ARAKODA- tafenoquine tablet, film coated
Arakoda by
Drug Labeling and Warnings
Arakoda by is a Prescription medication manufactured, distributed, or labeled by 60 Degrees Pharmaceuticals, INC., TENTAMUS INDIA PRIVATE LIMITED. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ARAKODA™ safely and effectively. See full prescribing information for ARAKODA™.
ARAKODA™ (tafenoquine) tablets, for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
ARAKODA is an antimalarial indicated for the prophylaxis of malaria in patients aged 18 years and older. (1)
DOSAGE AND ADMINISTRATION
- All patients must be tested for glucose-6-phosphate dehydrogenase (G6PD) deficiency prior to prescribing ARAKODA. (2.1)
- Pregnancy testing is recommended for females of reproductive potential prior to initiating treatment with ARAKODA. (2.1)
Regimen Name
Timing
Dosage
Loading regimen
For each of the 3 days before travel to a malarious area
200 mg (2 of the 100 mg tablets) once daily for 3 days
Maintenance regimen
While in the malarious area
200 mg (2 of the 100 mg tablets) once weekly – start 7 days after the last loading regimen dose
Terminal prophylaxis regimen
In the week following exit from the malarious area
200 mg (2 of the 100 mg tablets) one-time 7 days after the last maintenance dose
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg of tafenoquine (3)
CONTRAINDICATIONS
- G6PD deficiency or unknown G6PD status (4)
- Breastfeeding by a lactating woman when the infant is found to be G6PD deficient or if G6PD status is unknown (4, 8.2)
- Patients with a history of psychotic disorders or current psychotic symptoms (4, 5.4)
- Known hypersensitivity reactions to tafenoquine, other 8-aminoquinolines, or any component of ARAKODA. (4)
WARNINGS AND PRECAUTIONS
- Hemolytic Anemia: G6PD testing must be performed before prescribing ARAKODA due to the risk of hemolytic anemia. Monitor patients for signs or symptoms of hemolysis. (5.1)
- G6PD Deficiency in Pregnancy or Lactation: ARAKODA may cause fetal harm when administered to a pregnant woman with a G6PD-deficient fetus. ARAKODA is not recommended during pregnancy. A G6PD-deficient infant may be at risk for hemolytic anemia from exposure to ARAKODA through breast milk. Check infant’s G6PD status before breastfeeding begins. (5.2, 8.1, 8.2)
- Methemoglobinemia: Asymptomatic elevations in blood methemoglobin have been observed. Initiate appropriate therapy if signs or symptoms of methemoglobinemia occur. (5.3)
- Psychiatric Effects: Serious psychotic adverse reactions have been observed in patients with a history of psychosis or schizophrenia, at doses different from the approved dose. If psychotic symptoms (hallucinations, delusions, or grossly disorganized thinking or behavior) occur, consider discontinuation of ARAKODA therapy and, evaluation by a mental health professional as soon as possible. (5.4)
- Hypersensitivity Reactions: Serious hypersensitivity reactions have been observed with administration of ARAKODA. If hypersensitivity reactions occur, institute appropriate therapy. (5.5)
- Delayed Adverse Reactions: Due to the long half-life of ARAKODA (approximately 17 days), psychiatric effects, hemolytic anemia, methemoglobinemia, and hypersensitivity reactions may be delayed in onset and/or duration. (5.6, 12.3)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥1%) were: headache, dizziness, back pain, diarrhea, nausea, vomiting, increased alanine aminotransferase (ALT), motion sickness, insomnia, depression, abnormal dreams, anxiety. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact 60 Degrees Pharmaceuticals at 1-888-834-0225 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Avoid co-administration with drugs that are substrates of organic cation transporter-2 (OCT2) or multidrug and toxin extrusion (MATE) transporters (7.1)
USE IN SPECIFIC POPULATIONS
Lactation: Advise women not to breastfeed a G6PD-deficient infant or infant with unknown G6PD status during treatment and for 3 months after the last dose of ARAKODA. (5.2, 8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Tests to be Performed Prior to ARAKODA Dose Initiation
2.2 Recommended Dosage and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hemolytic Anemia
5.2 G6PD Deficiency in Pregnancy and Lactation
5.3 Methemoglobinemia
5.4 Psychiatric Effects
5.5 Hypersensitivity Reactions
5.6 Delayed Adverse Reactions, Including Hemolytic Anemia, Methemoglobinemia, Psychiatric Effects, and Hypersensitivity Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of ARAKODA on Organic Cation Transporter-2 (OCT2) and Multidrug and Toxin Extrusion (MATE) Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Tests to be Performed Prior to ARAKODA Dose Initiation
All patients must be tested for glucose-6-phosphate dehydrogenase (G6PD) deficiency prior to prescribing ARAKODA [see Contraindications (4), Warnings and Precautions (5.1)].
Pregnancy testing is recommended for females of reproductive potential prior to initiating treatment with ARAKODA [see Use in Specific Populations (8.1 and 8.3)].
2.2 Recommended Dosage and Administration Instructions
The recommended dosage of ARAKODA is described in Table 1 below. ARAKODA can be administered for up to 6 months of continuous dosing.
Table 1: Recommended Dosage of ARAKODA in Patients (18 Years of Age and Older) Regimen Name
Timing
Dosage
Loading regimen
For each of the 3 days before travel to a malarious area
200 mg (2 of the 100 mg tablets) once daily for 3 days
Maintenance regimen
While in the malarious area
200 mg (2 of the 100 mg tablets) once weekly – start 7 days after the last loading regimen dose
Terminal prophylaxis regimen
In the week following exit from the malarious area
200 mg (2 of the 100 mg tablets) taken one time, 7 days after the last maintenance dose
- Administer ARAKODA with food. [see Clinical Pharmacology (12.3)].
- Swallow the tablet whole. Do not break, crush or chew the tablets.
- Complete the full course of ARAKODA including the loading dose and the terminal dose.
Table 2: How to Replace Missed Doses of ARAKODA Dose(s) Missed
How to Replace Missed Dose(s):
1 Loading dose
1 dose of 200 mg (2 of the 100 mg tablets) so that a total of 3 daily loading doses have been taken. Begin maintenance dose 1 week after the last loading dose.
2 Loading doses
2 doses of 200 mg (2 of the 100 mg tablets) on 2 consecutive days so that a total of 3 daily loading doses have been taken. Begin maintenance dose 1 week after the last loading dose.
1 Maintenance (weekly) dose
1 dose of 200 mg (2 of the 100 mg tablets) on any day up to the time of the next scheduled weekly dose.
2 Maintenance (weekly) doses
1 dose of 200 mg (2 of the 100 mg tablets) on any day up to the time of the next scheduled weekly dose.
3 or more Maintenance (weekly) doses
2 doses of 200 mg (2 of the 100 mg tablets), taken as 200 mg (2 of the 100 mg tablets) once daily for 2 days up to the time of the next weekly dose.
Terminal prophylaxis dose
1 dose of 200 mg (2 of the 100 mg tablets) as soon as remembered.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ARAKODA is contraindicated in:
- patients with G6PD deficiency or unknown G6PD status due to the risk of hemolytic anemia [see Warnings and Precautions (5.2)].
- breastfeeding by a lactating woman when the infant is found to be G6PD deficient or if the G6PD status of the infant is unknown [see Warnings and Precautions (5.3), Use in Specific Populations (8.2)].
- patients with a history of psychotic disorders or current psychotic symptoms (i.e., hallucinations, delusions, and/or grossly disorganized behavior) [see Warnings and Precautions (5.4)]
- patients with known hypersensitivity reactions to tafenoquine, other 8-aminoquinolines, or any component of ARAKODA [see Warnings and Precautions (5.5)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hemolytic Anemia
Due to the risk of hemolytic anemia in patients with G6PD deficiency, G6PD testing must be performed before prescribing ARAKODA [see Contraindications (4)]. Due to the limitations with G6PD tests, physicians need to be aware of residual risk of hemolysis and adequate medical support and follow-up to manage hemolytic risk should be available. Treatment with ARAKODA is contraindicated in patients with G6PD deficiency or unknown G6PD status [see Contraindications (4)]. In clinical trials, declines in hemoglobin levels were reported in some G6PD-normal patients [see Adverse Reactions (6.1)]. Monitor patients for clinical signs or symptoms of hemolysis [see Warnings and Precautions (5.6)]. Advise patients to discontinue ARAKODA and seek medical attention if signs of hemolysis occur.
5.2 G6PD Deficiency in Pregnancy and Lactation
Potential Harm to the Fetus
The use of ARAKODA during pregnancy may cause hemolytic anemia in a G6PD-deficient fetus. Even if a pregnant woman has normal levels of G6PD, the fetus could be G6PD deficient. Advise females of reproductive potential that treatment with ARAKODA during pregnancy is not recommended and to avoid pregnancy or use effective contraception during treatment and for 3 months after the last dose of ARAKODA. If a pregnancy is detected during ARAKODA use, discontinue ARAKODA as soon as possible and switch to an alternative prophylactic drug for malaria during pregnancy [see Use in Specific Populations (8.1 and 8.3)].
Potential Harm to the Breastfeeding Infant
A G6PD-deficient infant may be at risk for hemolytic anemia from exposure to ARAKODA through breast milk. Infant G6PD status should be checked before breastfeeding begins. ARAKODA is contraindicated in breastfeeding women when the infant is found to be G6PD deficient or the G6PD status of the infant is unknown [see Contraindications (4)]. Advise the woman with a G6PD-deficient infant or if the G6PD status of the infant is unknown not to breastfeed during treatment with ARAKODA and for 3 months after the final dose [see Use in Specific Populations (8.2)].
5.3 Methemoglobinemia
Asymptomatic elevations in methemoglobin have been observed in the clinical trials of ARAKODA [see Adverse Reactions (6.1)]. Institute appropriate therapy if signs or symptoms of methemoglobinemia occur [see Warnings and Precautions (5.6)]. Carefully monitor individuals with nicotinamide adenine dinucleotide (NADH)-dependent methemoglobin reductase deficiency. Advise patients to discontinue ARAKODA and seek medical attention if signs of methemoglobinemia occur.
5.4 Psychiatric Effects
In patients receiving ARAKODA in clinical trials, psychiatric adverse reactions included sleep disturbances (2.5%), depression/depressed mood (0.3%), and anxiety (0.2%) [see Adverse Reactions (6.1)]. ARAKODA was discontinued in a subject with an adverse reaction of suicide attempt (0.1%). Subjects with a history of psychiatric disorders were excluded from three of five ARAKODA trials in which mefloquine was included as a comparator.
Psychosis was reported in three patients with a history of psychosis or schizophrenia who received tafenoquine doses (350 mg to 500 mg single dose, or 400 mg daily for 3 days) different from the approved ARAKODA regimen. Safety and effectiveness of ARAKODA have not been established at doses or regimens other than the approved regimen; use of ARAKODA at doses or regimens other than a 200-mg weekly dose is not approved by FDA.
ARAKODA is contraindicated in patients with a history of psychotic disorders or current psychotic symptoms [see Contraindication (4)]. If psychotic symptoms (hallucinations, delusions, or grossly disorganized thinking or behavior) occur, consider discontinuation of ARAKODA and prompt evaluation by a mental health professional as soon as possible. Other psychiatric symptoms, such as changes in mood, anxiety, insomnia, and nightmares, should be promptly evaluated by a medical professional if they are moderate and last more than three days or are severe [see Warnings and Precautions (5.6)].
5.5 Hypersensitivity Reactions
Serious hypersensitivity reactions (e.g., angioedema and urticaria) have been observed with administration of tafenoquine. Hypersensitivity reactions have been reported in clinical trials of ARAKODA [see Adverse Reactions (6.1)]. Discontinue prophylaxis with ARAKODA and institute appropriate therapy if hypersensitivity reactions occur [see Warnings and Precautions (5.6)]. ARAKODA is contraindicated in patients who develop hypersensitivity to tafenoquine or any component of ARAKODA or other 8-aminoquinolines [see Contraindications (4)].
5.6 Delayed Adverse Reactions, Including Hemolytic Anemia, Methemoglobinemia, Psychiatric Effects, and Hypersensitivity Reactions
Adverse reactions including hemolytic anemia, methemoglobinemia, psychiatric effects, and hypersensitivity reactions were reported with the use of ARAKODA or tafenoquine in clinical trials [see Warnings and Precautions (5.1, 5.3, 5.4, 5.5)]. Due to the long half-life of ARAKODA (approximately 17 days), psychiatric effects, hemolytic anemia, methemoglobinemia, and signs or symptoms of hypersensitivity reactions that may occur could be delayed in onset and/or duration. Advise patients to seek medical attention if signs of hypersensitivity occur [see Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions observed with ARAKODA are discussed in detail in the Warnings and Precautions section:
- Hemolytic Anemia [see Warnings and Precautions (5.2)]
- Methemoglobinemia [see Warnings and Precautions (5.3)]
- Psychiatric Effects [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of tafenoquine was studied in clinical trials at various doses and regimens in 3,184 subjects. The recommended ARAKODA regimen was evaluated in 825 subjects in 5 controlled clinical trials (Trials 1, Trial 2, Trial 3, Trial 4 and Trial 5). The mean duration of exposure to ARAKODA in these five clinical trials was 21 weeks (range 10-29 weeks). Trial 1, 2 and 4 were conducted in healthy semi-immune volunteers in Ghana or Kenya and were placebo-controlled; a mefloquine arm was included in Trials 2 and 4 as a benchmark. Trial 3, an active comparator (mefloquine) controlled trial was conducted in healthy soldiers deployed in East Timor (Timor Leste). A placebo-controlled Trial 5 was conducted in healthy volunteers in the United States and United Kingdom. The mean age of the subjects included in the five trials was 29 years (range 17 to 69 years); 84% were male.
Adverse Reactions Reported with ARAKODA in Trial 3 and Pooled Trials 1, 2, 4, and 5
Adverse reactions occurring in ≥1% of subjects in the ARAKODA group in the placebo-controlled pooled Trials 1, 2, 3, and 4 are presented in Table 3.
Table 3: Selected Adverse Reactions Occurring in ≥1% of Subjects Receiving ARAKODA in Pooled Trials 1, 2, 4, and 5 (Non-Deployed Subjects) Adverse Reaction ARAKODA1 (n=333)
%Placebo (n=295)
%Mefloquine2 (n=147)
%1 ARAKODA was administered as 200 mg daily for 3 days, then 200 mg weekly
2 Mefloquine was administered as 250 mg daily for 3 days, then 250 mg weekly
3 Includes headache, sinus headache, migraine and tension headache.
4 Includes dizziness and dizziness postural
5 Includes abnormal dreams, insomnia, nightmares, sleep disorder, and somnambulism.Nervous system Disorders
35
34
47
Headache3
32
32
44
Dizziness4
5
3
10
Musculoskeletal and connective tissue disorders
27
26
37
Back pain
14
9
11
Gastrointestinal disorders
31
33
46
Diarrhea
5
3
1
Nausea
5
2
2
Vomiting
2
2
1
Investigations
8
7
11
Alanine Aminotransferase (ALT) increased/abnormal
4
2
3
Psychiatric disorders
2
1
2
Any sleep symptom5
1
1
0
Insomnia
1
1
0
Depression/depressed mood
1
0
0
Adverse reactions occurring in ≥1% of subjects in the ARAKODA group in the active-control Trial 3 conducted in military personnel deployed to malaria endemic areas are presented in Table 4.
Table 4: Selected Adverse Reactions Occurring in ≥1% of Subjects Receiving ARAKODA in Trial 3 (Deployed Subjects) Adverse Reaction ARAKODA1 (n=492)
%Mefloquine2 (n=162)
%1 ARAKODA was administered as 200 mg daily for 3 days, then 200 mg weekly
2 Mefloquine was administered as 250 mg daily for 3 days, then 250 mg weekly
3 Includes headache, sinus headache, migraine and tension headache.
4 Includes dizziness and dizziness postural
5 Includes motion sickness, vertigo and vertigo positional.
6 Includes abnormal dreams, insomnia, nightmares, sleep disorder, and somnambulism.
7 Includes abnormal dreams, nightmares
8 Includes anxiety disorder, panic attack and stress.Nervous system Disorders
22
27
Headache3
15
19
Dizziness4
1
1
Ear and labyrinth Disorders
7
11
Motion sicknesss5
5
6
Musculoskeletal and connective tissue disorders
29
30
Back pain
14
15
Gastrointestinal disorders
36
41
Diarrhea
18
20
Nausea
7
9
Vomiting
5
6
Psychiatric disorders
5
4
Any sleep symptom6
4
4
Insomnia
2
1
Abnormal dreams7
2
2
Anxiety8
1
0
Clinically Significant Adverse Reactions in Trials 1 to 5 (Overall Safety Population)
Clinically significant adverse reactions with ARAKODA (200 mg daily for 3 days, followed by 200 mg weekly) in Trials 1 to 5 (n= 825) are described below:
Ocular Adverse Reactions
Vortex keratopathy was reported in 21% to 93% of subjects receiving ARAKODA in the trials which included ophthalmic evaluations (Trials 3, 5, and Trial 6 (NCT # 01290601, an active-control trial in patients from Thailand with P. vivax malaria. The keratopathy did not result in any apparent functional visual changes and resolved within one year after drug cessation in all patients. Retinal abnormalities were noted in less than 1% of subjects receiving ARAKODA.
A total of 7 serious ocular adverse reactions (SARs) were reported in ARAKODA-treated subjects in the trials which included ophthalmic evaluations: 5 reports of keratopathy and two reports of retinal disorders.
Laboratory Abnormalities
Methemoglobinemia: Asymptomatic methemoglobin elevations were observed in 13% of subjects receiving ARAKODA.
Hemoglobin decrease: Hemoglobin decreases of ≥ 3 g/dL were observed in 2.3% of subjects receiving ARAKODA.
Adverse Reactions Reported in < 1% of Subjects Receiving ARAKODA in Trials 1 to 5
The following selected adverse reactions were reported in subjects receiving ARAKODA in Trials 1 to 5 at a rate of less than 1%.
Blood and lymphatic system disorders: hemolytic anemia, anemia, thrombocytopenia
Ear and labyrinth disorders: hyperacusis, Meniere’s disease
Eye disorders: night blindness, photophobia, blurred vision, visual acuity reduced, visual impairment, vitreous floaters
Hepatobiliary disorders: hyperbilirubinemia, jaundice cholestatic
Immune system disorders: hypersensitivity
Investigations: blood bilirubin increased, blood creatinine increased, glomerular filtration rate decreased
Nervous system disorders: amnesia, coordination abnormal, hyperesthesia, hypoesthesia, somnolence, syncope, tremor, visual field defect
Psychiatric disorders: agitation, neurosis
Skin and subcutaneous tissue disorders: urticaria.
-
7 DRUG INTERACTIONS
7.1 Effect of ARAKODA on Organic Cation Transporter-2 (OCT2) and Multidrug and Toxin Extrusion (MATE) Substrates
The effect of coadministration of tafenoquine on the pharmacokinetics of OCT2 and MATE substrates in humans is unknown. However, in vitro observations suggest the potential for increased concentrations of these substrates [see Clinical Pharmacology (12.3)] which may increase the risk of toxicity of these drugs.
Avoid coadministration of ARAKODA with OCT2 and MATE substrates (e.g., dofetilide, metformin). If coadministration cannot be avoided, monitor for drug-related toxicities and consider dosage reduction if needed based on approved product labeling of the coadministered drug.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The use of ARAKODA during pregnancy may cause hemolytic anemia in a fetus who is G6PD-deficient. Treatment with ARAKODA during pregnancy is not recommended. If a pregnancy is detected during ARAKODA use, discontinue ARAKODA as soon as possible and switch to an alternative prophylactic drug for malaria during pregnancy [see Warnings and Precautions (5.2)]. Available data with use of ARAKODA in pregnant women are insufficient to establish a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal studies, there were increased abortions, with and without maternal toxicity when tafenoquine was given orally to pregnant rabbits at and above doses equivalent to about 0.4 times the clinical exposure based on body surface area comparisons. No fetotoxicity was observed at doses about 1.5 times the clinical exposure (based on body surface area comparisons) in a similar study in rats.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk:
Malaria during pregnancy increases the risk for adverse pregnancy outcomes, including maternal anemia, prematurity, spontaneous abortion and stillbirth.
Data
Animal Data:
Tafenoquine resulted in dose-related abortions when given orally to pregnant rabbits during organogenesis (Gestation Days 6 to 18), at doses of 7 mg/kg (about 0.4 times the clinical exposure based on body surface area comparisons) and above. Doses higher than 7 mg/kg were also associated with maternal toxicity (mortality and reduced body weight gain). In a similar study in rats, doses of 3, 10, or 30 mg/kg/day resulted in maternal toxicity (enlarged spleen, reduced body weight and reduced food intake) but no fetotoxicity at the high dose (about 1.5 times the clinical exposure based on body surface area comparisons). There was no evidence of malformations in either species. In a pre- and postnatal development study in rats, tafenoquine administered throughout pregnancy and lactation produced maternal toxicity and a reversible decrease in offspring body weight gain and decrease in motor activity at 18 mg/kg/day, which is equivalent to about 0.6 times the clinical dose based on body surface area comparisons.
8.2 Lactation
Risk Summary
A breastfed infant with G6PD deficiency is at risk for hemolytic anemia from exposure to ARAKODA. Infant G6PD status should be checked before breastfeeding begins. ARAKODA is contraindicated in breastfeeding women when the infant is found to be G6PD deficient or the G6PD status of the infant is unknown [see Contraindications (4) and Clinical Considerations].
There is no information regarding the presence of ARAKODA in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. In a breastfed infant with normal G6PD, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ARAKODA and any potential effects on the breastfed infant from ARAKODA or from the underlying maternal condition.
Clinical Considerations
Check the infant’s G6PD status before maternal breastfeeding commences. If an infant is G6PD-deficient, exposure to ARAKODA during breastfeeding may result in hemolytic anemia in the infant; therefore, advise the woman with an infant who has G6PD deficiency or whose G6PD status is unknown, not to breastfeed during treatment with ARAKODA and for 3 months after the final dose of ARAKODA.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating treatment with ARAKODA. [see Dosage and Administration (2.2), Warnings and Precautions, (5.2), and Use in Specific Populations (8.1)].
Contraception
ARAKODA may cause hemolytic anemia in a G6PD-deficient fetus [see Warnings and Precautions (5.2), Use in Specific Populations (8.1)]. Advise females of reproductive potential that treatment with ARAKODA during pregnancy is not recommended and to avoid pregnancy or use effective contraception for 3 months after the final dose of ARAKODA.
8.4 Pediatric Use
Safety and effectiveness of ARAKODA in pediatric patients have not been established.
8.5 Geriatric Use
Clinical trials of ARAKODA did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
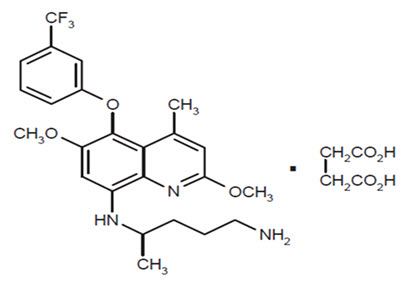
ARAKODA contains tafenoquine succinate, an antimalarial agent for oral administration. The structural formula of tafenoquine succinate is:
The chemical name of tafenoquine succinate is (±)-8-[(4-amino-1-methylbutyl) amino]-2,6-dimethoxy-4-methyl-5-[3-(trifluoromethyl) phenoxy]quinoline succinate. The molecular formula of tafenoquine succinate is C24H28F3N3O3∙C4H6O4 and its molecular weight is 581.6 as the succinate salt (463.49 as free base).
Each ARAKODA tablet contains 100 mg of tafenoquine (equivalent to 125.5 mg of tafenoquine succinate). Inactive ingredients include magnesium stearate, mannitol, and microcrystalline cellulose. The tablet film coating inactive ingredients include: hypromellose, iron oxide red, macrogol/polyethylene glycol and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tafenoquine is an 8-aminoquinoline antimalarial drug [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of tafenoquine on the QT interval was evaluated in a study of healthy adult subjects. In this study, subjects received once daily 400 mg (2 times the approved recommended dosage) doses of tafenoquine for 3 days. The results suggest that the mean increase in the QTcF interval for tafenoquine is less than 20 msec.
12.3 Pharmacokinetics
Absorption
A food effect study was not conducted with the 100 mg ARAKODA tablet. In majority of the clinical trials, tafenoquine was administered under fed conditions. Table 5 provides the pharmacokinetics of tafenoquine following single dose administration of 200 mg ARAKODA (two 100-mg ARAKODA tablets) in 65 healthy adult subjects under fed conditions. In this study, ARAKODA was administered with a high-calorie, high-fat meal (approximately 1000 calories with 19% protein, 31% carbohydrate, and 50% fat).
Table 5. Mean (%CV) Pharmacokinetic Parameters of Tafenoquine Following Single Oral Administration of Two 100-mg ARAKODA Tablets Under Fed Conditions in Healthy Adult Subjects (N=65) Parameter Value a Coefficient of Variance (CV)
b Median and (Range)
c Plasma tafenoquine AUCinf increased by 41% when tafenoquine was administered as an investigational capsule formulation with a high-calorie, high-fat meal compared with the fasted state.Cmax
147 ng/mL (20.7%)a
Tmax
14 hr (6 – 72 hr)b
AUCinf
70 hr*mcg/mL (24.6%)a, c
Following administration of a single dose of tafenoquine orally under fasted conditions in healthy adult subjects, AUC and Cmax increased dose proportionally over the dose range from 100 mg to 400 mg. When healthy adult subjects received once-weekly administrations of 200 mg tafenoquine orally for ten weeks without a loading dose under fasting conditions, the mean plasma accumulation ratio of tafenoquine was approximately 4.4.
Distribution
Tafenoquine is greater than 99.5% bound to protein in humans. The apparent volume of distribution of tafenoquine in healthy adult subjects is 2470 L [Inter-Individual Variability (IIV): 24.1 %].
Elimination
The apparent oral clearance of tafenoquine is approximately 4.2 L/hr (IIV: 23.6 %) in healthy adult subjects. The mean terminal half-life following administration of ARAKODA is approximately 16.5 days (range: 10.8 days to 27.3 days) in healthy adult subjects.
Metabolism
Negligible metabolism of tafenoquine was observed in vitro in human liver microsomes and hepatocytes. Following administration of tafenoquine orally, once daily for three days to healthy adult subjects, unchanged tafenoquine represented the only notable drug-related component in plasma at approximately 3 days following the first dose of tafenoquine.
Excretion
The full excretion profile of tafenoquine in humans is unknown.
Specific Populations
The pharmacokinetics of tafenoquine were not significantly impacted by age, sex, ethnicity, and body weight. The effect of renal or hepatic impairment on tafenoquine pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies
No clinically significant effects on the pharmacokinetics of substrates of cytochrome P450 isoenzymes (CYP)1A2 (caffeine), CYP2D6 (desipramine), CYP2C9 (flurbiprofen), or CYP3A4 (midazolam) were observed following coadministration with tafenoquine in healthy adult subjects.
In Vitro Studies Where Drug Interaction Potential Was Not Further Evaluated Clinically
Tafenoquine inhibited metformin transport via human OCT2, MATE1 and MATE2-K transporters [see Drug Interactions (7)].
Tafenoquine is not an inhibitor of human breast cancer resistance protein (BCRP), P-glycoprotein (P-gp), Organic anion transporter 1/3 (OAT1 or OAT3), Organic anion transporting polypeptide 1B1/1B3 (OATP1B1 or OATP1B3) mediated transport at clinically relevant concentrations. Tafenoquine is also not a substrate of human OATP1B1 or OATP1B3 at clinically relevant concentrations. It is inconclusive as to whether tafenoquine is a substrate of P-gp and/or BCRP mediated transport.
12.4 Microbiology
Mechanism of Action
Tafenoquine, an 8-aminoquinoline antimalarial, is active against all the stages of Plasmodium species that include the hypnozoite (dormant stage) in the liver. Studies in vitro with the erythrocytic forms of Plasmodium falciparum suggest that tafenoquine may exert its effect by inhibiting hematin polymerization and inducing apoptotic like death of the parasite. In addition to its effect on the parasite, tafenoquine causes red blood cell shrinkage in vitro. The molecular target of tafenoquine is not known.
Antimicrobial activity
Tafenoquine is active against pre-erythrocytic (liver) and erythrocytic (asexual) forms as well as gametocytes of Plasmodium species that include P. falciparum and P. vivax. The activity of tafenoquine against the pre-erythrocytic liver stages of the parasite, prevents the development of the erythrocytic forms of the parasite [see Clinical Studies (14)].
Resistance
A potential for development of resistance of Plasmodium species to tafenoquine was not evaluated.
Studies with the erythrocytic forms of P. falciparum strains/isolates suggest a potential for cross-resistance with primaquine, an 8-aminoquinoline. Clinical relevance of such findings is not known.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year oral carcinogenicity studies were conducted in rats and mice. Renal cell adenomas and carcinomas were increased in male rats at doses 1 mg/kg/day and above (0.5 times the clinical exposure based on AUC comparisons). Tafenoquine was not carcinogenic in mice. The relevance of these findings to a carcinogenic risk in humans is unclear.
Mutagenesis
Tafenoquine did not cause mutations or chromosomal damage in 2 definitive in vitro tests (bacterial mutation assay and mouse lymphoma L5178Y cell assay) or in an in vivo oral rat micronucleus test.
Impairment of Fertility
In a rat fertility study, tafenoquine was given orally at 1.5, 5, and 15 mg/kg/day (up to about 0.5 times the human dose based on body surface area comparisons) to males for at least 67 days, including 29 days prior to mating, and to females from 15 days prior to mating through early pregnancy. Tafenoquine resulted in reduced number of viable fetuses, implantation sites, and corpora lutea at 15 mg/kg in the presence of maternal toxicity (mortality, piloerection, rough coat, and reduced body weight).
-
14 CLINICAL STUDIES
Clinical Trials 1, 2, and 3
Three double-blind, randomized, controlled studies have been performed to evaluate the efficacy of ARAKODA.
Trial 1 (NCT #02491606) was a Phase IIb, placebo-controlled study conducted in Kenya, an area of holoendemic P. falciparum malaria. After taking a three-day presumptive course of halofantrine to eliminate any existing parasitemia, subjects were randomized into one of four groups (placebo and three different ARAKODA dosing groups; one group received 200 mg once daily for 3 days, then a maintenance regimen of weekly dose of 200 mg for 10-15 weeks). Sixty-one percent of subjects were male. The mean age was 32.4 years (range 17-55). Subjects were evaluated for parasitemia by weekly blood smears. Protective efficacy at 15 weeks was defined based on the reduced incidence of parasitemia during the prophylaxis phase relative to placebo. The results in the intention-to-treat population, which included all subjects who received three doses of halofantrine and were randomized, are shown in Table 6 below.
Table 6: Incidence of Parasitemia and Protective Efficacy of ARAKODA at 15 weeks for Trial 1 Placebo ARAKODA1 1 200 mg once daily for 3 days, then 200 mg weekly for 10-15 weeks
2 Protective efficacy is reduced incidence of parasitemia relative to placebo (0: no protection; 1: full protection); CI: confidence interval. Bonferroni adjustment was used for multiple comparisons. Missing outcome was considered a failure due to parasitemia for this analysis.Number of subjects
62
61
Subjects free of parasitemia
5 (8.1%)
46 (75.4)
Subjects with parasitemia
54 (87.1%)
7 (11.5%)
Subjects with missing data
3 (4.8%)
8 (13.1%)
Protective efficacy
[98.3% CI]2–
73.3%
[54.0%, 84.5%]Trial 2 (NCT #02488902) was a comparison of tafenoquine to placebo for prophylaxis in healthy semi-immune residents of a malarious region in Ghana. After treating existing parasitemia with quinine/doxycycline/primaquine, subjects were randomized into prophylactic groups including ARAKODA and placebo. Patients were administered a loading regimen of daily drug or placebo for 3 days followed by a maintenance regimen of weekly drug or placebo for 12 weeks. For the ARAKODA and placebo groups, males were 65% of the total population. The mean age was 38.4 years and 53.5 years for males and females, respectively, as women in reproductive ages were excluded from the study. The mean weight was 55.4 kg and 47.5 kg for males and females, respectively. Subjects were evaluated for parasitemia by weekly blood smears. Parasitemia required a blood smear positive for asexual stage of P. falciparum. The incidence of parasitemia at week 12 for all randomized subjects who received at least one dose of ARAKODA or placebo is presented in Table 7 below.
Table 7: Incidence of Parasitemia and Protective Efficacy of ARAKODA at Week 12 for Trial 2 Placebo ARAKODA1 1 200 mg once daily for 3 days, then 200 mg weekly for 12 weeks
2 Protective efficacy is reduced incidence of parasitemia relative to placebo; CI: confidence interval. Bonferroni adjustment was used for multiple comparisons. Missing outcome was considered a failure due to parasitemia for this analysis.Number of subjects
94
93
Subjects free of parasitemia
6 (6.4%)
68 (73.1%)
Subjects with parasitemia
86 (91.5%)
12 (12.9%)
Subjects with missing data
2 (2.1%)
13 (14.0%)
Protective efficacy
[98.75% CI]2–
71.3%
[55.8%, 81.4%]Trial 3 compared ARAKODA with mefloquine for the prophylaxis of both P. falciparum and P. vivax malaria in healthy non-immune soldiers deployed to East Timor (now Timor-Leste). No subject developed malaria during the 26-week prophylactic phase. Subjects were exposed to P. vivax and there is a high likelihood that the study subjects were also exposed to P. falciparum. Since the precise degree of exposure to malaria in study subjects is unknown, this study provides only supportive evidence of efficacy.
Clinical Trial 7
In a randomized, double-blind, placebo-controlled trial (Trial 7) in healthy, non-immune volunteers, ARAKODA was shown to have prophylactic activity directed against blood-stage P. falciparum parasites. Twelve subjects received ARAKODA (200 mg once daily for 3 days, then 200 mg on 10 day) and 4 subjects received placebo. On Day 13, subjects were inoculated with erythrocytes containing viable P. falciparum parasites. Fifteen subjects (93.8%) were of white race. The mean age was 27.5 years (range 20-42). The mean body weight was 72.3 kg (range 56-97.7). The efficacy endpoint was parasitemia by Day 34; parasitemia was based on detection of P. falciparum 18S ribosomal DNA by real time polymerase chain reaction assay (PCR). There was a statistically significant difference in malaria incidence between the two groups; 4/4 (100%) subjects in the placebo group had detectable parasites from Day 17 compared to 0/12 (0%) subjects on ARAKODA were PCR negative at all visits (p<0.0005).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
ARAKODA tablets contain 100 mg of tafenoquine (equivalent to 125.5 mg of tafenoquine succinate) and are dark pink, film-coated, capsule-shaped, and debossed with ‘TQ100’ on one side.
ARAKODA tablets are packed in polyamide aluminum and PVC formable laminate backed blisters with a polyethylene terephthalate aluminum foil cover. Each blister card contains 8 tablets. Each package contains 2 blister cards (16 tablets) housed in a contiguous outer paperboard child-resistant carton component (NDC: 71475-257-01).
Storage
Store at 20°C to 25°C (68°F to 77°F). Temperature excursions are permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Protect from moisture. Dispense only in the original carton.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
G6PD Testing and Hemolytic Anemia
Inform patients of the need for testing for G6PD deficiency before starting ARAKODA. Advise patients on the symptoms of hemolytic anemia and instruct them to seek medical advice promptly if such symptoms occur. Patients should contact their health care provider if they have darker lips or urine as these may be signs of hemolysis or methemoglobinemia [see Warnings and Precautions (5.1)].
Important Administration Instructions
- Advise patients to take ARAKODA with food.
- Advise patients to swallow the tablet whole and not to break, crush or chew it.
- Advise patients to complete the full course of ARAKODA including the loading dose, maintenance dose and terminal dose.
Potential Harm to the Fetus
Advise females of reproductive potential of the potential risk of ARAKODA to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.2) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to avoid pregnancy or use effective contraception during treatment with ARAKODA and for 3 months after the final dose [see Use in Specific Populations (8.3)].
Lactation
Advise women with a G6PD-deficient infant, or if they do not know the G6PD status of their infant, not to breastfeed during treatment with ARAKODA and for 3 months after the final dose [see Contraindication (4), Warnings and Precautions (5.2), Use in Specific Populations (8.2)].
Methemoglobinemia
Inform patients that methemoglobinemia has occurred with ARAKODA. Advise patients on the symptoms of methemoglobinemia and instruct them to seek medical advice promptly if such symptoms occur [see Warnings and Precautions (5.3)].
Psychiatric Symptoms
Advise patients who experience hallucinations, delusions, or confused thinking while taking ARAKODA to seek medical attention as soon as possible. Other psychiatric symptoms, such as changes in mood, anxiety, insomnia, and nightmares, should be promptly evaluated by a medical professional if they last more than three days or severe [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions have occurred with ARAKODA. Advise patients on the symptoms of hypersensitivity reactions and instruct them to seek medical advice promptly if such symptoms occur [see Warnings and Precautions (5.5)].
Manufactured For:
60 Degrees Pharmaceuticals LLC,
1025 Connecticut Avenue NW, Suite 1000,
Washington DC 200361217a
-
Medication Guide
This Medication Guide has been approved by the U.S. Food and Drug Administration
Issued: August 2018
-
PRINCIPAL DISPLAY PANEL - Carton Label
Rx only
NDC : 71475-257-01
KEEP OUT OF REACH OF CHILDREN
ATTENTION: Dispense the enclosed
Medication Guide to each patientArakoda™
(tafenoquine) tablets,
for oral use100 mg
16 (2 x 8)
Unit Dose TabletsSixty
Degrees
PharmaEach tablet contains 100 mg tafenoquine (equivalent to
125.5 mg tafenoquine succinate). Store at 20°C to 25°C (68°F to 77°F);
excursions permitted to 15 to 30°C (59 to 86°F).Protect from moisture. Dispense only in the original carton.
See Prescribing Information for Dosage and Administration information.
Two blister packs are contained in each carton. Each blister
pack contains eight 100 mg tablets of Arakoda™ (tafenoquine),
for oral use.Manufactured for:
60° Pharmaceuticals, LLC.
1025 Connecticut Ave., NW, Suite 1000
Washington DC 20036Country of Origin: India
Sixty
Degrees
PharmaArakoda™
(tafenoquine) tablets,
for oral useLot
EXP MMMYYYY
GTIN
SN -
INGREDIENTS AND APPEARANCE
ARAKODA
tafenoquine tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71475-257 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TAFENOQUINE (UNII: 262P8GS9L9) (TAFENOQUINE - UNII:262P8GS9L9) TAFENOQUINE 100 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color PINK (Dark) Score no score Shape OVAL Size 15mm Flavor Imprint Code TQ100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71475-257-01 2 in 1 CARTON 08/20/2018 1 8 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210607 08/20/2018 Labeler - 60 Degrees Pharmaceuticals, LLC (079146968) Establishment Name Address ID/FEI Business Operations Megsan Labs Private Limited 650801215 ANALYSIS(71475-257)


Trademark Results [Arakoda]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ARAKODA 87688137 5950691 Live/Registered |
60° Pharmaceuticals, LLC 2017-11-16 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.