain on ARCALYST 160 mg weekly or to receive placebo. Patients were then given the option to enroll in a 24-week, open-label treatment extension phase in which all patients were treated with ARCALYST 160 mg weekly.
Using a daily diary questionnaire, patients rated the following five signs and symptoms of CAPS: joint pain, rash, feeling of fever/chills, eye redness/pain, and fatigue, each on a scale of 0 (none, no severity) to 10 (very severe). The study evaluated the mean symptom score using the change from baseline to the end of treatment.
The changes in mean symptom scores for the randomized parallel-group period (Part A) and the randomized withdrawal period (Part B) of the study are shown in Table 2. ARCALYST-treated patients had a larger reduction in the mean symptom score in Part A compared to placebo-treated patients. In Part B, mean symptom scores increased more in patients withdrawn to placebo compared to patients who remained on ARCALYST.
Table 2: Mean Symptom Scores Part A Placebo
(n=24)ARCALYST
(n=23)Part B Placebo
(n=23)ARCALYST
(n=22)- * Differences are adjusted using an analysis of covariance model with terms for treatment and Part A baseline.
- † A confidence interval lying entirely below zero indicates a statistical difference favoring ARCALYST versus placebo.
Pre-treatment Baseline Period
(Weeks -3 to 0)2.4 3.1 Active ARCALYST Baseline Period
(Weeks 13 to 15)0.2 0.3 Endpoint Period
(Weeks 4 to 6)2.1 0.5 Endpoint Period
(Weeks 22 to 24)1.2 0.4 LS* Mean Change from Baseline to Endpoint -0.5 -2.4 LS* Mean Change from Baseline to Endpoint 0.9 0.1 95% confidence interval for difference between treatment groups (-2.4, -1.3)† 95% confidence interval for difference between treatment groups (-1.3, -0.4)† Daily mean symptom scores over time for Part A are shown in Figure 1.
Figure 1: Group Mean Daily Symptom Scores by Treatment Group in Part A and Single-blind ARCALYST Treatment Phase from Week -3 to Week 15
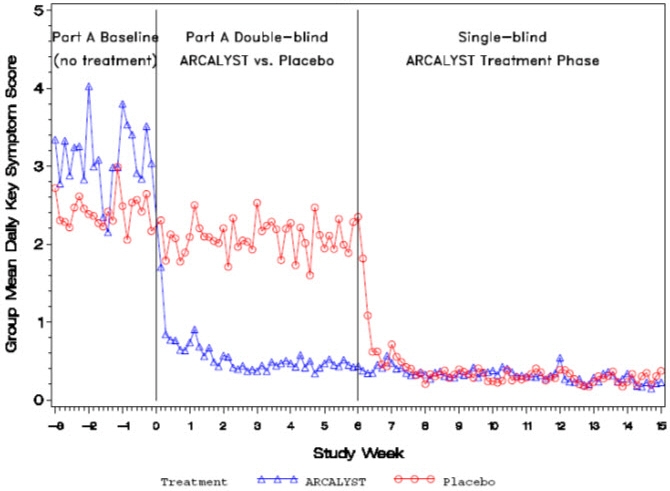
Improvement in symptom scores was noted within several days of initiation of ARCALYST therapy in most patients.
In Part A, patients treated with ARCALYST experienced more improvement in each of the five components of the composite endpoint (joint pain, rash, feeling of fever/chills, eye redness/pain, and fatigue) than placebo-treated patients.
In Part A, a higher proportion of patients in the ARCALYST group experienced improvement from baseline in the composite score by at least 30% (96% vs. 29% of patients), by at least 50% (87% vs. 8%) and by at least 75% (70% vs. 0%) compared to the placebo group.
Serum Amyloid A (SAA) and C-Reactive Protein (CRP) levels are acute phase reactants that are typically elevated in patients with CAPS with active disease. During Part A, mean levels of CRP decreased versus baseline for the ARCALYST treated patients, while there was no change for those on placebo (Table 3). ARCALYST also led to a decrease in SAA versus baseline to levels within the normal range.
Table 3. Mean Serum Amyloid A and C-Reactive Protein Levels Over Time in Part A Part A ARCALYST Placebo SAA
(normal range: 0.7 – 6.4 mg/L)(n=22) (n=24) Pre-treatment Baseline 60 110 Week 6 4 110 CRP
(normal range: 0.0 – 8.4 mg/L)(n= 21) (n=24) Pre-treatment Baseline 22 30 Week 6 2 28 During the open-label extension, reductions in mean symptom scores, serum CRP, and serum SAA levels were maintained for up to one year.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each 20-mL glass vial of ARCALYST contains a sterile, white to off-white, preservative-free, lyophilized powder. ARCALYST is supplied in a carton containing four vials (NDC: 61755-001-01).
The lyophilized ARCALYST product is to be stored refrigerated at 2° to 8°C (36° to 46°F) inside the original carton to protect from light. Do not use beyond the date stamped on the label. After reconstitution, ARCALYST may be kept at room temperature, should be kept from light, and should be used within three hours of reconstitution. ARCALYST does not contain preservatives; therefore, unused portions of ARCALYST should be discarded. Discard the vial after a single withdrawal of drug.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling.
The first injection of ARCALYST should be performed under the supervision of a qualified healthcare professional. If a patient or caregiver is to administer ARCALYST, he/she should be instructed on aseptic reconstitution of the lyophilized product and injection technique. The ability to inject subcutaneously should be assessed to ensure proper administration of ARCALYST, including rotation of injection sites. (See Patient Information Leaflet for ARCALYST®). ARCALYST should be reconstituted with preservative-free Sterile Water for Injection to be provided by the pharmacy. A sharps disposal container should be used for disposal of vials, needles and syringes. Patients or caregivers should be instructed in proper vial, syringe, and needle disposal, and should be cautioned against reuse of these items.
Injection-site Reactions: Physicians should explain to patients that almost half of the patients in the clinical trials experienced a reaction at the injection site. Injection-site reactions may include pain, erythema, swelling, pruritus, bruising, mass, inflammation, dermatitis, edema, urticaria, vesicles, warmth, and hemorrhage. Patients should be cautioned to avoid injecting into an area that is already swollen or red. Any persistent reaction should be brought to the attention of the prescribing physician.
Infections: Patients should be cautioned that ARCALYST has been associated with serious, life-threatening infections, and not to initiate treatment with ARCALYST if they have a chronic or active infection. Patients should be counseled to contact their healthcare professional immediately if they develop an infection after starting ARCALYST. Treatment with ARCALYST should be discontinued if a patient develops a serious infection. Patients should be counseled not to take any IL-1 blocking drug, including ARCALYST, if they are also taking a drug that blocks TNF such as etanercept, infliximab, or adalimumab. Use of ARCALYST with other IL-1 blocking agents, such as anakinra, is not recommended.
Vaccinations: Prior to initiation of therapy with ARCALYST physicians should review with adult and pediatric patients their vaccination history relative to current medical guidelines for vaccine use, including taking into account the potential of increased risk of infection during treatment with ARCALYST.
-
SPL UNCLASSIFIED SECTION
Manufactured and distributed by:
Regeneron Pharmaceuticals, Inc.
777 Old Saw Mill River Road,
Tarrytown, NY 10591-6707, 1-877-REGN-777 (1-877-734-6777)
U.S. License Number 1760
NDC: 61755-001-01
© 2020, Regeneron Pharmaceuticals, Inc.
All rights reserved.
-
PATIENT PACKAGE INSERT
Patient Information
ARCALYST® (ARK-a-list)
(rilonacept)
Injection for Subcutaneous UseRead the Patient Information that comes with ARCALYST before you start taking it and each time you get a refill. There may be new information. This Patient Information leaflet does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about ARCALYST?
ARCALYST can affect your immune system. ARCALYST can lower the ability of your immune system to fight infections. Serious infections, including life-threatening infections and death have happened in patients taking ARCALYST. Taking ARCALYST can make you more likely to get infections, including life-threatening serious infections, or may make any infection that you have worse.
You should not begin treatment with ARCALYST if you have an infection or have infections that keep coming back (chronic infection).
After starting ARCALYST, if you get an infection, any sign of an infection including a fever, cough, flu-like symptoms, or have any open sores on your body, call your healthcare provider right away. Treatment with ARCALYST should be stopped if you get a serious infection.
You should not take medicines that block tumor necrosis factor (TNF), such as Enbrel® (etanercept), Humira® (adalimumab), or Remicade® (infliximab), while you are taking ARCALYST. You should also not take other medicines that block interleukin-1 (IL-1), such as Kineret® (anakinra), while taking ARCALYST. Taking ARCALYST with any of these medicines may increase your risk of getting a serious infection.
Before starting treatment with ARCALYST, tell your healthcare provider if you:
- think you have an infection.
- are being treated for an infection.
- have signs of an infection, such as fever, cough, or flu-like symptoms.
- have any open sores on your body.
- have a history of infections that keep coming back.
- have asthma. Patients with asthma may have an increased risk of infection.
- have diabetes or an immune system problem. People with these problems have a higher chance for infections.
- have tuberculosis (TB), or if you have been in close contact with someone who has had tuberculosis.
- have or have had HIV, Hepatitis B, or Hepatitis C.
- take other medicines that affect your immune system.
Before you begin treatment with ARCALYST, talk with your healthcare provider about your vaccine history. Ask your healthcare provider whether you should receive any vaccines, including the pneumonia vaccine and flu vaccine, before you begin treatment with ARCALYST.
What is ARCALYST?
- ARCALYST is a prescription medicine called an interleukin-1 (IL-1) blocker.
- ARCALYST is used to treat adults and children 12 years and older with Cryopyrin-Associated Periodic Syndromes (CAPS), including Familial Cold Auto-inflammatory Syndrome (FCAS) and Muckle Wells Syndrome (MWS).
It is not known if ARCALYST is safe and effective in children under 12 years of age.
What should I tell my healthcare provider before taking ARCALYST? ARCALYST may not be right for you. Before taking ARCALYST, tell your healthcare provider about all of your medical conditions, including if you:
- are scheduled to receive any vaccines. You should not receive live vaccines if you take ARCALYST. See "What is the most important information I should know about ARCALYST?"
- are pregnant or plan to become pregnant. It is not known if ARCALYST will harm your unborn child. Tell your healthcare provider right away if you become pregnant while taking ARCALYST.
- are breastfeeding or plan to breastfeed. It is not known if ARCALYST passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with ARCALYST.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take other medicines that affect your immune system, such as:
- See "What is the most important information I should know about ARCALYST?"
- corticosteroids
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist every time you get a new prescription.
If you have any questions about any of this information, ask your healthcare provider.
How should I take ARCALYST?
See the Instructions for Use at the end of this Patient Information leaflet.
- Take ARCALYST exactly as prescribed by your healthcare provider.
- ARCALYST is given by injection under the skin (subcutaneous injection) 1 time each week.
- Your healthcare provider will tell you how much ARCALYST to take and show you or your caregiver how to prepare and give the injection.
- Do not try to give ARCALYST injections until you are sure that you or your caregiver understands how to prepare and inject your dose. Call your healthcare provider or pharmacist if you have any questions, or if you or your caregiver would like more training.
- If you take too much ARCALYST, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of ARCALYST?
ARCALYST can cause serious side effects, including:
- See "What is the most important information I should know about taking ARCALYST?"
- Allergic Reaction. Stop taking ARCALYST and call your healthcare provider or get emergency care right away if you get any of the following symptoms of an allergic reaction while taking ARCALYST:
- rash
- swollen face
- trouble breathing
- Changes in your blood cholesterol and triglycerides (lipids). Your healthcare provider will check blood tests for this.
The most common side effects of ARCALYST include:
- Injection-site reactions including: pain, redness, swelling, itching, bruising, lumps, inflammation, skin rash, blisters, warmth, and bleeding at the injection site.
- Upper respiratory infection.
These are not all the possible side effects of ARCALYST. Tell your healthcare provider if you have any side effects that bothers you or that does not go away. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ARCALYST?
- Keep ARCALYST in the carton it comes in to protect from light.
- Store ARCALYST in the refrigerator between 36°F to 46°F (2°C to 8°C). Call your pharmacy if you have any questions.
- Refrigerated ARCALYST can be used until the expiration date printed on the vial and carton.
- ARCALYST may be kept at room temperature after mixing and should be used within 3 hours of mixing. Keep ARCALYST away from light.
Keep ARCALYST, injection supplies, and all other medicines out of reach of children.
General Information about the safe and effective use of ARCALYST.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ARCALYST for a condition for which it was not prescribed. Do not give ARCALYST to other people even if they have the same symptoms that you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about ARCALYST that is written for health professionals
What are the ingredients in ARCALYST?
Active ingredient: rilonacept.
Inactive ingredients: histidine, arginine, polyethylene glycol 3350, sucrose, and glycine.
-
Instructions for UseARCALYST® (ARK-a-list)(rilonacept)Injection for Subcutaneous Use
It is important that you read, understand and follow these instructions before using ARCALYST so that you prepare and inject the medicine the right way.
Do not try to inject ARACLYST until you have been shown the right way to give the injections by your healthcare provider.
How do I prepare and give an ARCALYST injection?
Step 1: Setting up for an injection
- Choose a table or other flat area to set up the supplies for your injection. Be sure the area is clean or clean it with an antiseptic or soap and water.
- Wash your hands well with soap and water, and dry with a clean towel.
- Put the following supplies on the cleaned area for each injection (see Figure 1):
Supplies needed to give your ARCALYST injection:
- 1 vial of ARCALYST (powder for mixing)
Additional supplies needed (available from the pharmacy):
- 1 vial of preservative-free Sterile Water for Injection
- 2 sterile, 3-milliliter (mL) disposable syringes with markings at each 0.1 mL (see Figure 2):
- 1 syringe for mixing ARCALYST
- 1 syringe for injection
- 2 sterile disposable needles with needle covers (27-gauge, ½-inch)
- 1 needle for mixing ARCALYST
- 1 needle for injection
- 4 alcohol wipes
- 1 gauze pad
- 1 sharps disposal container for throwing away (disposing of) used needles, syringes, and vials (see Figure 22)
Note:
- Do not use Sterile Water for Injection, syringes or needles other than those provided by your pharmacy. Contact your pharmacy if you need replacement syringes or needles.
- Do not touch the needles or the rubber stoppers on the vials with your hands. If you touch the rubber stopper, clean it with a new alcohol wipe.
- If you touch a needle or the needle touches any surface, throw away the entire syringe in the sharps disposal container and use a new syringe.
- Do not reuse needles or syringes.
- To protect yourself and others from possible needle sticks, it is very important to throw away each syringe with the needle attached, in the sharps disposal container right after use. Do not try to recap the needle.
Step 2: Preparing the vials
- Check the expiration date on the carton of ARCALYST. Do not use the vial if the expiration date has passed. Contact your pharmacy for assistance.
- Check the expiration date on the vial of Sterile Water for Injection. Do not use the vial if the expiration date has passed. Contact your pharmacy for assistance.
- Remove the protective plastic cap from both vials.
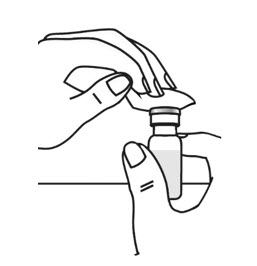
- Clean the top of each vial with an alcohol wipe. Use 1 wipe for each vial and wipe in 1 direction around the top of the vial (see Figure 3).
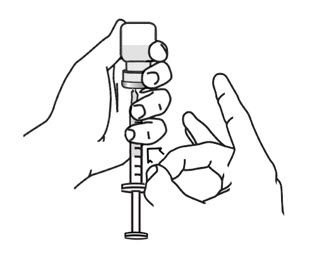
- Open the wrapper that contains the needle by pulling apart the tabs and set it aside for later use. Do not remove the needle cover. This needle will be used to mix the water with ARCALYST powder in the vial. Open the wrapper that contains the syringe by pulling apart the tabs (see Figure 4). Hold the barrel of the syringe with 1 hand and use your other hand to twist the needle onto the tip of the syringe until it fits firmly (see Figure 4).
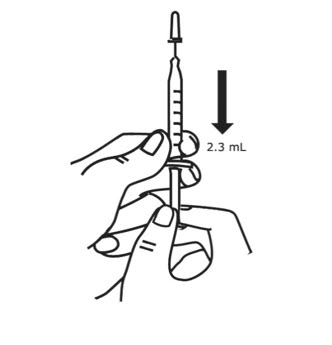
- Hold the syringe at eye level. With the needle cover still on the needle, pull back the plunger to the 2.3 mL mark to fill the syringe with air (see Figure 5).
- Hold the syringe in 1 hand and use the other hand to pull the needle cover straight off. Do not twist the needle as you pull off the cover. Place the needle cover aside. Hold the syringe in the hand that you will use to mix your medicine. Hold the Sterile Water for Injection vial on a firm surface with your other hand. Slowly insert the needle straight through the rubber stopper. Do not bend the needle. Push the plunger in all the way to push the air into the vial (see Figure 6).
- Hold the vial in 1 hand and the syringe in the other hand and carefully turn the vial upside down so that the needle is pointing straight up.
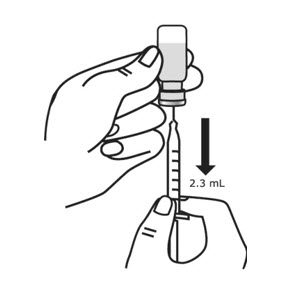
- Make sure the tip of the needle is covered by the liquid and slowly pull back on the plunger to the 2.3 mL mark to withdraw the Sterile Water for Injection from the vial (see Figure 7).
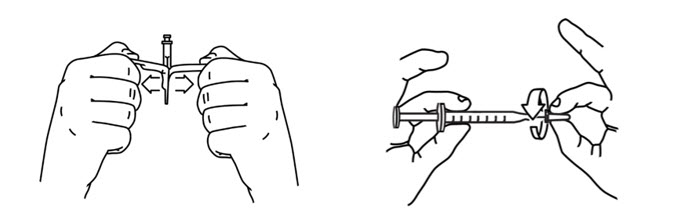
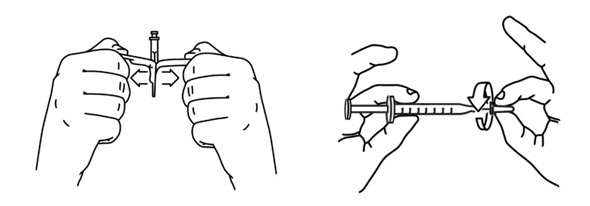
- Keep the vial upside down and tap the syringe with your fingers until any air bubbles rise to the top of the syringe.
- To remove the air bubbles, gently push in the plunger so only the air is pushed out of the syringe and back into the bottle.
- After removing the air bubbles, check the syringe to be sure that the right amount of Sterile Water for Injection has been drawn into the syringe (see Figure 8).
- Carefully remove the syringe with the needle from the Sterile Water for Injection vial. Do not touch the needle.
Step 3: Mixing ARCALYST
- With 1 hand, hold the ARCALYST vial on a firm surface.
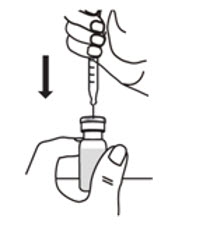
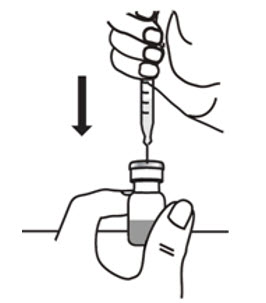
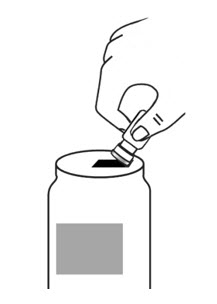
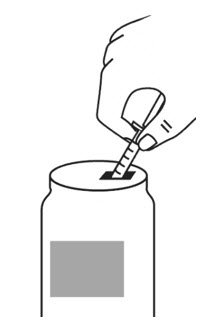
- With the other hand, take the syringe with the Sterile Water for Injection and the same needle, and slowly insert the needle straight down through the rubber stopper of the ARCALYST vial. Push the plunger in all the way to inject the Sterile Water for Injection into the vial.
- Aim the stream of Sterile Water for Injection to gently go down the side of the vial into the powder (see Figure 9).
- Remove the syringe and needle from the stopper and throw away the needle, syringe, and Sterile Water for Injection vial in the sharps disposal container. Do not try to put the needle cover back on the needle.
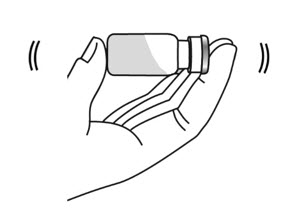
- Hold the vial containing the ARCALYST and Sterile Water for Injection sideways (not upright) with your thumb and a finger at the top and bottom of the vial, and quickly shake the vial back and forth (side-to-side) for about 1 minute (see Figure 10).
- Put the vial back on the table and let the vial sit for about 1 minute.
- Check the vial for any particles or clumps of powder which have not dissolved.
- If the powder has not completely dissolved, shake the vial quickly back and forth for 30 seconds more. Let the vial sit for about 1 minute.
- Repeat Step 8 until the powder is completely dissolved and the solution is clear (see Figure 11).
- The mixed ARCALYST should be thick, clear, and colorless to pale yellow. Do not use the mixed liquid if it is discolored or cloudy, or if particles are in it.
Note: Contact your pharmacy to report any mixed ARCALYST that is discolored, cloudy or contains particles. - ARCALYST may be kept at room temperature after mixing. ARCALYST should be used within 3 hours of mixing. Keep ARCALYST away from light.
Step 4: Preparing the injection
- Hold the ARCALYST vial on a firm surface and wipe the top of the ARCALYST vial with a new alcohol wipe (see Figure 11).
- Take a new sterile, disposable needle and attach it firmly to a new syringe without removing the needle cover (see Figure 12).
- The amount of air you draw into the syringe should equal the amount of mixed ARCALYST that your healthcare provider has prescribed for you to inject.
- To draw air into the syringe, hold the syringe at eye level. Do not remove the needle cover. Pull back the plunger on the syringe to the mark that is equal to the amount of mixed ARCALYST that your healthcare provider has prescribed for you to inject (see Figure 13).
- Remove the needle cover and be careful not to touch the needle. Keep the ARCALYST vial on a flat surface and slowly insert the needle straight down through the stopper. Push the plunger down and inject all of the air into the vial (see Figure 14).
Figure 14
- Hold the vial in 1 hand and the syringe in the other hand and carefully turn the vial upside down so that the needle is pointing straight up. Hold the vial at eye level.
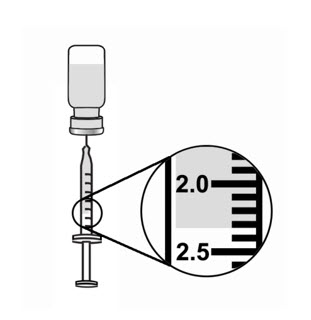
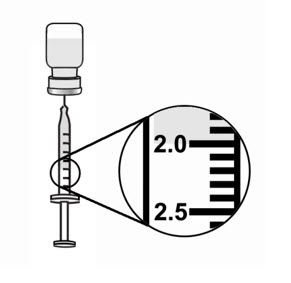
- Keep the tip of the needle in the liquid and slowly pull back on the plunger to the mark on the syringe that matches the amount of medicine prescribed by your healthcare provider (see Figure 15).
Note: The maximum adult dose of ARCALYST is 2 mL. - Keep the vial upside down with the needle straight up, and gently tap the syringe until any air bubbles rise to the top of the syringe (see Figure 16).
It is important to remove air bubbles so that you withdraw the right amount of medicine from the vial. - To remove the air bubbles, slowly and gently push in the plunger so only the air is pushed through the needle.
- Check to make sure that you have the amount of medicine prescribed by your healthcare provider in the syringe.
- Throw away the ARCALYST vial in the sharps disposal container even if there is any medicine left in the vial (see Figure 17). Do not use any vial of ARCALYST more than 1 time.
Step 5: Giving the Injection
- ARCALYST is given by injection into the tissue directly under the skin (subcutaneous injection). Do not inject ARCALYST into any muscle, vein, or artery.
You should change (rotate) the injection site each time you inject ARCALYST.
Changing injection sites helps to prevent irritation and allows the medicine to be completely absorbed. Ask your healthcare provider if you have any questions about changing injection sites.- Do not inject into skin that is tender, red, or hard. If an area is tender or feels hardened, choose another site for injection until the tenderness or hardening goes away.
- Tell your healthcare provider about any skin reactions including redness, swelling, or hardening of the skin.
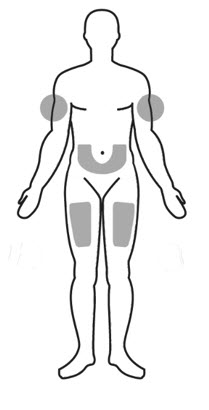
- Areas where you may inject ARCALYST include the left and right sides of the abdomen, and the left and right thighs. If someone else is giving the injection, the upper left and right arms may also be used for injection (see Figure 18):
Do not inject within a 2-inch area around the belly button.
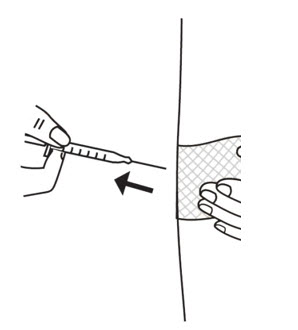
- Choose the area for the injection. Clean the area in a circular motion with a new alcohol wipe. Begin at the center of the injection site and move outward. Let the alcohol air dry completely.
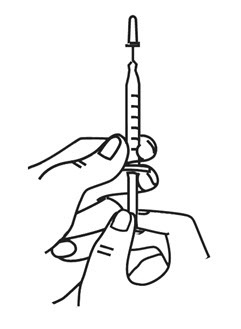
- Hold the syringe in 1 hand like you would hold a pencil.
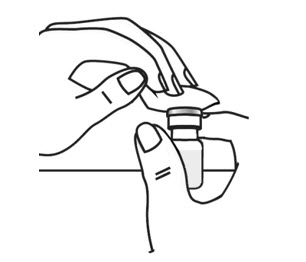
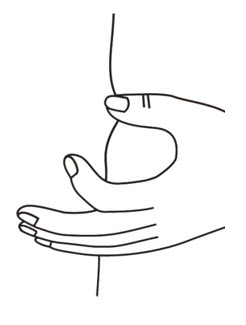
- With the other hand gently pinch a fold of skin at the cleaned injection site (see Figure 19).
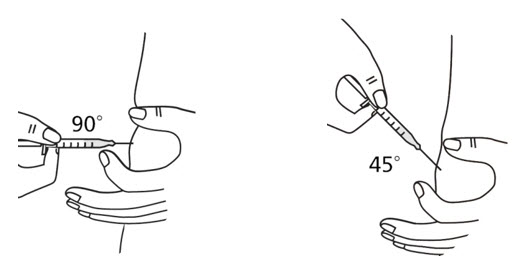
- Use a quick "dart like" motion to insert the needle straight into the skin at a 90 degree angle (see Figure 20). Do not push down on the plunger while inserting the needle into the skin.
For small children or people with little fat under the skin, you may need to hold the syringe and needle at a 45 degree angle (see Figure 20). - After the needle is completely in the skin, let go of the pinched skin.
- With your free hand hold the syringe near the bottom. Gently pull back the plunger. If blood comes into the syringe, the needle has entered a blood vessel. Remove the needle and throw away (discard) the syringe and needle. Start over with "Step 1: Setting up for an injection" using new supplies (syringes, needles, vials, alcohol swabs and gauze pad).
- If no blood comes into the syringe, inject all the medicine in the syringe at a slow, steady rate, pushing the plunger all the way down. It may take up to 30 seconds to inject the entire dose.
- Pull the needle out of the skin and hold a gauze pad over the injection site for several seconds (see Figure 21).
- Do not replace the needle cover. Throw away the vials, used syringes and needles in a FDA-cleared sharps disposal container (see Figure 22). Do not throw away vials, needles, or syringes in the household trash or recycle.
If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal- Do not reuse or share your syringes with other people.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
- Keep the sharps disposal container out of the reach of children.
- Used alcohol wipes can be thrown away in the household trash.
Contact your healthcare provider right away with any questions or concerns about ARCALYST.
This Patient Information and Instructions for Use have been approved by the U.S. Food and Drug Administration.
Notes: 1. Enbrel®, Humira®, Kineret®, and Remicade®, respectively, are trademarks of Immunex Corporation, AbbVie Biotechnology Ltd., Amgen Inc., and Janssen Biotech, Inc., respectively.
Manufactured and distributed by:
Regeneron Pharmaceuticals, Inc.
777 Old Saw Mill River Road
Tarrytown, NY 10591-6707U.S. License Number 1760
NDC: 61755-001-01
For more information about ARCALYST, call 1-877-REGN-777 (1-877-734-6777), or visit
www.ARCALYST.com.© 2020, Regeneron Pharmaceuticals, Inc.
All rights reserved.
Revised: 02/2020
-
PRINCIPAL DISPLAY PANEL - 220 mg Vial Carton
NDC: 61755-001-01
Arcalyst®
(rilonacept)
Injection for Subcutaneous Use220 mg sterile powder
for reconstitutionStore at 2-8°C (36-46°F) until use.
Protect from light.Contents: four (4) single-use vials
Rx ONLY
REGENERON
-
INGREDIENTS AND APPEARANCE
ARCALYST
rilonacept injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61755-001 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength rilonacept (UNII: 8K80YB5GMG) (rilonacept - UNII:8K80YB5GMG) rilonacept 160 mg in 2 mL Inactive Ingredients Ingredient Name Strength Histidine (UNII: 4QD397987E) Arginine (UNII: 94ZLA3W45F) Polyethylene glycol 3350 (UNII: G2M7P15E5P) Sucrose (UNII: C151H8M554) Glycine (UNII: TE7660XO1C) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61755-001-01 4 in 1 CARTON 03/24/2008 1 2 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125249 02/27/2008 Labeler - Regeneron Pharmaceuticals, Inc. (194873139) Establishment Name Address ID/FEI Business Operations Regeneron Pharmaceuticals, Inc. 945589711 ANALYSIS(61755-001) , API MANUFACTURE(61755-001) , LABEL(61755-001)
Trademark Results [Arcalyst]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ARCALYST 88851874 not registered Live/Pending |
Regeneron Pharmaceuticals, Inc. 2020-03-29 |
 ARCALYST 85497724 4189984 Live/Registered |
Regeneron Pharmaceuticals, Inc. 2011-12-16 |
 ARCALYST 78980233 3451501 Live/Registered |
Regeneron Pharmaceuticals, Inc. 2006-06-22 |
 ARCALYST 78914043 not registered Dead/Abandoned |
Regeneron Pharmaceuticals, Inc. 2006-06-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.