YARGESA- miglustat capsule
Yargesa by
Drug Labeling and Warnings
Yargesa by is a Prescription medication manufactured, distributed, or labeled by Edenbridge Pharmaceuticals LLC.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use YARGESA safely and effectively.
See full prescribing information for YARGESA.
YARGESA (miglustat) capsules, for oral use
Initial U.S. Approval: 2003INDICATIONS AND USAGE
YARGESA is a glucosylceramide synthase inhibitor indicated as monotherapy for treatment of adult patients with mild/moderate type 1 Gaucher disease for whom enzyme replacement therapy is not a therapeutic option ( 1.1).
DOSAGE AND ADMINISTRATION
Recommended dosage is 100 mg administered orally three times a day at regular intervals ( 2.1).
May reduce dosage to 100 mg once or twice a day in some patients due to tremor or diarrhea ( 2.1).
Adjust in patients with renal impairment ( 2.2):
Renal Impairment Adjusted Creatinine
Clearance (in mL/min/1.73 m 2)Recommendations Mild 50 – 70 Start dose at 100 mg twice a day Moderate 30 – 50 Start dose at 100 mg once a day Severe < 30 Use is not recommended
DOSAGE FORMS AND STRENGTHS
Capsules: 100 mg ( 3)
CONTRAINDICATIONS
None ( 4)
WARNINGS AND PRECAUTIONS
Peripheral neuropathy: Perform baseline and follow-up neurological evaluations at 6-month intervals in all patients ( 5.1).
Tremor: Reduce dose to ameliorate tremor or discontinue treatment if tremor does not resolve within days of dose reduction ( 5.2).
Diarrhea and weight loss: Evaluate for underlying gastrointestinal disease in patients who do not respond to usual interventions (e.g. diet modification) ( 5.3).
Reductions in Platelet count: Mild reductions in platelet counts without association with bleeding were observed in some patients. Monitoring of platelet counts is recommended. ( 5.4)ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ to 5%) are: diarrhea, weight loss, stomach pain, gas, nausea and vomiting, headache including migraine, tremor, leg cramps, dizziness, weakness, vision problems, thrombocytopenia, muscle cramps, back pain, constipation, dry mouth, heaviness in arms and legs, memory loss, unsteady walking, anorexia, indigestion, paresthesia, stomach bloating, stomach pain not related to food, and menstrual changes ( 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Edenbridge Pharmaceuticals, LLC at 1-877-381-3336 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Co-administration of YARGESA and imiglucerase may lead to increased clearance of imiglucerase ( 7).
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Type 1 Gaucher Disease
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Administration
2.2 Patients with Renal Insufficiency
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Peripheral Neuropathy
5.2 Tremor
5.3 Diarrhea and Weight Loss
5.4 Reductions in Platelet Count
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Administration
Therapy should be directed by physicians who are knowledgeable in the management of Gaucher disease.
The recommended dose for the treatment of adult patients with type 1 Gaucher disease is one 100 mg capsule administered orally three times a day at regular intervals. If a dose is missed, the next YARGESA capsule should be taken at the next scheduled time.
It may be necessary to reduce the dose to one 100 mg capsule once or twice a day in some patients due to adverse reactions, such as tremor or diarrhea.2.2 Patients with Renal Insufficiency
In patients with mild renal impairment (adjusted creatinine clearance 50-70 mL/min/1.73 m 2), initiate YARGESA treatment at a dose of 100 mg twice per day. In patients with moderate renal impairment (adjusted creatinine clearance of 30-50 mL/min/1.73 m 2), initiate YARGESA treatment at a dose of one 100 mg capsule per day. YARGESA is not recommended for use in patients with severe renal impairment (creatinine clearance <30 mL/min/1.73 m 2) [ see Use in Specific Populations (8.6)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Peripheral Neuropathy
In clinical trials, cases of peripheral neuropathy have been reported in 3% of Gaucher‘s patients treated with miglustat capsules. All patients receiving miglustat capsules treatment should undergo baseline and repeat neurological evaluations at approximately 6-month intervals. Patients who develop symptoms of peripheral neuropathy such as pain, weakness, numbness and tingling should have a careful re-assessment of the risk/benefit of miglustat therapy, and cessation of treatment may be considered.
5.2 Tremor
Approximately 30% of patients have reported tremor or exacerbation of existing tremor on treatment. These tremors were described as an exaggerated physiological tremor of the hands. Tremor usually began within the first month of therapy and in many cases resolved between 1 to 3 months during treatment. Reduce dose to ameliorate tremor or discontinue treatment if tremor does not resolve within days of dose reduction.
5.3 Diarrhea and Weight Loss
Diarrhea and weight loss were common in clinical studies of patients treated with miglustat capsules, occurring in approximately 85% and up to 65% of treated patients, respectively. Diarrhea appears to be the result of the inhibitory activity of miglustat on intestinal disaccharidases such as sucrase-isomaltase in the gastrointestinal tract leading to reduced absorption of dietary disaccharides in the small intestine, with a resultant osmotic diarrhea. It is unclear if weight loss results from the diarrhea and associated gastrointestinal complaints, a decrease in food intake, or a combination of these or other factors. The incidence of weight loss was most evident in the first 12 months of treatment. Diarrhea decreased over time with continued miglustat treatment, and may respond to individualized diet modification (e.g., reduction of sucrose, lactose and other carbohydrate intake), to taking miglustat capsules between meals, and/or to anti-diarrheal medications, most commonly loperamide. Patients may be instructed to avoid high carbohydrate content foods during treatment with miglustat capsules if they present with diarrhea.
Patients with persistent gastrointestinal events that continue during treatment with miglustat capsules, and who do not respond to usual interventions (e.g. diet modification), should be evaluated to determine whether significant underlying gastrointestinal disease is present. The safety of treatment with miglustat capsules has not been evaluated in patients with significant gastrointestinal disease, such as inflammatory bowel disease, and continued treatment of these patients with miglustat capsules should occur only after consideration of the risks and benefits of continued treatment.5.4 Reductions in Platelet Count
In clinical trials evaluating the use of miglustat capsules for treatment of indications other than type 1 Gaucher disease, mild reductions in platelet counts without association with bleeding were observed in some patients; approximately 40% of patients in this trial had low platelet counts (defined as below 150 × 10 9/L) before starting treatment with miglustat capsules. Monitoring of platelet counts is recommended in patients with type 1 Gaucher
disease. Mild reductions in platelet counts without association with bleeding were observed in patients with type 1 Gaucher disease who were switched from enzyme replacement therapy (ERT) to miglustat capsules. -
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
Peripheral Neuropathy [ see Warnings and Precautions (5.1)]
Tremor [ see Warnings and Precautions (5.2)]
Diarrhea and weight loss [ see Warnings and Precautions (5.3)]
Reductions in platelet count [ see Warnings and Precautions (5.4)]6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure of 80 patients with type 1 Gaucher disease in two open-label, uncontrolled, monotherapy trials, one open-label, active-controlled trial, and two extensions, who received miglustat capsules at doses ranging from 50 mg to 200 mg three times daily. Patients were aged 18 to 69 years at first treatment. The population was evenly distributed by gender.
The most common serious adverse reaction reported with miglustat capsules treatment in clinical trials was peripheral neuropathy [ see Warnings and Precautions (5.1)].
The most commonly reported adverse reactions in patients treated with miglustat capsules (occurring in ≥ to 5%) that were considered related to miglustat capsules are shown in Tables 1 and 2. [ see Warnings and Precautions (5.2, 5.3)].
The most common adverse reactions requiring intervention were diarrhea and tremor. [ see Warnings and Precautions (5.2, 5.3)].
In two open-label, uncontrolled monotherapy trials, adult type 1 Gaucher disease patients were treated with miglustat capsules at a starting dose of 100 mg three times daily (dose range 100 to 200 mg three times daily) for up to 12 months in 28 patients [Study 1], or at a dose of 50 mg three times daily for up to 6 months in 18 patients [Study 2]. Table 1 below lists adverse reactions that occurred during the trials in ≥ to 5% of patients.Table 1: Adverse Reactions in greater than or equal to 5% of Patients in Two Open-Label, Uncontrolled Monotherapy Trials of miglustat capsules Incidence of adverse reactions Study 1
(starting dose 100mg three times daily)Study 2
(50mg three times daily)Patients entered in Study (n) 28 18 Body System – Preferred Term % of patients reporting % of patients reporting Gastrointestinal System Diarrhea 89 89 Flatulence 29 44 Abdominal Pain 18 50 Nausea 14 22 Vomiting 4 11 Bloating 0 6 Anorexia 7 0 Dyspepsia 7 0 Epigastric pain not food-related 0 6 Metabolic and Nutritional Disorders Weight Decrease 39 67 Central and Peripheral Nervous System Headache 21 22 Tremor 11 11 Dizziness 0 11 Leg cramps 4 11 Paresthesia 7 0 Migraine 0 6 Vision Disorders Visual Disturbance 0 17 Musculoskeletal Disorders Cramps 0 11 Platelet, Bleeding and Clotting Disorders Thrombocytopenia 7 6 Reproductive disorders, female Menstrual disorder 0 6 In an open-label, active-controlled study, 36 adult type 1 Gaucher disease patients were treated with miglustat capsules, imiglucerase, or miglustat capsules plus imiglucerase [Study 3] for up to 12 months. Table 2 lists adverse reactions that occurred during the trial in greater than or equal to 5% of patients.
Table 2: Adverse Reactions in greater than or equal to 5% of Patients in Open-Label Active Controlled Study Incidence of adverse reactions Miglustat capsules alone Imiglucerase alone Patients entered in Study (n) 12 12 Body System – Preferred Term % of patients reporting % of patients reporting Gastrointestinal System Diarrhea 100 0 Abdominal Pain 67 0 Flatulence 50 0 Constipation 8 0 Nausea 8 0 Dry Mouth 8 0 Body as Whole Pain 0 8 Generalized weakness 17 0 Abdominal distension 8 0 Back pain 8 0 Heaviness in limbs 8 0 Metabolic and Nutritional Disorders Weight Decrease 67 0 Central Peripheral Nervous System Tremor 17 0 Dizziness 8 0 Leg cramps 8 0 Unsteady gait 8 0 Psychiatric disorders Memory loss 8 0 -
7 DRUG INTERACTIONS
While co-administration of miglustat capsules appeared to increase the clearance of imiglucerase by 70%, these results are not conclusive because of the small number of patients studied and because patients took variable doses of imiglucerase [ see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, miglustat capsules may cause fetal harm when administered to a pregnant woman. Available data from postmarketing case reports with miglustat capsules use in pregnancy are insufficient to assess a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks associated with symptomatic Type I Gaucher disease in pregnancy, including hepatosplenomegaly and thrombocytopenia (see Clinical Considerations). Advise pregnant women of the potential risks to the fetus.In animal reproduction studies, miglustat was maternally toxic in rabbits at exposures near the expected human therapeutic dose and caused embryo-fetal toxicities in rats at doses twice the recommended human dose. No adverse developmental outcomes were observed with administration of miglustat to pregnant rats at dose levels 6 times the recommended human dose. (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo-fetal risk
Pregnancy may exacerbate existing Type 1 Gaucher disease symptoms or result in new disease manifestations. Type 1 Gaucher disease manifestations may lead to adverse pregnancy outcomes including, hepatosplenomegaly which can interfere with the normal growth of a pregnancy and thrombocytopenia which can lead to increased bleeding and possible hemorrhage.
Data
Animal Data
In female rats given miglustat by oral gavage at doses of 20, 60, 180 mg/kg/day beginning 14 days before mating and continuing through gestation day 17 (organogenesis), increased post implantation loss, decreased embryo-fetal survival and decreased fetal and pup weights were observed at doses greater than or equal to 60 mg/kg/day (greater than or equal to 2 times the human therapeutic dose on a mg/m 2basis). Miglustat was also administered to pregnant rats by oral gavage at doses of 20, 60, 180 mg/kg/day from gestation day 6 through lactation (postpartum day 20). Delayed and prolonged parturition with decreased live births were observed at doses greater than or equal to 60 mg/kg/day (greater than or equal
to 2 times the human therapeutic dose on a mg/m 2basis).
In pregnant rabbits given miglustat by oral gavage at doses of 15, 30, 45 mg/kg/day during gestation days 6-18 (organogenesis), maternal toxicity, including maternal deaths (all doses), reduced food consumption (30 and 45 mg/kg/day), and decreased body weight gain (15 and 30 mg/kg/day), was observed. The 15 mg/kg/day dose level was 0.97 times the human therapeutic dose on a mg/m 2basis.
A pre- and postnatal development study in rats, miglustat was administered by oral gavage at doses of 20, 60, 180 mg/kg/day from gestation day 6 through day 20 of lactation, decreased live births were observed in dams, as well as decreased body weight gain in the offspring at doses greater than or equal to 60 mg/kg/day (greater than or equal to 2 times the human therapeutic dose on a mg/m 2basis). There was no effect on behavioral and learning assessments, sexual maturation or reproductive performance of the offspring at doses up to 180 mg/kg/day (about 6 times the human therapeutic dose on a mg/m 2basis).8.2 Lactation
Risk Summary
There are no available data on the presence of miglustat in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. Based on the physical properties of miglustat, miglustat capsules is likely to be present in breast milk. Because of the potential for serious adverse reactions in breastfed infants, advise women that breastfeeding is not recommended.8.3 Females and Males of Reproductive Potential
Infertility
Findings from a small clinical study in seven healthy adult males who received miglustat for six weeks did not indicate effects on male fertility. Studies in male rats have shown that miglustat decreased fertility but findings were reversible. Studies in female rats have shown increased post-implantation loss and decreased embryo-fetal survival [see Use in Specific Populations (8.1), Nonclinical Toxicology (13.1)].8.4 Pediatric Use
The safety and effectiveness of miglustat capsules in pediatric patients have not been established.
In a combined clinical trial safety data set of 45 patients less than 18 years of age exposed to miglustat capsules in indications other than type 1 Gaucher disease, the median weight and height percentiles adjusted for age and gender decreased during the first year of treatment but then stabilized. The mean length of exposure in these studies ranged from 2 to 2.6 years; some pediatric patients were exposed for up to 4 years. However, the effect of miglustat capsules on long-term gain in weight and height in pediatric patients is unclear.8.5 Geriatric Use
Clinical studies of miglustat capsules did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger patients. Other reported clinical experience has not identified differences in responses between elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, and cardiac function and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Miglustat is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function [ see Clinical Pharmacology (12.3)].
In patients with mild renal impairment (adjusted creatinine clearance 50-70 mL/min/1.73 m 2), miglustat capsules administration should commence at a dose of 100 mg twice per day.
In patients with moderate renal impairment (adjusted creatinine clearance of 30-50 mL/min/1.73 m 2), miglustat capsules administration should commence at a dose of 100 mg once a day.
Use of miglustat capsules in patients with severe renal impairment (creatinine clearance less than 30 mL/min/1.73 m 2) is not recommended.
Since elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function. The impact of hemodialysis on the disposition of miglustat capsules has not been investigated. -
11 DESCRIPTION
YARGESA (miglustat capsules) is an inhibitor of the enzyme glucosylceramide synthase, which is a glucosyl transferase enzyme responsible for the first step in the synthesis of most glycosphingolipids. YARGESA is an N-alkylated imino sugar, a synthetic analog of D-glucose. The chemical name for miglustat is 1,5-(butylimino)-1,5-dideoxy-D-glucitol with the chemical formula C 10H 21NO 4and a molecular weight of 219.28.
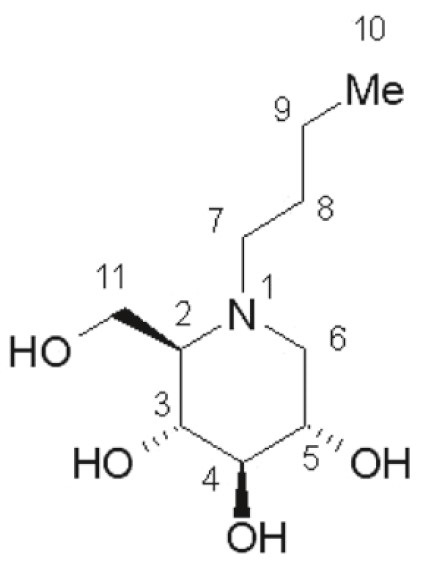
Miglustat is a white to off-white crystalline solid and has a bitter taste. It is highly soluble in water (greater than 1000 mg/mL as a free base).
YARGESA is supplied in hard gelatin capsules each containing 100 mg miglustat for oral administration. Each YARGESA 100 mg capsule also contains sodium starch glycolate (type A, potato), povidone (K-29/32), and magnesium stearate. Ingredients in the capsule shell include gelatin and titanium dioxide, and the shells are printed with edible ink consisting of black iron oxide, shellac, and propylene glycol. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Type 1 Gaucher disease is caused by a functional deficiency of glucocerebrosidase, the enzyme that mediates the degradation of the glycosphingolipid glucosylceramide.
Miglustat functions as a competitive and reversible inhibitor of the enzyme glucosylceramide synthase, the initial enzyme in a series of reactions which results in the synthesis of most glycosphingolipids.
Miglustat capsules helps reduce the rate of glycosphingolipid biosynthesis so that the amount of glycosphingolipid substrate is reduced to a level which allows the residual activity of the deficient glucocerebrosidase enzyme to be more effective (substrate reduction therapy). In vitroand in vivostudies have shown that miglustat can reduce the synthesis of glucosylceramide-based glycosphingolipids.12.3 Pharmacokinetics
Absorption:
After a 100 mg oral dose, the time to maximum observed plasma concentration of miglustat (t max) ranged from 2 to 2.5 hours in Gaucher patients. Plasma concentrations show a biexponential decline, characterized by a short distribution phase and a longer elimination phase. The effective half-life of miglustat is approximately 6 to 7 hours, which predicts that steady-state will be achieved by 1.5 to 2 days following the start of three times daily dosing. Miglustat, dosed at 50 and 100 mg three times daily in Gaucher patients, exhibits dose-proportional pharmacokinetics. The pharmacokinetics of miglustat were not altered after repeated dosing three times daily for up to 12 months.
In healthy subjects, co-administration of miglustat capsules with food results in a decrease in the rate of absorption of miglustat (maximum plasma concentration [C max] was decreased by 36% and t maxdelayed 2 h) but had no statistically significant effect on the extent of absorption of miglustat (area-under-the-plasma-concentration time curve [AUC] was decreased by 14%). The mean oral bioavailability of a 100-mg miglustat capsule is about 97% relative to an oral solution administered under fasting conditions. The pharmacokinetics of miglustat were similar between adult type 1 Gaucher disease patients and healthy subjects after a single dose administration of miglustat 100 mg.
Distribution:
Miglustat does not bind to plasma proteins. Mean apparent volume of distribution of miglustat is 83-105 liters in Gaucher patients. At steady state, the concentration of miglustat in cerebrospinal fluid of six non-Gaucher patients was 31.4-67.2% of that in plasma, indicating that miglustat crosses the blood brain barrier.
Metabolism and Excretion:
The major route of excretion of miglustat is via kidney. Following administration of a single dose of 100 mg 14C-miglustat to healthy volunteers, 83% of the radioactivity was recovered in urine and 12% in feces. In healthy subjects, 67% of the administered dose was excreted unchanged in urine over 72 hours. The most abundant metabolite in urine was miglustat glucuronide accounting for 5% of the dose. The terminal half-life of radioactivity in plasma was 150 hours, suggesting the presence of one or more metabolites with a prolonged half-life. The metabolite accounting for this observation has not been identified but may accumulate and reach concentrations exceeding those of miglustat at steady state.Specific Populations
Gender: There was no statistically significant gender difference in miglustat pharmacokinetics, based on pooled data analysis.
Race: Ethnic differences in miglustat pharmacokinetics have not been evaluated in Gaucher patients. However, apparent oral clearance of miglustat in patients of Ashkenazi Jewish descent was not statistically different to that in others (1 Asian and 15 Caucasians), based on a cross-study analysis.
Hepatic Impairment:No studies have been performed to assess the pharmacokinetics of miglustat in patients with hepatic impairment.
Renal Impairment:Limited data in non-Gaucher patients with impaired renal function indicate that the apparent oral clearance (CL/F) of miglustat decreases with decreasing renal function. While the number of subjects with mild and moderate renal impairment was very small, the data suggest an approximate decrease in the apparent oral clearance of 40% and 60% respectively, in mild and moderate renal impairment, justifying the need to decrease the dosing of miglustat in such patients dependent upon creatinine clearance levels [ see Dosage and Administration (2.2)].
Data in severe renal impairment are limited to two patients with creatinine clearances in the range 18-29 mL/min and cannot be extrapolated below this range. These data suggest a decrease in CL/F by at least 70% in patients with severe renal impairment [ see Dosage and Administration (2.2)and Use in Specific Populations (8.6)].Drug Interaction Studies
Miglustat does not inhibit the metabolism of various substrates of cytochrome P450 enzymes including, CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4 and CYP4A11 in vitro; consequently significant interactions via inhibition of these enzymes are unlikely with drugs that are substrates of cytochrome P450 enzymes.Drug interaction between miglustat capsules (miglustat 100 mg orally three times daily) and imiglucerase 7.5 or 15 U/kg/day was assessed in imiglucerase-stabilized patients after one month of co-administration. There was no significant effect of imiglucerase on the pharmacokinetics of miglustat, with the co-administration of imiglucerase and miglustat resulting in a 22% reduction in Cmax and a 14% reduction in the AUC for miglustat. While miglustat capsules appeared to increase the clearance of imiglucerase by 70%, these results are not conclusive because of the small number of subjects studied and because patients took variable doses of imiglucerase [see Drug Interactions (7)]. Concomitant therapy with loperamide during clinical trials did not appear to significantly alter the pharmacokinetics
of miglustat. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenesis:
Two-year carcinogenicity studies have been conducted with miglustat in CD-1 mice at oral doses up to 500 mg/kg/day and in Sprague Dawley rats at oral doses up to 180 mg/kg/day. Oral administration of miglustat for 104 weeks produced mucinous adenocarcinomas of the large intestine at 210, 420 and 500 mg/kg/day (about 3, 6 and 7 times the recommended human dose, respectively, based on the body surface area) in male mice and at 420 and 500 mg/kg/day (about 6 and 7 times the recommended human dose, based on the body surface area) in female mice. The adenocarcinomas were considered rare in CD-1 mice and occurred in the presence of inflammatory and hyperplastic lesions in the large intestine of both males and females. In rats, oral administration of miglustat for 100 weeks produced increased incidences of interstitial cell adenomas of the testis at 30, 60 and 180 mg/kg/day (about 1, 2 and 5 times the recommended human dose, respectively, based on the body surface area).Mutagenesis:
Miglustat was not mutagenic or clastogenic in a battery of in vitro and in vivo assays including the bacterial reverse mutation (Ames), chromosomal aberration (in human lymphocytes), gene mutation in mammalian cells (Chinese hamster ovary), and mouse micronucleus assays.Impairment of Fertility:
Miglustat was administered by oral gavage to male rats at doses of 20, 60, 180 mg/kg/day beginning at least 14 days prior to mating and continuing through mating. Effects on sperm parameters (concentration, motility and morphology) resulting in decreased fertility were observed at all dose levels (the lowest dose level was 0.65 times the human therapeutic dose on a mg/m 2basis). Reversibility was demonstrated following 6 weeks of drug withdrawal. Findings of, seminiferous tubule degeneration and atrophy were observed in the testes in rat repeat-dose toxicity studies.Miglustat was administered by oral gavage to female rats at doses of 20, 60, 180 mg/kg/day beginning 14 days before mating and continuing through gestation day 17. There were no effects on mating performance or fertility in female rats at doses up to 180 mg/kg/day. However, increased post implantation loss and decreased embryo-fetal survival were observed at doses greater than or equal to 60 mg/kg/day (≥ 2 times the human therapeutic dose on a mg/m 2basis).
13.2 Animal Toxicology and/or Pharmacology
Histopathology findings in the absence of clinical signs in the central nervous system of the monkey (brain, spine) that included vascular mineralization, in addition to mineralization and necrosis of white matter were observed at greater than 750 mg/kg/day (4 times the human therapeutic systemic exposure based on area-under-the-plasma-concentration curve [AUC] comparisons) in a 52-week oral toxicity study using doses of 750 and 2000 mg/kg/d. Vacuolization of white matter was observed in rats dosed orally by gavage at greater than or equal to 180 mg/kg/d (6 times the human therapeutic exposure based on surface area comparisons, mg/m 2) in a 4-week study using doses of 180, 840, and 4200 mg/kg/d.
Vacuolization can sometimes occur as an artifact of tissue processing. Findings in dogs included tremor and absent corneal reflexes at 105 mg/kg/day (10 times the human therapeutic systemic exposure, based on body surface area comparisons, mg/m 2) after a 4-week oral gavage toxicity study using doses of 35, 70, 105, and 140 mg/kg/d. Ataxia, diminished/absent pupillary, palpebral, or patellar reflexes were observed in a dog at greater than or equal to 495 mg/kg/day (50 times the human therapeutic systemic exposure based on body surface area comparisons, mg/m2), in a 2-week oral gavage toxicity study using doses of 85, 165, 495, and 825 mg/kg/d.
Cataracts were observed in rats at greater than or equal to 180 mg/kg/day (4 times the human therapeutic systemic exposure, based on AUC) in a 52-week oral gavage toxicity study using doses of 180, 420, 840, and 1680 mg/kg/d.
Gastrointestinal necrosis, inflammation, and hemorrhage were observed in dogs at greater than or equal to 85 mg/kg/day (9 times the human therapeutic systemic exposure based on body surface area comparisons, mg/m 2) after a 2-week oral (capsule) toxicity study using doses of 85, 165, 495, and 825 mg/kg/d. Similar GI toxicity occurred in rats at 1200 mg/kg/day (7 times the human therapeutic systemic exposure, based on AUC) in a 26-week oral gavage toxicity study using doses of 300, 600, and 1200 mg/kg/d. In monkeys, similar GI toxicity occurred at greater than or equal to 750 mg/kg/day (6 times the human therapeutic systemic exposure based on AUC) following a 52-week oral gavage toxicity study using doses of 750 and 2000 mg/kg/d. -
14 CLINICAL STUDIES
The efficacy of miglustat capsules in type 1 Gaucher disease has been investigated in two open-label, uncontrolled trials and one randomized, open-label, active-controlled trial with enzyme replacement given as imiglucerase. Patients who received miglustat capsules were treated with doses ranging from 100 to 600 mg a day, although the majority of patients were maintained on doses between 200 to 300 mg a day. Efficacy parameters included the evaluation of liver and spleen organ volume, hemoglobin concentration, and platelet count. A total of 80 patients were exposed to miglustat capsules during the three trials and their extension period.
Open-Label Uncontrolled Monotherapy Trials
In Study 1, miglustat capsules was administered at a starting dose of 100 mg three times daily for 12 months (dose range of 100 once-daily to 200 mg three times daily) to 28 adult patients with type 1 Gaucher disease, who were unable to receive enzyme replacement therapy and who had not taken enzyme replacement therapy in the preceding 6 months. Twenty-two patients completed the trial. After 12 months of treatment, the results showed significant mean percent reductions from baseline in liver volume of 12% and spleen volume of 19%, a non-significant increase from baseline in mean absolute hemoglobin concentration of 0.26 g/dL and a mean absolute increase from baseline in platelet counts of 8 × 109/L (See Tables 3-6).
In Study 2, miglustat capsules was administered at a dose of 50 mg three times daily for 6 months to 18 adult patients with type 1 Gaucher disease who were unable to receive enzyme replacement therapy and who had not taken enzyme replacement therapy in the preceding 6 months. Seventeen patients completed the trial. After 6 months of treatment, the results showed significant mean percent reductions from baseline in liver volume of 6% and spleen volume of 5%. There was a non-significant mean absolute decrease from baseline in hemoglobin concentration of 0.13 g/dL and a non-significant mean absolute increase from baseline in platelet counts of 5 × 10 9/L (See Tables 3-6).Extension Period
Eighteen patients were enrolled in a 12-month extension to Study 1. A subset of patients continuing in the extension had larger mean baseline liver volumes, and lower mean baseline platelet counts and hemoglobin concentrations than the original study population (See Tables 3-6). After a total of 24 months of treatment, there were significant mean decreases from baseline in liver and spleen organ volumes of 15% and 27%, respectively, and significant mean absolute increases from baseline in hemoglobin concentration and platelet count of 0.9 g/dL and 14 × 10 9/L, respectively (See Tables 3-6).
Sixteen patients were enrolled in a 6-month extension to Study 2. After a total of 12 months of treatment, there was a mean decrease from baseline in spleen organ volume of 10%, whereas the mean percent decrease in liver organ volume remained at 6%. There were no significant changes in hemoglobin concentrations or platelet counts (See Tables 3-6).
Liver volume results from Studies 1 and 2 and their extensions are summarized in Table 3:
Table 3: Liver Volume Changes in Two Open-Label Uncontrolled Monotherapy Trials of miglustat capsules with Extension Period
Liver Volume
n
Absolute Mean (L)
(2-sided 95% CI)
Percent Mean (%)
(2-sided 95% CI)
Study 1 (starting dose miglustat capsules 100mg three times daily)
Baseline (Month 0)
21
2.39
Month 12 Change from baseline
-0.28 (-0.38, -0.18)
-12.1% (-16.4, 7.9)
Study 1 Extension Phase
Baseline (Month 0)
12
2.54
Month 24 Change from baseline
-0.36 (-0.48, -0.24)
-14.5% (-19.3, 9.7)
Study 2 (miglustat capsules 50mg three times daily)
Baseline (Month 0)
17
2.45
Month 6 Change from baseline
-0.14 (-0.25, -0.03)
-5.9% (-9.9, -1.9)
Study 2 Extension Phase
Baseline (Month 0)
13
2.35
Month 12 Change from baseline
-0.17 (-0.3, -0.0)
-6.2% (-12.0, -0.5)
Spleen volume results from Studies 1 and 2 and their extensions are summarized in Table 4:
Table 4: Spleen Volume Changes in Two Open-Label Uncontrolled Monotherapy Trials of miglustat capsules with Extension Period
Spleen Volume
n
Absolute Mean (L)
(2-sided 95% CI)
Percent Mean (%)
(2-sided 95% CI)
Study 1 (starting dose miglustat capsules 100mg three times daily)
Baseline (Month 0)
18
1.64
Month 12 Change from baseline
-0.32 (-0.42, -0.22)
-19.0% (-23.7, -14.3)
Study 1 Extension Phase
Baseline (Month 0)
10
1.56
Month 24 Change from baseline
-0.42 (-0.53, -0.30)
-26.4% (-30.4, -22.4)
Study 2 (miglustat capsules 50 mg three times daily)
Baseline (Month 0)
11
1.98
Month 6 Change from baseline
-0.09 (-0.18, -0.01)
-4.5% (-8.2, -0.7)
Study 2 Extension Phase
Baseline (Month 0)
9
1.98
Month 12 Change from baseline
-0.23 (-0.46, 0.00)
-10.1% (-20.1, -0.1)
Hemoglobin concentration results from Studies 1 and 2 and their extensions are summarized in Table 5:
Table 5: Hemoglobin Concentration Changes in Two Open-Label Uncontrolled Monotherapy Trials of miglustat capsules with Extension Period
Hemoglobin Concentration
n
Absolute Mean (g/dL)
(2-sided 95% CI)
Percent Mean (%)
(2-sided 95% CI)
Study 1 (starting dose miglustat capsules 100mg three times daily)
Baseline (Month 0)
22
11.94
Month 12 Change from baseline
0.26 (-0.05, 0.57)
2.6% (-0.5, 5.7)
Study 1 Extension Phase
Baseline (Month 0)
13
11.03
Month 24 Change from baseline
0.91 (0.30, 1.53)
9.1% (2.9, 15.2)
Study 2 (miglustat capsules 50 mg three times daily)
Baseline (Month 0)
17
11.60
Month 6 Change from baseline
-0.13 (-0.51, 0.24)
-1.3% (-4.4, 1.8)
Study 2 Extension Phase
Baseline (Month 0)
13
11.94
Month 12 Change from baseline
0.06 (-0.73, 0.85)
1.2% (-5.2, 7.7)
Platelet count results from Studies 1 and 2 and their extensions are summarized in Table 6:
Table 6: Platelet Count Changes in Two Open-Label Uncontrolled Monotherapy Trials of miglustat capsules with Extension Period
Platelet Count
n
Absolute Mean (10 9/L)
(2-sided 95% CI)
Percent Mean (%)
(2-sided 95% CI)
Study 1 (starting dose miglustat capsules 100mg three times daily)
Baseline (Month 0)
22
76.58
Month 12 Change from baseline
8.28 (1.88, 14.69)
16.0% (-0.8, 32.8)
Study 1 Extension Phase
Baseline (Month 0)
13
72.35
Month 24 Change from baseline
13.58 (7.72, 19.43)
26.1% (14.7, 37.5)
Study 2 (miglustat capsules 50 mg three times daily)
Baseline (Month 0)
17
116.47
Month 6 Change from baseline
5.35 (-6.31, 17.02)
2.0% (-6.9, 10.8)
Study 2 Extension Phase
Baseline (Month 0)
13
122.15
Month 12 Change from baseline
14.0 (-3.4, 31.4)
14.7% (-1.4, 30.7)
Open-Label Active-Controlled Trial
Study 3 was an open-label, randomized, active-controlled study of 36 adult patients with type 1 Gaucher disease, who had been receiving enzyme replacement therapy with imiglucerase for a minimum of 2 years prior to study entry. Patients were randomized 1:1:1 to one of three treatment groups, as follows:
miglustat capsules 100 mg three times daily alone
imiglucerase (patient‘s usual dose) alone
miglustat capsules 100 mg three times daily and imiglucerase (usual dose)
Patients were treated for 6 months, and 33 patients completed the study. Because miglustat capsules is only indicated as monotherapy, the results for the monotherapy arms are described below. At Month 6, the results showed a decrease in mean percent change in liver volume in the miglustat capsules treatment group compared to the imiglucerase alone group. There were no significant differences between the groups for mean absolute changes in liver and spleen volume and hemoglobin concentration. However, there was a significant difference between the miglustat capsules alone and imiglucerase alone groups in platelet counts at Month 6, with the miglustat capsules alone group having a mean absolute decrease in platelet count of 21.6 × 10 9/L and the imiglucerase alone group having a mean absolute increase in platelet count of 10.1 × 10 9/L (See Tables 7-10).Extension period
Twenty-nine patients were enrolled in a 6-month extension to Study 3. In the extension phase, all 29 patients had withdrawn from imiglucerase and received open-label miglustat capsules 100 mg three times daily monotherapy. At Month 12, the results showed non-significant decreases in platelet counts from baseline in all the treatment groups (by original randomization). There was a significant decrease in platelet counts from Month 6 to Month 12 in the group originally randomized to treatment with imiglucerase, and a continued decrease in platelet counts in the group originally randomized to miglustat capsules alone. There were no significant changes in any treatment group for liver volume, spleen volume, or hemoglobin concentration (See Tables 7-10).Liver volume results from Study 3 and extension are summarized in Table 7:
Table 7: Liver Volume Changes from Study 3 and Extension Phase
Imiglucerase Alone
miglustat capsules Alone
Study 3
n = 11
n = 10
Month 0
1.81
1.58
Month 6 Change (L)
0.04
-0.05
Month 6 % Change
3.6%
-2.9%
Adjusted Mean Difference from Imiglucerase (95% CI)
-4.5% (-13.2, 4.2)
Extension Phase*
n = 10
n = 8
Month 0
1.94
1.60
Month 12 Change (L)
-0.05
-0.01
Month 12 % Change
-0.7%
-0.8%
* All patients received miglustat capsules 100mg three times daily monotherapy from Month 6 to Month 12.
Spleen volume results from Study 3 and extension are summarized in Table 8:
Table 8: Spleen Volume Changes from Study 3 and Extension Phase
Imiglucerase Alone
miglustat capsules Alone
Study 3
n = 8
n = 7
Month 0
0.61
0.69
Month 6 Change (L)
-0.02
-0.03
Month 6 % Change
-2.1%
-4.8%
Adjusted % Difference from Imiglucerase (95% CI)
-5.8% (-22.1, 10.5)
Extension Phase*
n = 7
n = 6
Month 0
0.83
0.57
Month 12 Change (L)
0.04
-0.05
Month 12 % Change
1.5%
-6.1%
* All patients received miglustat capsules 100mg three times daily monotherapy from Month 6 to Month 12.
Hemoglobin concentration results from Study 3 and extension are summarized in Table 9:
Table 9: Hemoglobin Concentration Changes from Study 3 and Extension Phase
Imiglucerase Alone
miglustat capsules Alone
Study 3
n = 12
n = 10
Month 0
13.18
12.44
Month 6 Change g/dL
-0.15
-0.31
Month 6 % Change
-1.2%
-2.4%
Adjusted % Difference from Imiglucerase (95% CI)
-1.9% (-6.4, 2.6)
Extension Phase*
n = 10
n = 9
Month 0
13.39
12.46
Month 12 Change g/dL
-0.48
-0.13
Month 12 % Change
-3.1%
-1.1%
* All patients received miglustat capsules 100mg three times daily monotherapy from Month 6 to Month 12.
Platelet count results from Study 3 and extension are summarized in Table 10:
Table 10: Platelet Count Changes from Study 3 and Extension Phase
Imiglucerase Alone
miglustat capsules Alone
Study 3
n = 12
n = 10
Month 0
165.75
170.55
Month 6 Change (10 9/L)
15.29
-21.60
Month 6 % Change
10.1%
-9.6%
Adjusted % Difference from Imiglucerase (95% CI)
-17.1% (-32.9, -1.3)
Extension Phase*
n = 10
n = 9
Month 0
170.05
184.83
Month 12 Change (10 9/L)
-3.75
-27.39
Month 12 % Change
-3.2%
-10.4%
* All patients received miglustat capsules 100mg three times daily monotherapy from Month 6 to Month 12.
Patients with platelet counts above 150 × 10 9/L at baseline who were randomized to miglustat capsules treatment had significant decreases in platelet counts at Month 12.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
YARGESA is supplied in hard gelatin capsules containing 100 mg miglustat. YARGESA 100 mg capsules are white opaque with “709” printed in black on the body.
YARGESA are packed in blister cards.
Six blister cards of 15 capsules are supplied in each carton.
NDC: 42799-709-01: carton containing 90 capsules.
One blister card of 15 capsules is supplied in each carton.
NDC: 42799-709-15: carton containing 15 capsules.Storage:
Store at 20°C to 25°C (68°F to 77°F). Excursions are permitted between 15°C to 30°C (59°F to 86°F) (see USP Controlled Room Temperature).
Keep out of reach of children -
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information)
Information for Patients
Advise patients that the most common serious adverse reactions reported with YARGESA are peripheral neuropathy. Advise patients to promptly report any numbness, tingling, pain, or burning in the hands and feet [see Warnings and Precautions (5.1.)]
Advise patients that other adverse reactions include tremor and reductions in platelet counts. Advise patients to promptly report the development of tremor or worsening in an existing tremor. [see Warnings and Precautions ( 5.2, 5.4)]
Advise patients that other serious adverse reactions include diarrhea and weight loss. Advise patients to adhere to dietary instructions [see Warnings and Precautions (5.3)].
Advise patients to take the next YARGESA capsule at the next scheduled time if a dose is missed.
Inform patients of the potential risks and benefits of YARGESA and of alternative modes of therapy.
Pregnancy
Advise pregnant women and females of reproductive potential of the potential risk to a fetus, based on animal data. Advise patients who may become pregnant to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].Lactation
Advise women not to breastfeed if they are taking YARGESA [see Use in Specific Populations (8.2)].
Manufactured for:
Edenbridge Pharmaceuticals, LLCDBA Dexcel Pharma USA
Parsippany, NJ 07054
877-381-3336Issued: July 2024
-
Patient Information
YARGESA (yär ges uh)
(miglustat)
Capsules
Read this Patient Information before you start taking YARGESA and each time you get a refill. There may be new information.
What is YARGESA ?
YARGESA is a prescription medicine used alone to treat adults with mild to moderate type 1 Gaucher disease.YARGESA is used only in people who cannot be treated with enzyme replacement therapy.
It is not known if YARGESA is safe and effective in children under 18 years of age.
Before taking YARGESA , tell your healthcare provider about all of your medical conditions, including if you:
have kidney problems
are pregnant or plan to become pregnant. Yargesa may harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with YARGESA
are breastfeeding or plan to breastfeed. It is not known if YARGESA passes into your breast milk and may harm your baby. Do not breastfeed during treatment with YARGESA. Talk to your healthcare provider about the best way to feed your baby during treatment with YARGESA.Tell your Healthcare Provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. YARGESA may affect how other medicines work.
How should I take YARGESA ?
Take YARGESA exactly as your healthcare provider tells you to.
Take YARGESA at the same time each day.
If you miss a dose of YARGESA, skip that dose. Take the next miglustat capsule at the usual time.What are the possible side effects of YARGESA ?
YARGESA may cause serious side effects including:
Numbness, tingling, pain, or burning in your hands or feet (peripheral neuropathy). Call your healthcare provider right away if you get numbness, tingling, pain, or burning in your hands or feet.
Your healthcare provider may test your nerves (neurological exam) before you start YARGESA and during treatment with YARGESA.
New or worsening hand tremors (shaky movements).Tremors are common with YARGESA and may begin within the first month of starting treatment. Sometimes the tremors may go away between 1 to 3 months with continued treatment. Your healthcare provider may lower your dose or stop YARGESA if you develop new or worsening hand tremors. Call your healthcare provider right away if you get new hand tremors during treatment with YARGESA or if the hand tremors you already have get worse.
Diarrheais common with YARGESA and sometimes can be serious. Your healthcare provider may prescribe another medicine (anti-diarrheal) to treat diarrhea if it is a problem for you and may recommend changes to your diet, such as avoiding foods high in carbohydrates. Talk with your healthcare provider about your diet if you have diarrhea.
Weight lossis common with YARGESA and sometimes can be serious. You may lose weight when you start treatment with YARGESA.
Low platelet countis common with YARGESA and can be serious. Your healthcare provider may do blood tests to monitor your blood platelet count.
The most common side effects of YARGESA include:
weight loss
stomach pain
gas
nausea and vomiting
headache, including migraine
back pain
constipation
dry mouth
heaviness in arms and legs
memory loss
unsteady walking
leg cramps
dizziness
weakness
vision problems
muscle cramps
loss of appetite
indigestion
numbness, tingling, pain, or burning of your skin
stomach bloating
stomach pain not related to food
menstrual changesThese are not all the possible side effects of YARGESA. For more information, ask your healthcare provider or pharmacist.Call your doctor for medical advice about side effects.
You may report side effects to FDA at 1-800-FDA-1088.
How should I store YARGESA ?
Store YARGESA at room temperature between 68ºF to 77ºF (20ºC to 25ºC)
Keep YARGESA and all medicines out of the reach of children
General information about the safe and effective use of YARGESA .
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet.
Do not use YARGESA for a condition for which it was not prescribed. Do not give YARGESA to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about YARGESA that is written for health professionals.
What are the ingredients in YARGESA ?
Active ingredient : miglustat
Inactive ingredients:sodium starch glycolate (type A, potato), povidone (K-29/32), and magnesium stearate.
The capsule shell contains: gelatin and titanium dioxide; the edible printing ink contains black iron oxide, shellac,
and propylene glycol.This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured for:
Edenbridge Pharmaceuticals, LLCDBA Dexcel Pharma USA
Parsippany, NJ 07054
877-381-3336Revised: 07/2024
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
YARGESA
miglustat capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 42799-709 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MIGLUSTAT (UNII: ADN3S497AZ) (MIGLUSTAT - UNII:ADN3S497AZ) MIGLUSTAT 100 mg Inactive Ingredients Ingredient Name Strength SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) POVIDONE (UNII: FZ989GH94E) MAGNESIUM STEARATE (UNII: 70097M6I30) GELATIN (UNII: 2G86QN327L) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) SHELLAC (UNII: 46N107B71O) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) Product Characteristics Color white Score no score Shape CAPSULE Size 14mm Flavor Imprint Code 709 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 42799-709-01 6 in 1 CARTON 08/06/2020 08/06/2020 1 15 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC: 42799-709-15 1 in 1 CARTON 10/31/2023 2 15 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA209821 08/06/2020 Labeler - Edenbridge Pharmaceuticals LLC. (948715060)

Trademark Results [Yargesa]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 YARGESA 97221004 not registered Live/Pending |
Edenbridge Pharmaceuticals, LLC 2022-01-14 |
 YARGESA 87918216 not registered Live/Pending |
Edenbridge Pharmaceuticals, LLC 2018-05-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.